

Chromatin organization is critical to the regulation of most cellular activities. Direct three-dimensional visualization by electron cryotomography shows that budding yeast chromatin is best explained by a polymer melt interspersed with small nucleosome clusters, with no evidence of 30-nm fibers or condensed chromosomes in G1 or mitosis.
Abstract
Chromatin organization has an important role in the regulation of eukaryotic systems. Although recent studies have refined the three-dimensional models of chromatin organization with high resolution at the genome sequence level, little is known about how the most fundamental units of chromatin—nucleosomes—are positioned in three dimensions in vivo. Here we use electron cryotomography to study chromatin organization in the budding yeast Saccharomyces cerevisiae. Direct visualization of yeast nuclear densities shows no evidence of 30-nm fibers. Aside from preribosomes and spindle microtubules, few nuclear structures are larger than a tetranucleosome. Yeast chromatin does not form compact structures in interphase or mitosis and is consistent with being in an “open” configuration that is conducive to high levels of transcription. From our study and those of others, we propose that yeast can regulate its transcription using local nucleosome–nucleosome associations.
INTRODUCTION
Eukaryotic nuclear DNA is packaged to 1/10,000th of its contour length but must remain accessible to intranuclear machinery. The nucleosome is the first level of chromatin organization: 146 base pairs of double-stranded DNA wrap around a histone octamer that is composed of two copies each of histones H2A, H2B, H3, and H4 (Luger et al., 1997). Chromatin organization beyond the nucleosome has been intensively studied for nearly half a century. One noteworthy traditional electron microscopy (EM) study of purified chromatin proposed that sequential nucleosomes are arranged into compact ∼30-nm-diameter helical fibers (herein referred to as 30-nm fibers, even though the actual diameter is variable; Finch and Klug, 1976). Further studies proposed at least two broad classes of models of 30-nm fibers: the one-start solenoid (Robinson et al., 2006) and the two-start zigzag (Schalch et al., 2005; Song et al., 2014). In these 30-nm-fiber models, the nucleosomes pack so closely that the chromatin takes on the appearance of a discrete particle. The majority of these chromatin studies, however, were done in vitro at low ionic strength, making it unclear whether the resultant models reflect chromatin organization in the crowded, metabolically active interior of a cell's nucleus (Maeshima et al., 2010, 2016; Hansen, 2012).
Although traditional EM revealed the overall organization of purified chromatin (Olins and Olins, 1974; Finch et al., 1975), it has provided limited insights into chromatin structure in vivo because macromolecular structure is highly sensitive to sample preparation parameters: buffer conditions, chemical fixation, dehydration, and heavy metal staining (Dubochet et al., 1988; Maeshima et al., 2010). Recently high-throughput-sequencing–based chromatin-conformation capture (3C, 5C, Hi-C, etc.; herein abbreviated 3C) has been used as a complementary method to study chromatin structure in fixed cells (Dekker et al., 2013; Smallwood and Ren, 2013; Pombo and Dillon, 2015). These 3C approaches reveal the most probable pairwise chromatin contacts from a population of cells. The detected contacts are distance constraints that can be used to infer three-dimensional (3D) chromatin models. Single-celled 3C is also possible, but the number of detected contacts is so sparse that the resultant models are limited to larger higher-order structures such as topologically associating domains (Nagano et al., 2013). Owing to the dynamic nature of cells, 3C models are also susceptible to potential biases in nucleosome accessibility and fixation artifacts.
Electron cryomicroscopy (cryo-EM) permits the direct visualization of macromolecular densities in a near-native state. Furthermore, cryo-EM can provide relatively “noninvasive” windows onto how macromolecular complexes interact inside of organelles and cells. For example, cryo-EM studies of vitreous sections showed that in isolated chicken erythrocyte nuclei and partially lysed starfish and sea cucumber sperm, chromatin is condensed into 30-nm fibers (Woodcock, 1994; Scheffer et al., 2011). In contrast, studies of vitreous sections of intact HeLa and CHO cells did not reveal evidence of the 30-nm fibers (McDowall et al., 1986; Eltsov et al., 2008); instead, nucleosome densities were packed in an irregular state akin to the polymer-melt–like structure model (Maeshima et al., 2014b). Electron spectroscopic imaging of mouse cells also did not reveal any 30-nm fibers (Fussner et al., 2012).
Electron cryotomography (cryo-ET) makes it possible to address structural cell-biology problems at molecular resolution in three dimensions in a near-native state (Dubrovsky et al., 2015). Limitations imposed by electron scattering physics have nevertheless restricted the vast majority of such advances to bacteria, which are thin enough to be plunge-frozen and then imaged in toto (Pilhofer and Jensen, 2013). Cryo-ET studies of eukaryotes—most of which are much thicker than bacteria—require that intact cells be thinned in the frozen-hydrated state. This challenge can be surmounted by cryomicrotomy (Al-Amoudi et al., 2004). Using this approach, we previously showed that marine picoplankton chromatin is also organized like a polymer melt (Gan et al., 2013). Despite this growing body of evidence that the 30-nm fiber is not the predominant form of chromatin packing, most studies continue to assume that chromatin packs into 30-nm fibers in vivo. This confusion is also perpetuated because very few cryo-ET studies have been done on intact model eukaryotic cells.
The budding yeast Saccharomyces cerevisiae (herein referred to as yeast) is an important model system for chromatin studies. Fluorescence microscopy imaging of certain genomic loci (Bystricky et al., 2004) and high-resolution nucleosome-positioning studies (Brogaard et al., 2012) produced models of yeast chromatin that were consistent with 30-nm fibers. This conclusion is controversial because 3C studies did not detect any evidence of 30-nm fibers (Dekker, 2008; Hsieh et al., 2015). Although light microscopy– and high-throughput-sequencing–based approaches have produced important advances in our understanding of chromatin structure, no study has directly visualized nucleosomes within the crowded molecular environment of intact yeast.
To understand how chromatin organization might influence transcription, we directly visualized the nuclear densities of yeast in three dimensions using cryo-ET of vitreous sections. We controlled for sample preparation artifacts using known chromatin structures. Our analysis of cryotomograms of G1- and metaphase-arrested yeast did not uncover any evidence of 30-nm fibers. Instead, nucleosomes have an irregular organization and do not adopt any higher-order structures. Nucleosomes do frequently pack close enough to form small clusters. Given the low frequency of introns in yeast and the nucleosome occupancy data showing nucleosome depletion near the transcription start site (Lee et al., 2007), we propose that some of the small clusters of nucleosomes may in fact contain the coding regions of genes.
