

Summary
Objectives
Disease comorbidity is a pervasive phenomenon impacting patients’ health outcomes, disease management, and clinical decisions. This review presents past, current and future research directions leveraging both phenotypic and molecular information to uncover disease similarity underpinning the biology and etiology of disease comorbidity.
Methods
We retrieved ~130 publications and retained 59, ranging from 2006 to 2015, that comprise a minimum number of five diseases and at least one type of biomolecule. We surveyed their methods, disease similarity metrics, and calculation of comorbidities in the electronic health records, if present.
Results
Among the surveyed studies, 44% generated or validated disease similarity metrics in context of comorbidity, with 60% being published in the last two years. As inputs, 87% of studies utilized intragenic loci and proteins while 13% employed RNA (mRNA, LncRNA or miRNA). Network modeling was predominantly used (35%) followed by statistics (28%) to impute similarity between these biomolecules and diseases. Studies with large numbers of biomolecules and diseases used network models or naïve overlap of disease-molecule associations, while machine learning, statistics, and information retrieval were utilized in smaller and moderate sized studies. Multiscale computations comprising shared function, network topology, and phenotypes were performed exclusively on proteins.
Conclusion
This review highlighted the growing methods for identifying the molecular mechanisms underpinning comorbidities that leverage multiscale molecular information and patterns from electronic health records. The survey unveiled that intergenic polymorphisms have been overlooked for similarity imputation compared to their intragenic counterparts, offering new opportunities to bridge the mechanistic and similarity gaps of comorbidity.
Keywords: Comorbidity, disease similarity, electronic health records, phenome, biomolecules, function, omics, modularity, integrative data analysis
1 Introduction
The last century has witnessed some of the greatest medical, scientific, and technological advances, substantially increasing life expectancy by 40% through better health care and prevention [1]. Global public health has been instrumental in reducing neonatal and transmissible disease mortality [1, 2]. However, in this common age of industrialization and urbanization, living longer increases the risk of developing non-communicable diseases, often chronic, complex, and comorbid [[3-5]. Disease comorbidity is becoming widely pervasive counting for 35% to 80% of case reports among 20 to 75 year-old patients [3, 6]. Although a growing body of research studies used associative analysis to identify risk factors of disease comorbidity, approaches leveraging both phenotypic and molecular patient information to understand the biological underpinnings of comorbidity remain elusive.
Disease comorbidity includes illnesses that mutually or gradually occur during one’s lifetime [3]. In the United States, with over 1.7 million sampled electronic health records, 42.2% of patients have been reported with at least one other morbidity [7]. Among individuals living with multiple diseases, there are those with two or more co-existing diseases (e.g. diabetes, Alzheimer’s, and cancer) and those with associated diseases developed as secondary conditions (e.g., diabetics with hypertension or retinopathy) [3, 8]. Further, disease comorbidity can be deleterious or protective [9-11], for example, immune-mediated inflammatory disorders increase the odds of infections and developing lymphoma, [12] while solid tumors in Down Syndrome patients lower the risk of developing leukemia and testicular cancer (inverse comorbidity) [11].
The sequence of events and the type of diseases developed during the course of time impact disease comorbidity management [3, 13]. Furthermore, disease comorbidity can limit the use of standard therapeutic options and medical interventions, e.g., a) the use of oral corticosteroids to treat patients with chronic obstructive pulmonary disease (COPD) patients and diabetes mellitus [14], b) the use of beta-blockers to treat asthmatic patients who are hypertensive [13], or c) resecting malignant lung tumors in patients with severe COPD [15]. Often, cancer patients with comorbidity found their survival outcomes compromised with a 5-year mortality hazard ratio between 1.1 and 5.8 years due to suboptimal or non-adequate chemotherapeutics [16, 17]. Different risk factors of comorbidity have been identified, such as frailty due to aging, drug side effects, and poor socio-economic status [3, 6, 18]. However, epidemiology alone has been insufficient to understand fully the causes of developing multiple diseases and inform on adequate healthcare solutions.
In the last decade, the biological underpinning of disease comorbidity is coming into focus to complement epidemiology studies and bridge the mechanistic gap between phenotypes and genotypes. Increasingly, studies are looking at disease overlap and similarity based on disease-shared cellular and molecular mechanisms [6, 10, 19-23] such as shared disease genes, genetic variants, associated proteins [24, 25], or biological pathways, [26] etc. The probability of developing a secondary disease is about 3-fold and 1.8-fold if the primary disease shares the same genes or metabolic fluxes, respectively [24, 27]. Biologically informed disease similarity metrics have been developed using computational and statistical methods ranging from simple overlap and machine learning to network-based methods. Goh et al. and others constructed a disease network where diseases are connected given their associated molecular modules [19, 21, 28-30] including both at the topological (e.g. distance in protein-protein interactions (PPI) [31]) and functional levels (e.g. enzyme functions or apoptosis [19, 28]. The overlaps of disease modules can be significantly enriched with disease genetic variants [32, 33] supportive of shared pathogenesis between diseases and their comorbidity [19, 24, 27, 34].
In this review, we primarily focused on studies that incorporated both detailed phenotypes and biomolecular information to uncover disease similarity and understand the underpinning of comorbidity.
2 Methods
We retrieved publications using combinations of keywords queried in PubMed and Google database search engines such as comorbidity, phenotypic information, electronic health records (EHR), patient heath records (PHR), diseasome, genome, gene expression, data integration, computational methods, gene ontology, disease-disease similarity, disease network, shared mechanisms, molecular network, PPI, etc. We selected over 135 publications and studies including 59 that were surveyed based on the number of phenotypes, type of biomolecules, methods, and similarity metrics used and analyzed in the context of multiple diseases and/or comorbidity. The 59 studies, ranging from 2006 to 2015, were selected under the following criteria, (i) as an input, at least five diseases or traits and one type of biomolecule (single nucleotide polymorphism, (SNP) gene, RNA and/or proteins), (ii) at least used one of the five methods based on network, statistics, machine learning, information retrieval, and overlap, and (iii) at least produced one of the five disease similarity metrics based on shared loci, function, phenotype, both function and topology and/or jointly function, topology and phenotype (Fig 1 and Fig 2).
Fig. 1.