RESULTS
The 30-nm fibers are compact and stable
The diversity of model systems and cryo-EM techniques makes it challenging to understand the most reproducible structural features of chromatin. Chromatin inside cells cannot be effectively imaged by cryo-EM unless the cells are first thinned in cryogenic conditions; cryomicrotomy can produce such vitreous sections (Dubochet et al., 1988). To account for the effects of cryo-EM sample preparation, we needed a form of chromatin that is thin enough to visualize after either plunge-freezing or cryomicrotomy. We therefore used purified chicken erythrocyte oligonucleosomes, which can be stabilized as 30-nm fibers, as a positive control (Supplemental Figure S1). We stabilized the 30-nm fibers in dialysis buffer plus 2 mM Mg2+ (Widom, 1989), both with and without cryoprotectant, and then performed cryo-ET on these samples prepared either by plunge-freezing or cryomicrotomy (Figure 1 and Supplemental Figure S2). If the sample preparation disrupted the 30-nm fibers, we might expect to see either a beads-on-a-string or a zigzag motif such as when purified chromatin is suspended in a buffer that inhibits 30-nm-fiber formation (Bednar et al., 1998). Pairwise comparisons of the resultant cryotomograms lead to the following conclusions: 1) 30-nm fibers are recognizable as compact particles regardless of sample preparation technique; 2) these 30-nm fibers are so compact that it is difficult to distinguish individual nucleosome densities when the chromatin aggregates; and 3) in cryosections, 30-nm fibers remain intact and are compressed along the cutting direction, as expected. Having controlled for the technical aspects of cryo-ET samples, we next combined cryomicrotomy with automated cryo-ET to image many yeast cells to ensure that our observations were reproducible (Table 1 and Supplemental Table S1).
FIGURE 1:

The 30-nm fibers are compact and remain intact in cryosections. (A) Projection image of chicken erythrocyte 30-nm fibers plunge-frozen in dialysis buffer plus 2 mM Mg2+. (B) Tomographic slice (12 nm thick) of the position in A. Arrowheads indicate examples of 30-nm fibers. (C) Projection image of chicken erythrocyte 30-nm fibers plunge-frozen in the presence of dextran. Note that due to the low dose (∼2 electrons/Å2 per projection) and relatively small defocus (−6 μm), the 30-nm fibers are difficult to see. The dark, punctate densities are 10-nm gold fiducials. The curved dashed line marks the edge of the holey carbon support. (D) Tomographic slice (12 nm thick) of the position in C. Arrowheads indicate examples of 30-nm fibers. (E) Projection image of a frozen-hydrated section containing 30-nm fibers. Knife marks are thin linear features that are parallel to the cutting direction, as indicated by the black line. (F) Tomographic slice (12 nm thick) of the same area as in E. Arrowheads indicate examples of 30-nm fibers. The alternating light-dark background bands running from the lower left to upper right of C and D are crevasse artifacts, which are visible due to the proximity of the tomographic slice to the cryosection surface. Scale bars, 100 nm (black), 30 nm (white).
TABLE 1:
Summary of chromatin conformations observed.
| Sample | Treatment | Observations | Data | Notes |
|---|---|---|---|---|
| CEN + 2 mM Mg2+ | PF, no dextran | Disperse 30-nm fiber | Figure 1, Supplemental Figure S2 | + control |
| CEN + 2 mM Mg2+ | PF, dextran | Aggregates of 30-fibers | Figure 1, Supplemental Figure S2 | + control |
| CEN + 2 mM Mg2+ | CEMOVIS, dextran | Aggregates, compressed | Figure 1, Supplemental Figure S2 | + control |
| Wild-type cells | CEMOVIS, fixed, dextrana | Irregular chromatin | Figure 4, Supplemental Figure S4C | + control, in vivo |
| G1 cells | CEMOVIS, dextrana | Irregular chromatin | Figures 2–5, Supplemental Figures S4A and S5 | In vivo |
| Metaphase cells | CEMOVIS, dextrana | Irregular chromatin | Figures 2 and 3, Supplemental Figure S4B, Supplemental Movie S1 | In vivo |
CEMOVIS, cryo-EM of vitreous sections; CEN, chicken-erythrocyte nuclei chromatin; PF, plunge freezing.
aNote that the cell wall prevents dextran from entering, and so chromatin inside cells is not perturbed by dextran.
Chromatin does not have long-range order in yeast
In many eukaryotes, chromosomes undergo global reorganization from an “open” interphase state to a condensed mitotic state. Although it is controversial how much yeast mitotic chromatin condenses (see Discussion), yeast chromatin might form more 30-nm fibers in mitosis than in interphase. To test for this condensation, we arrested cells at both G1 and metaphase (Supplemental Figure S3) and then imaged them by cryo-ET of cryosections. Nucleosome-like densities were abundant inside the nuclei of both kinds of cells, but higher-order chromatin structures that resemble 30-nm fibers or highly compact arrays were absent (Figure 2, A–C and E, and Supplemental Movie S1; more examples are shown in Supplemental Figure S4, A and B). In fact, we did not see any assemblies of nucleosome-sized particles that have long-range order of any kind. Ribosome-like particles—most likely preribosomes (Tschochner and Hurt, 2003)—were also present in the nucleus (Figure 2, A, B, G, and H). In addition, in metaphase cells, spindle microtubules, which have a 25-nm diameter, could be seen inside the nucleus (Supplemental Figure S4B). Visualization of intranuclear macromolecular complexes of size comparable to 30-nm fibers further demonstrates that our data have enough contrast to reveal these structures. In summary, these data show that G1 and metaphase yeast chromatin does not have features consistent with 30-nm fibers or compact chromatin structures of any kind.
FIGURE 2:

Chromatin is not organized as 30-nm fibers in yeast. Tomographic slices (30 nm thick) of yeast nuclei in (A) G1 and (B) metaphase (M) cells. The nuclei (Nuc) and mitochondria (Mi) are labeled. Parallel white bars mark inner and outer nuclear membranes. Black arrowheads point to cytoplasmic ribosomes. Scale bars, 200 nm. (C, E) Enlargements (threefold) of the intranuclear positions enclosed by blue boxes in A and B, respectively. (D, F) Enlargements (threefold) of cytoplasmic ribosomes enclosed by red boxes in A and B, respectively. Scale bars, 30 nm. (G, H) Examples of intranuclear ribosome-sized densities boxed out (gray) from A and B, respectively, and enlarged sixfold. Scale bars, 10 nm. (I, J) Rotationally averaged power spectra of chromatin- and ribosome-rich positions from C and D, and E and F, respectively. Arrows point to 30- and 10-nm spacings.
Fourier analysis is a well-established method to detect the presence of densely packed regular particles like nucleosomes and 30-nm fibers (Eltsov et al., 2008; Scheffer et al., 2011). To detect and characterize any regular motifs that may be present, we performed Fourier analysis on positions within the nuclei (Figure 2, C and E). A broad peak at ∼10-nm spacing stood out in both G1 and metaphase cells (blue plots in Figure 2, I and J). This signal is expected from loosely packed nucleosomes, which are 6 nm thick and 11 nm in diameter (Joti et al., 2012; Nishino et al., 2012). In contrast, we did not observe a peak at ∼30-nm spacing, which would be expected of a nucleus enriched with 30-nm fibers (Scheffer et al., 2011). These observations were reproducible in all 19 of our cryosectioned yeast samples (Supplemental Table S1).
As an internal control, we analyzed the cytoplasms of both G1 and metaphase cells. Many of these positions are densely packed with ribosomes (Figure 2, D and F), which produced the expected broad peak at ∼30-nm spacing (red plots in Figure 2, I and J). To eliminate even the remotest possibility that the effects of microscope underfocus conditions caused us to miss the 30-nm fibers, we also acquired several tilt series much closer to focus (Supplemental Figure S5). These close-to-focus data did not show any evidence of 30-nm fibers.
The high contrast in our best tomograms allowed us to render the nuclear volumes as isosurfaces so that the nucleosome-like densities could be visualized in three dimensions (Figure 3). This rendering style enables the direct inspection for 3D arrangements of chromatin structures that we might otherwise have missed when inspecting two-dimensional tomographic slices. As expected, the isosurfaces confirmed the crowded and irregular nature of the yeast nuclear structures (Figure 3, B, C, F, and G). This crowdedness was even more evident when we increased the thickness to 70 nm (Figure 3, D and H). Our cryo-ET data therefore show that the vast majority of yeast chromatin does not organize as 30-nm fibers or any periodic higher-order structures in vivo.
FIGURE 3:

Yeast nuclei are crowded but do not have highly ordered chromatin complexes. Tomographic slices (10 nm thick) of nuclei in (A) G1 and (E) metaphase (M) cells. Scale bars, 200 nm. (B, F) Enlargements (threefold) of the intranuclear positions enclosed by boxes in A and E, respectively. (C, G) Isosurface rendering of a 10-nm-thick volume of the region in B and F, respectively. Note that some of the smaller densities are from tomographic slices just “above” and “below” the selected volume. (D, H) Isosurface rendering of a 70-nm-thick volume centered on the same region in B and F, respectively.
Fixed cells also have disorganized chromatin
A recent study used the new 3C variant called “Micro-C” to study chromatin structure in formaldehyde-fixed yeast cells (Hsieh et al., 2015). That study concluded that yeast chromatin does not form 30-nm fibers but instead packs into tetranucleosome clusters. Because 3C approaches are believed to capture native chromatin interactions, they could inform our observations if the formaldehyde fixation step does not seriously disrupt nuclear structure. We therefore tested whether fixed cells are significantly perturbed and if they have the proposed oligonucleosome structures. We fixed log-phase wild-type cells (US1363) in formaldehyde using published protocols (Hsieh et al., 2015) and then high-pressure froze, cryosectioned, imaged, and generated cryotomograms using the same conditions as for unfixed cells. Overall we did not see any gross distortions to cellular morphology except to the mitochondrial membranes (Figure 4). Macromolecular complexes such as ribosomes did not form aggregates either. Although the contrast was not as high as in unfixed cells, we could see that the nucleosome-like densities were still organized irregularly (Figure 4C). Some nucleosome-like densities were also close enough to form contacts (Figure 4D), but we did not see any highly compacted structures reminiscent of tetranucleosomes in crystals (Schalch et al., 2005) or in glutaraldehyde-fixed chromatin arrays (Song et al., 2014). These observations were reproducible in all of our fixed yeast samples (Supplemental Figure S4C).
FIGURE 4:

Fixation does not seriously perturb nuclear structure. (A) A tomographic slice (10 nm thick) of an unfixed US1363 G1-arrested cell. (B) A 15-fold enlargement of the nuclear densities boxed in A. (C) A tomographic slice (10 nm thick) of a formaldehyde-fixed US1363 cell. (D) A 15-fold enlargement of the nuclear densities boxed in in C. Arrows point to the mitochondrial membranes. Scale bars, 200 nm.
Local chromatin structure in yeast
If groups of nucleosomes form abundant, well-defined complexes, they must also appear frequently as clusters of intranuclear densities in our cryotomograms. This notion has been used in the study of 30-nm fibers in chicken erythrocytes (Scheffer et al., 2011), ATPases in mitochondria (Davies et al., 2011), and polysomes in Escherichia coli and neurons (Brandt et al., 2009, 2010). We could clearly see small clusters of nucleosome-like densities in many of our best cryotomograms. These clusters may, for example, be tetranucleosomes, albeit not as densely packed as in the aforementioned crystal and cryo-EM structures (Schalch et al., 2005; Robinson et al., 2006; Song et al., 2014). To locate these candidate oligonucleosome structures in a more objective and automated way, we searched using template matching (Figure 5A). Not knowing exactly how the nucleosomes would be positioned next to each other, we used as references simple clusters of spheres packed to ∼10-nm center-to-center distance (Figure 5B). Very few template-matching hits looked exactly like the reference models, further supporting our observation that yeast chromatin is irregular. Of the best-correlating hits, the positions of the nucleosome-like densities varied so much that further analysis by subtomogram averaging was not feasible (Figure 5B). Furthermore, fewer than ∼10 clusters of each “class” could be found in the search volume. If we extrapolated to the entire yeast nucleus, then there would be fewer than ∼1000 of each of these classes of oligonucleosome clusters. Although it was tempting to template match with other arrangements of closely packed spheres, many of these would yield overlapping hits because the clusters share similar motifs (e.g., two nucleosomes in a row). This analysis shows that although clusters of nucleosomes exist in yeast, they are arranged in many different configurations.
FIGURE 5:

Oligonucleosome-like densities are heterogeneous. (A) A tomographic slice (10 nm thick) of the interior of a G1 nucleus. The few template-matching hits of a candidate tetranucleosomes are circled, but only part of the density is within this 10-nm-thick volume. (B) Top, four different templates showing potential clustering of nucleosomes, enlarged sixfold relative to A. Bottom rows, examples of extracted and aligned template-matching hits (10-nm-thick tomographic slices), showing the heterogeneous nature of these particles. The missing wedge causes some of the densities to appear elongated along one direction.
DISCUSSION
Vitreous sections reveal details of in vivo chromatin organization
Our study reveals that although the yeast nucleus is crowded with macromolecular complexes, the vast majority of chromatin cannot be explained by a 30-nm-fiber model or anything that could be a densely packed chromatin structure. If 30-nm fibers were abundant, we would have detected a peak at ∼30-nm spacing in the rotationally averaged power spectra taken from the nuclei (Figure 2, I and J, and Supplemental Figures S4, A and B, and S5C). If chromatin formed rare 30-nm fibers, then we would have observed compact particles like those purified from chicken erythrocytes (Figure 1 and Supplemental Figure S2) instead of dispersed punctate, nucleosome-like densities. The absence of periodic chromatin structures in yeast is reminiscent of the picoplankton Ostreococcus tauri (Gan et al., 2013) and HeLa cells (Mahamid et al., 2016), the only other eukaryotes studied intact by cryo-ET. More cryo-ET studies of eukaryotes are needed to establish whether 30-nm fibers are the exception or the rule.
Yeast chromatin does not form condensed structures
Yeast has been studied extensively using EM of fixed and stained cells, but there has been no evidence of mitotic chromosome condensation, perhaps as a result of sample preparation artifacts (Winey et al., 1995; Robinow and Marak, 1966; O’Toole et al., 1999). In contrast, both fluorescence in situ hybridization and Lac-operator-array–tagging experiments have shown that distant sequences on the same chromosome move closer together during mitosis (Guacci et al., 1994; Vas et al., 2007). Cryo-EM of cryosections can reveal condensed chromosomes like those in metaphase HeLa cells because the local concentration of nucleosomes increases at positions corresponding to chromatids (Eltsov et al., 2008). In our cryotomograms of metaphase-arrested cells, we could not detect any condensed chromosomes. Condensation would have produced large zones in which the large, ribosome-like bodies are excluded, as in the exclusion of ribosomes from mitotic chromosomes in HeLa cells (Eltsov et al., 2008), or a large shift in the peaks of the rotationally averaged power spectra to smaller spacings in comparison to G1 cells. Therefore yeast undergoes mitotic condensation without increasing local nucleosome concentration. The simplest explanation is that yeast chromosomes condense by means of looping interactions, perhaps as proposed by a recent computational study (Cheng et al., 2015). Alternatively, the chromosomes may pack via hierarchical looping, analogous to “rope flaking,” as proposed from recent studies using electron microscopy–assisted nucleosome interaction capture plus modeling (Grigoryev et al., 2016). These looping-type models allow for chromosome condensation without the need for longitudinal compaction, which is the primary mechanism of 30-nm-fiber–based chromosome condensation.
The Micro-C approach was recently developed to probe the 3C “blind spot,” making it possible to detect chromatin structures corresponding to a few nucleosomes (Hsieh et al., 2015). This study also did not find evidence for 30-nm fibers in yeast. However, the mapping of Micro-C data onto a 3D chromatin model depends on some assumptions that have not yet been controlled for (Pombo and Dillon, 2015). Perhaps the most critical factor is how much the nucleus is perturbed by the fixation step. We have now shown that formaldehyde fixation used in 3C does not seriously perturb the nucleus. Therefore cryo-ET and 3C yield the same conclusion, that there is no evidence of 30-nm fibers or large-scale nucleosome assemblies in yeast in vivo. As cryo-ET and 3C further improve, we will gain more complementary insights into chromatin structure. Chromatin structural models of yeast can be further improved via the integration of superresolution fluorescence in situ hybridization (Boettiger et al., 2016) and fluorescence microscopies (Ricci et al., 2015).
The meaning of higher-order chromatin
The prevailing model of higher-order chromatin is based on a hierarchy of helices. Nucleosomes are packed as highly ordered ∼30-nm helical fibers (Figure 6A). These 30-nm fibers can then fold into ∼130-nm-thick coiled structures called chromonema fibers, which fold into yet-larger structures such as mitotic chromosomes (Belmont and Bruce, 1994). None of these structures has been observed in the two small eukaryotes studied intact by cryo-ET: S. cerevisiae and the picoplankton O. tauri. Furthermore, computer modeling has shown that S. cerevisiae chromosomes can pack in the nucleus without forming 30-nm fibers (Kimura et al., 2013). Although most of the nucleosome densities can be explained by polymer-melt–like chromatin (Figure 6B), some densities pack into smaller oligomers containing fewer than ∼5 nucleosomes. Some of these densities appear in a linear series, perhaps analogous to the face-to-face stacking motif seen in purified starfish sperm chromatin (Scheffer et al., 2012). We therefore propose that, at least in smaller eukaryotes, chromatin is regulated by “lower-order” structures. These lower-order structures are heterogeneous: chromatin organization in yeast cannot be explained by just a few nucleosome-packing motifs.
FIGURE 6:
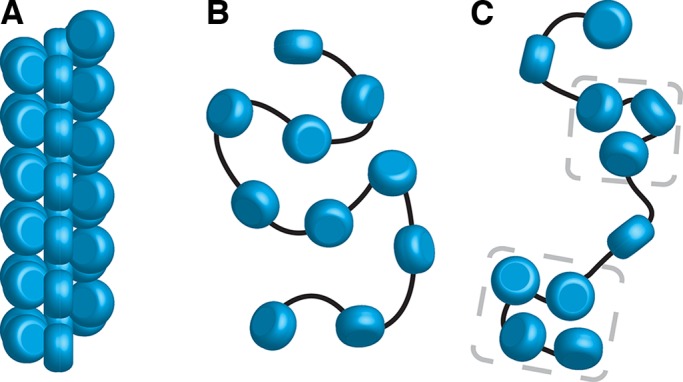
Yeast chromatin has a polymer melt–like structure with local nucleosome clustering. Three levels of chromatin organization: (A) regular 30-nm fiber, (B) disordered polymer melt–like chromatin, and (C) polymer melt interspersed with local nucleosome clusters (bracketed by gray dashes). Our cryo-ET data are more consistent with nucleosomes (blue cylinders) packing without long-range order (B, C). Black lines indicate how linker DNA might connect adjacent nucleosomes.
S. cerevisiae has an ultrahigh gene density and ultralow abundance of introns (Derelle et al., 2006), meaning that, on average, the coding region of each gene spans fewer than ∼10 nucleosomes. Micro-C reveals that the most probable interchromatin interactions are along the diagonal of the contact map (Hsieh et al., 2015), that is, between sequential nucleosomes. Nucleosome occupancy data show that eukaryotic genes have a nucleosome-depleted region at the transcription start site (Lee et al., 2007). Combination of these high-throughput-sequencing–based models and our direct imaging data suggests that the coding regions of genes can fold into oligonucleosome clusters. In the absence of 30-nm fibers, the overall nucleosomal arrangement is likely to be zigzag, punctuated by short stretches of extended linker DNA (Figure 6C).
Biological factors that regulate higher-order chromatin
Together with the previous reports, our study raises questions about the control of higher-order structure, at least in proliferating cells. Indeed, the only two cell types shown to have 30-nm fibers—by cryo-EM—are starfish sperm and chicken erythrocytes (Woodcock, 1994; Scheffer et al., 2011), both of which are terminally differentiated cells that have minimal transcriptional activity. In vitro, 30-nm fibers can be stabilized by changing the ionic environment (Maeshima et al., 2016). What factors, then, control 30-nm-fiber formation in vivo? Yeast chromatin is highly acetylated (Clarke et al., 1993), which would destabilize the critical ionic interaction between the histone H4 N-terminal tail and the acidic patch on adjacent nucleosomes (Shogren-Knaak et al., 2006). Hence the extent of acetylation may be important for modulating chromosome compaction. Another factor that may influence 30-nm fiber formation is that yeast has an unconventional linker histone compared with those in chickens (Harshman et al., 2013). It will therefore be valuable to image the chromatin of cells that have low levels of acetylation and proliferating cells that have conventional linker histones.
Biological consequences of nuclear architecture
The absence of 30-nm fibers and chromatin condensation in yeast leads to profound consequences because highly compact chromatin (Figure 6A) exposes less sequence to transcriptional machinery than loosely packed chromatin (Figure 6, B and C). Transcriptional repression would depend solely on either mononucleosomes or oligonucleosome clusters. DNA sequence accessibility would have to be increased by transient exposure of short sequences via nucleosome sliding or partial unwrapping. More sequence could be exposed by nucleosomal eviction, such as those found at steady state in nucleosome-depleted regions. Our data, in combination with 3C, suggest that yeast chromatin is best characterized as polymer-melt like, with small oligonucleosome clusters that do not pack into regular structures (Figure 6C). The local compaction of coding regions could be a mechanism that suppresses aberrant transcriptional initiation (Struhl, 2007).
MATERIALS AND METHODS
Yeast strains
The yeast strains used in this study are given in Table 2.
TABLE 2:
Yeast strains used in this work.
| Strain | Genotype | Source |
|---|---|---|
| US1363 | MATa bar1(unmarked) his3 leu2 trp1 ura3 | Surana lab |
| US1375 | MATa ura3 his3 cdc20Δ: LEU2 GAL-CDC20::TRP1 | Surana lab |
Chicken erythrocyte chromatin preparation
Fresh chicken blood was purchased from Nippon Bio-Test Laboratories (Tokyo, Japan). Erythrocyte nuclei were prepared as before (Langmore and Schutt, 1980; Maeshima et al., 2014a). Briefly, 1 ml of fresh chicken blood was lysed with 10 ml of MLB (60 mM KCl, 15 mM NaCl, 15 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.3, 2 mM MgCl2, 0.1% NP-40, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 10 min on ice. After centrifugation at 1000 × g at 4°C for 5 min, the supernatant was removed and resuspended in 10 ml of MLB. This step was repeated three times, and then samples were ready for chromatin purification. Chromatin purification was carried out as described by Ura and Kaneda (2001), with some modifications. The nuclei (equivalent to ∼2 mg of DNA) in nuclei isolation buffer (10 mM Tris-HCl, pH 7.5, 1.5 mM MgCl2, 1.0 mM CaCl2, 0.25 M sucrose, 0.1 mM PMSF) were digested with 50 U of micrococcal nuclease (Worthington, Lakewood, NJ) at 35°C for 2 min. The reaction was stopped by addition of ethylene glycol tetraacetic acid to 2 mM final concentration. After being washed with nuclei isolation buffer, the nuclei were lysed with lysis buffer (10 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.1 mM PMSF). The lysate was dialyzed against dialysis buffer (10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 0.1 mM PMSF) at 4°C overnight. The dialyzed lysate was centrifuged at 1000 × g at 4°C for 5 min. The supernatant was recovered and used as a purified chromatin fraction. The purity and integrity of the chromatin protein components were verified by SDS–PAGE (Supplemental Figure S1A). To examine average DNA length of the purified chromatin, DNA was isolated from the chromatin fraction and electrophoresed in 0.7% agarose gel (Supplemental Figure S1B).
S. cerevisiae cell culture
All strains were derived from W303. Wild-type strain US1363 was grown in yeast extract/peptone medium (YEP) supplemented with 2% glucose and 0.05% adenine (Lim et al., 1996). A 50-ml starter culture was shaken at 200 rpm in a 24°C water bath overnight. To arrest cells in G1, the culture was diluted to OD600 of ∼0.5, at which point α-factor was added to a final concentration of 0.8 μg/ml. After 3-h incubation at 24°C, the majority of cells were synchronized at G1 as confirmed by their “shmoo” morphology (Yeong et al., 2000).
To enrich for metaphase cells, the cdc20Δ GAL-CDC20 strain US1375 (Liang et al., 2012) was incubated in YEP supplemented with 2% raffinose and 2% galactose in a 24°C water bath overnight. On the next day, the cells were synchronized in G1 with α-factor (final concentration, 5 μg/ml). Then the culture was filtered and washed free of α-factor with two volumes of YEP medium and subsequently incubated in fresh YEP supplemented with 2% glucose to inhibit CDC20. After 3.5 h, the majority of cells were arrested with short spindles and undivided nuclei, confirming that the cells were synchronized at metaphase.
Fluorescence microscopy
Microtubules were stained as before (Lim et al., 1996): S. cerevisiae was collected by centrifuge at 13,000 rpm (15,871 × g) for 1 min and fixed in 1 ml of 0.1 M K2HPO4, pH 6.4, and 3.7% formaldehyde at 4°C overnight. Cells were then washed and resuspended in 0.2 ml of 1.2 M sorb/phos/cit (1.2 M sorbitol, 0.1 M phosphate-citrate, pH 5.9). Next the cells were spheroplasted with 20 μl of glusulase (glucuronidase, >90,000 U/ml, and sulfatase, >10,000 U/ml) and 2 μl of 10 mg/ml lyticase at 30°C for 75 min. Then the cells were washed and resuspended in 1.2 M sorb/phos/cit. A 4-μl sample was added to a 30-well slide pretreated with 0.1% poly-l-lysine (Sigma-Aldrich, St. Louis, MO). Tubulin was stained with the rat monoclonal anti–α-tubulin YOL1/34 primary antibody (MCA78G; AbD Serotec, Bio-Rad, Hercules, CA) and Alexa Fluor 594–conjugated goat anti-rat immunoglobulin G secondary antibody (A11007; Invitrogen, Thermo Fisher Scientific, Waltham, MA). DNA was counterstained with Vectashield-DAPI (Vector Laboratories, Burlingame, CA). The cells were imaged using a Zeiss AxioImager (Zeiss, Jena, Germany) upright motorized microscope with Plan Apochromat 100× objective equipped with EXFO 120W metal-halide illuminator. Images were recorded on a Photometrics CoolSNAP HQ2 (Photometrics, Tucson, AZ) charge-coupled device camera controlled by MetaMorph version 7.7.10.0 software (Molecular Devices, Sunnyvale, CA).
High-pressure freezing
Yeast pellet or purified chicken erythrocyte chromatin sample was mixed with 40-kDa dextran (Sigma-Aldrich) to a final concentration of 20% as an extracellular cryoprotectant. The sample/dextran mixture was loaded into a copper tube (0.45-mm outer diameter, 0.3-mm inner diameter) by using a syringe-type filler device (Part 733-1; Engineering Office M. Wohlwend, Sennwald, Switzerland). The tube was sealed at one end and high-pressure frozen using an HPF Compact 01 machine (Engineering Office M. Wohlwend). Once frozen, the tube was stored in liquid nitrogen.
Preparation of fixed yeast for vitreous sections
Wild-type yeast (US1363, 50 ml) were grown to OD600 of 0.36 in a shaker at 200 rpm at 24°C. The cells were then fixed by addition of 4.41 ml of 37% formaldehyde (final concentration, 3%) and incubated with shaking at 200 rpm for 15 min at 30°C. Cells were collected by centrifugation at 4600 rpm (1987 × g) for 2 min at 4°C. The supernatant was removed, and the cells were resuspended in 1 ml of YEP/dextrose (YEPD). The cells were washed a second time by centrifugation at 4600 rpm (1987 × g) for 2 min. Supernatant was removed, and dextran (in YEPD medium) was added to a final concentration of 20% as extracellular cryoprotectant. The cells were then quick-spun to 3000 rpm (845 × g) to remove bubbles and then immediately high-pressure frozen as described.
Vitreous sectioning
Vitreous sections were cut using the strategy proposed by Ladinsky (2010). Frozen-hydrated samples were cut using a Leica UC7/FC7 cryo-ultramicrotome (Leica Microsystems, Vienna, Austria) at −150°C. First a 100 × 100 × 30–μm mesa-shaped block was trimmed using a Trimtool 20 diamond blade (Diatome, Hatfield, PA). Sections were then cut using a Cryo 25° diamond knife (Diatome). The nominal thickness was set to 50–100 nm and cutting speed to 1 mm/s. A customized micromanipulator (MN-151S, model EDMS12-260; Narishige, Tokyo, Japan) was used to control the cryoribbon. A Crion electrostatic charging device was operated in discharge mode to prevent the sections from sticking to the diamond blade (Pierson et al., 2010). Once the ribbon was 2–3 mm long, it was transferred onto an EM grid (Protochips C-flat [Protochips, Morrisville, NC] or EMS continuous carbon grid, CF-200-CU-50 [EMS, Hatfield, PA]) to which 10-nm gold fiducials (EM.GC10; BBI Solutions, Cardiff, UK) were already added. This transfer was initiated by operating the Crion in charge mode. Subsequently, the ribbon was physically pressed with a 10-mm laser window glass (65-855; Edmund Optics, Barrington, NJ) to ensure firm attachment. The grid was stored in liquid nitrogen until imaging.
Plunge freezing
Plunge freezing was done using a Vitrobot MARK IV (FEI, Thermo Fisher Scientific, Waltham, MA) operated at 20°C with 100% humidity. Purified chromatin sample was mixed with 10-nm gold fiducials (BBI), and 3 μl of this mixture was applied onto freshly glow-discharged EM grids (C-flat CF-22-2C-T; Protochips). The grid was blotted once and then plunged into a liquid propane–ethane mixture (Tivol et al., 2008).
Electron cryotomography
Tilt series were collected using Leginon (Suloway et al., 2009) or FEI TOMO 3 & 4 on a Titan Krios cryo-TEM (FEI) operating at 300 keV. Tomography data were recorded on either Falcon I or Falcon II direct detection cameras. Imaging parameters for each sample type are listed in Supplemental Table S2. Image alignment, contrast transfer function correction, 3D reconstruction, and visualization were done using the IMOD software package (Kremer et al., 1996; Mastronarde, 1997; Xiong et al., 2009). Default settings were used, except that the low-pass-filter cutoff was set to 0.3.
Fourier analysis
Tomographic slices were imported into ImageJ, 1.49v (Schneider et al., 2012). The Fourier transform was calculated using the FFT function. The radial plot of the Fourier transform was generated using the Radial Profile Angle plug-in (rsbweb.nih.gov/ij/plugins/radial-profile-ext.html). The plot values (radius and normalized intensities) were saved. Radius values in inverse pixels were converted into real-space values based on the pixel size at the specimen level. The plot was generated using Excel (version 14.1.0; Microsoft, Redmond, WA) and saved as an image.
Three-dimensional density visualization
Isosurface rendering was done with the UCSF Chimera package (Pettersen et al., 2004). Subtomograms were normalized to a mean of 0 and SD of 1 using EMAN2 (Tang et al., 2007), and the contour level was set to 1.5σ. Isosurface densities <6 nm were removed using the “hide dust” function.
Template matching
Template matching was done using Particle Estimation for Electron Tomography (PEET), which accounts for the tomographic missing wedge (Nicastro et al., 2006; Heumann, 2016). Oligonucleosome reference models were generated using Bsoft (Heymann and Belnap, 2007). To minimize the effects of adjacent densities in the highly crowded intranuclear environment, we applied either a squat or an elongated cylindrical mask (depending on the aspect ratio of the reference) around the oligonucleosome reference. Overlapping hits were automatically subjected to duplicate removal at the end of template matching. The top 10% of hits were visually inspected to remove the remaining spurious hits.
Figures and media
All figures were composed in Adobe Photoshop or Illustrator; Supplemental Movie S1 was assembled and rendered with Adobe Premiere Pro (Adobe Systems, San Jose, CA).
Data sharing
An example tomogram (corresponding to Figure 2B) has been deposited at the Electron Microscopy Data Bank (EMD-8157). Tilt series (raw) data and IMOD reconstruction parameters of all samples presented in this article have been made publicly accessible in the EMPIAR online database (EMPIAR-10062; Iudin et al., 2016). Details of the corresponding figure and sample are summarized in Supplemental Table S1.
Supplementary Material
Acknowledgments
We thank the Centre for BioImaging Sciences Cryo-EM staff for support and especially G. L. Loy for microtomy training; Mark Ladinsky for cryomicrotomy advice; and Benoît Zuber, Danny Studer, and Dmitri Vanhecke for cryomicrotomy training at the Institute of Anatomy of the University of Bern. We thank John Heumann and David Mastronarde for advice on PEET and IMOD and Matthijn Vos for Titan Krios training. We thank our Centre for BioImaging Sciences colleagues and the Gan group for feedback on the manuscript. C.C. and L.G. were supported by National University of Singapore Startup Grants R-154-000-515-133, R-154-000-524-651, and D-E12-303-154-217, with equipment support from National University of Singapore Grants YIA R-154-000-558-133 and MOE T2 R-154-000-624-112. H.H.L. and U.S. were funded by the Biomedical Research Council of A*STAR (Agency of Science Technology and Research), Singapore. S.T. and K.M. were supported by a Japan Science and Technology Agency CREST grant and a Ministry of Education, Culture, Sports, Science and Technology KAKENHI grant (23115005).
Abbreviations used:
- 3C
chromatin conformation capture
- CEMOVIS
cryo-EM of vitreous sections
- CEN
chicken-erythrocyte nuclei chromatin
- Cryo-EM
electron cryomicroscopy
- Cryo-ET
electron cryotomography.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0506) on September 7, 2016.
REFERENCES
- Al-Amoudi A, Norlen LP, Dubochet J. Cryo-electron microscopy of vitreous sections of native biological cells and tissues. J Struct Biol. 2004;148:131–135. doi: 10.1016/j.jsb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL. Nucleosomes, linker DNA, linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci USA. 1998;95:14173–14178. doi: 10.1073/pnas.95.24.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont AS, Bruce K. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J Cell Biol. 1994;127:287–302. doi: 10.1083/jcb.127.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529:418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt F, Carlson L-A, Hartl FU, Baumeister W, Grünewald K. The three-dimensional organization of polyribosomes in intact human cells. Mol Cell. 2010;39:560–569. doi: 10.1016/j.molcel.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Brandt F, Etchells SA, Ortiz JO, Elcock AH, Hartl FU, Baumeister W. The native 3D organization of bacterial polysomes. Cell. 2009;136:261–271. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang J-P, Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystricky K, Heun P, Gehlen L, Langowski J, Gasser SM. Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. Proc Natl Acad Sci USA. 2004;101:16495–16500. doi: 10.1073/pnas.0402766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TM, Heeger S, Chaleil RA, Matthews N, Stewart A, Wright J, Lim C, Bates PA, Uhlmann F. A simple biophysical model emulates budding yeast chromosome condensation. Elife. 2015;4:e05565. doi: 10.7554/eLife.05565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DJ, O’Neill LP, Turner BM. Selective use of H4 acetylation sites in the yeast Saccharomyces cerevisiae. Biochem J. 1993;294:557–561. doi: 10.1042/bj2940557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kühlbrandt W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA. 2011;108:14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. Mapping in vivo chromatin interactions in yeast suggests an extended chromatin fiber with regional variation in compaction. J Biol Chem. 2008;283:34532–34540. doi: 10.1074/jbc.M806479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derelle E, Ferraz C, Rombauts S, Rouzé P, Worden AZ, Robbens S, Partensky F, Degroeve S, Echeynié S, Cooke R. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci USA. 2006;103:11647–11652. doi: 10.1073/pnas.0604795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Dubrovsky A, Sorrentino S, Harapin J, Sapra KT, Medalia O. Developments in cryo-electron tomography for in situ structural analysis. Arch Biochem Biophys. 2015;581:78–85. doi: 10.1016/j.abb.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Eltsov M, MacLellan KM, Maeshima K, Frangakis AS, Dubochet J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc Natl Acad Sci USA. 2008;105:19732–19737. doi: 10.1073/pnas.0810057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch JT, Klug A. Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci USA. 1976;73:1897–1901. doi: 10.1073/pnas.73.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch JT, Noll M, Kornberg RD. Electron microscopy of defined lengths of chromatin. Proc Natl Acad Sci USA. 1975;72:3320–3322. doi: 10.1073/pnas.72.9.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussner E, Strauss M, Djuric U, Li R, Ahmed K, Hart M, Ellis J, Bazett-Jones DP. Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 2012;13:992–996. doi: 10.1038/embor.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Ladinsky MS, Jensen GJ. Chromatin in a marine picoeukaryote is a disordered assemblage of nucleosomes. Chromosoma. 2013;122:377–386. doi: 10.1007/s00412-013-0423-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev SA, Bascom G, Buckwalter JM, Schubert MB, Woodcock CL, Schlick T. Hierarchical looping of zigzag nucleosome chains in metaphase chromosomes. Proc Natl Acad Sci USA. 2016;113:1238–1243. doi: 10.1073/pnas.1518280113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V, Hogan E, Koshland D. Chromosome condensation and sister chromatid pairing in budding yeast. J Cell Biol. 1994;125:517–530. doi: 10.1083/jcb.125.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JC. Human mitotic chromosome structure: what happened to the 30-nm fibre. EMBO J. 2012;31:1621–1623. doi: 10.1038/emboj.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshman SW, Young NL, Parthun MR, Freitas MA. H1 histones: current perspectives and challenges. Nucleic Acids Res. 2013;41:9593–9609. doi: 10.1093/nar/gkt700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumann JM. Particle Estimation for Electron Tomography (PEET) 2016. University of Colorado Boulder. http://bio3d.colorado.edu/PEET/ (accessed 18 January 2016)
- Heymann JB, Belnap DM. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Iudin A, Korir PK, Salavert-Torres J, Kleywegt GJ, Patwardhan A. EMPIAR: a public archive for raw electron microscopy image data. Nat Methods. 2016;13:387–388. doi: 10.1038/nmeth.3806. [DOI] [PubMed] [Google Scholar]
- Joti Y, Hikima T, Nishino Y, Kamada F, Hihara S, Takata H, Ishikawa T, Maeshima K. Chromosomes without a 30-nm chromatin fiber. Nucleus. 2012;3:404–410. doi: 10.4161/nucl.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Shimooka Y, Nishikawa J, Miura O, Sugiyama S, Yamada S, Ohyama T. The genome folding mechanism in yeast. J Biochem. 2013;154:137–147. doi: 10.1093/jb/mvt033. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Ladinsky MS. Micromanipulator-assisted vitreous cryosectioning and sample preparation by high-pressure freezing. Methods Enzymol. 2010;481:165–194. doi: 10.1016/S0076-6879(10)81008-0. [DOI] [PubMed] [Google Scholar]
- Langmore JP, Schutt C. The higher order structure of chicken erythrocyte chromosomes in vivo. Nature. 1980;288:620–622. doi: 10.1038/288620a0. [DOI] [PubMed] [Google Scholar]
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- Liang H, Lim HH, Venkitaraman A, Surana U. Cdk1 promotes kinetochore bi‐orientation and regulates Cdc20 expression during recovery from spindle checkpoint arrest. EMBO J. 2012;31:403–416. doi: 10.1038/emboj.2011.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Goh P-Y, Surana U. Spindle pole body separation in Saccharomyces cerevisiae requires dephosphorylation of the tyrosine 19 residue of Cdc28. Mol Cell Biol. 1996;16:6385–6397. doi: 10.1128/mcb.16.11.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Hihara S, Eltsov M. Chromatin structure: does the 30-nm fibre exist in vivo. Curr Opin Cell Biol. 2010;22:291–297. doi: 10.1016/j.ceb.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Imai R, Hikima T, Joti Y. Chromatin structure revealed by X-ray scattering analysis and computational modeling. Methods. 2014a;70:154–161. doi: 10.1016/j.ymeth.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Imai R, Tamura S, Nozaki T. Chromatin as dynamic 10-nm fibers. Chromosoma. 2014b;123:225–237. doi: 10.1007/s00412-014-0460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K, Rogge R, Tamura S, Joti Y, Hikima T, Szerlong H, Krause C, Herman J, Seidel E, DeLuca J, et al. Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J. 2016;35:1115–1132. doi: 10.15252/embj.201592660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Cuellar LK, Forster F, Hyman AA, Plitzko JM, Baumeister W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 2016;351:969–972. doi: 10.1126/science.aad8857. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol. 1997;120:343–352. doi: 10.1006/jsbi.1997.3919. [DOI] [PubMed] [Google Scholar]
- McDowall AW, Smith JM, Dubochet J. Cryo-electron microscopy of vitrified chromosomes in situ. EMBO J. 1986;5:1395–1402. doi: 10.1002/j.1460-2075.1986.tb04373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313:944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- Nishino Y, Eltsov M, Joti Y, Ito K, Takata H, Takahashi Y, Hihara S, Frangakis AS, Imamoto N, Ishikawa T, Maeshima K. Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J. 2012;31:1644–1653. doi: 10.1038/emboj.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olins AL, Olins DE. Spheroid chromatin units (v bodies) Science. 1974;183:330–332. doi: 10.1126/science.183.4122.330. [DOI] [PubMed] [Google Scholar]
- O’Toole ET, Winey M, McIntosh JR. High-voltage electron tomography of spindle pole bodies and early mitotic spindles in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:2017–2031. doi: 10.1091/mbc.10.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pierson J, Fernández JJ, Bos E, Amini S, Gnaegi H, Vos M, Bel B, Adolfsen F, Carrascosa JL, Peters PJ. Improving the technique of vitreous cryo-sectioning for cryo-electron tomography: electrostatic charging for section attachment and implementation of an anti-contamination glove box. J Struct Biol. 2010;169:219–225. doi: 10.1016/j.jsb.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Pilhofer M, Jensen GJ. The bacterial cytoskeleton: more than twisted filaments. Curr Opin Cell Biol. 2013;25:125–133. doi: 10.1016/j.ceb.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–257. doi: 10.1038/nrm3965. [DOI] [PubMed] [Google Scholar]
- Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M, Cosma MP. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160:1145–1158. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- Robinow CF, Marak J. A fiber apparatus in the nucleus of the yeast cell. J Cell Biol. 1966;29:129–151. doi: 10.1083/jcb.29.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PJ, Fairall L, Huynh VA, Rhodes D. EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci USA. 2006;103:6506–6511. doi: 10.1073/pnas.0601212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- Scheffer MP, Eltsov M, Bednar J, Frangakis AS. Nucleosomes stacked with aligned dyad axes are found in native compact chromatin in vitro. J Struct Biol. 2012;178:207–214. doi: 10.1016/j.jsb.2011.11.020. [DOI] [PubMed] [Google Scholar]
- Scheffer MP, Eltsov M, Frangakis AS. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc Natl Acad Sci USA. 2011;108:16992–16997. doi: 10.1073/pnas.1108268108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Smallwood A, Ren B. Genome organization and long-range regulation of gene expression by enhancers. Curr Opin Cell Biol. 2013;25:387–394. doi: 10.1016/j.ceb.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu R-M, Zhu P, Li G. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014;344:376–380. doi: 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007;14:103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- Suloway C, Shi J, Cheng A, Pulokas J, Carragher B, Potter CS, Zheng SQ, Agard DA, Jensen GJ. Fully automated, sequential tilt-series acquisition with Leginon. J Struct Biol. 2009;167:11–18. doi: 10.1016/j.jsb.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Tivol WF, Briegel A, Jensen GJ. An improved cryogen for plunge freezing. Microsc Microanal. 2008;14:375–379. doi: 10.1017/S1431927608080781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschochner H, Hurt E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 2003;13:255–263. doi: 10.1016/s0962-8924(03)00054-0. [DOI] [PubMed] [Google Scholar]
- Ura K, Kaneda Y. Reconstitution of chromatin in vitro. Methods Mol Biol. 2001;181:309–325. doi: 10.1385/1-59259-211-2:309. [DOI] [PubMed] [Google Scholar]
- Vas AC, Andrews CA, Kirkland Matesky K, Clarke DJ. In vivo analysis of chromosome condensation in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:557–568. doi: 10.1091/mbc.E06-05-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T-HS, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell. 2015;162:108–119. doi: 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom J. Toward a unified model of chromatin folding. Annu Rev Biophys Biophys Chem. 1989;18:365–395. doi: 10.1146/annurev.bb.18.060189.002053. [DOI] [PubMed] [Google Scholar]
- Winey M, Mamay CL, O’Toole ET, Mastronarde DN, Giddings TH, Jr, McDonald KL, McIntosh JR. Three-dimensional ultrastructural analysis of the Saccharomyces cerevisiae mitotic spindle. J Cell Biol. 1995;129:1601–1615. doi: 10.1083/jcb.129.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock CL. Chromatin fibers observed in situ in frozen hydrated sections. Native fiber diameter is not correlated with nucleosome repeat length. J Cell Biol. 1994;125:11–19. doi: 10.1083/jcb.125.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN. CTF determination and correction for low dose tomographic tilt series. J Struct Biol. 2009;168:378–387. doi: 10.1016/j.jsb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong FM, Lim HH, Padmashree CG, Surana U. Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28-Clb2 mitotic kinase and the role of Cdc20. Mol Cell. 2000;5:501–511. doi: 10.1016/s1097-2765(00)80444-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.