Survey criteria overview. We surveyed the literature to identify publications that used methods linking phenotypic and molecular information with disease comorbidity. The inclusion criteria required a minimum of five diseases and one type of analyzed biomolecule in a single publication or study (input). We categorized these studies according to the methods and type of molecular and phenotypic similarity metric between diseases (output).
Fig. 2.

Publication survey based on the biomolecules, methods, and metrics used and generated without and with assessing comorbidity. A. Histogram illustrating number of publications analyzing disease similarity without and with assessing comorbidity. B. The studies are grouped based on the input biomolecules, mathematical framework, and the similarity metrics, which highlight the extensive lack of contribution of RNA species and the use of FTP metric for comorbidity analysis. Each pie chart presents the number of distinct studies for each captured element. The legend describes (clockwise starting at the top) the segments of the pie from lighter color to darker. In some cases, a subset of studies includes comorbidities (c), thus leading to two segments with the same color. For example, SNPs with and without comorbidities are relevant to 12% and 2% of the studied papers, respectively.
We also produced two plots reflective of the distribution of the methods used (Fig 3) as well as the similarity metrics generated (Fig 4) across the 59 surveyed publications and based on the type and number of biomolecules relatively to the number of phenotype used as an input. Abbreviations, key concepts and databases are outlined and defined in Table 1.
Fig. 3.
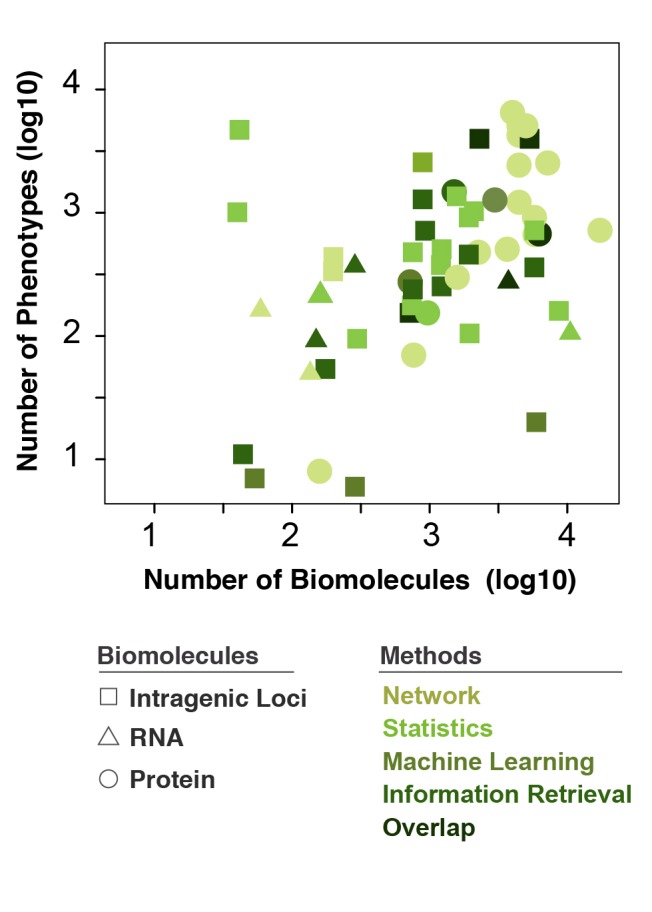
Computational methods based on the number of biomolecular and phenotypic inputs. Distribution of the types of methods widely changes according to the number of biomolecular and phenotypic/disease inputs. A few patterns are recognizable: (i) the predominant method utilizing proteins (circles) is “networks”, (ii) very few methods employ RNA (triangles), (iii) big datasets with ~104 biomolecules and as many phenotypes predominantly rely on proteins and to a lesser extent intragenic loci, and (iv) no studies comprised intergenic loci for imputation of similarity thus none shown in the legend.
Fig. 4.

Distribution of the utilization of the five major similarity metrics defined in this study across different numbers of input biomolecules and diseases. As expected, proteins were not utilized in shared intragenic loci (top left) and minimally in the two other first row metrics; however, they are predominantly employed in the leftmost two metrics of the second row. Utilization of RNA is mostly found in “Shared Function” (top middle). No studies comprised intergenic loci for imputation of similarity thus none shown in the legend.
Table 1.
Abbreviations and key concepts used in this review
| A. Phenotypic database and ontology | ||
|---|---|---|
| Abbreviations or concepts | Type | Definition |
| Phenotype | Disease Database | Clinical phenotypes: diseases, traits, digital database of medical and/or personal health records of patients (Electronic Health Records, EHR) which include age, gender, race, clinical laboratory test results, response to therapy, medications, signs and symptoms, medical imaging, quantitative risk factors (endophenotypes), etc. |
| OMIM | Disease ontology | Online Mendelian Inheritance in Man: collection of human genes and genetic phenotypes, which includes gene-phenotype interactions |
| PubMed | Literature Database | Collection of citations for biomedical literature |
| B. Molecular database, ontology and association study | ||
| Abbreviations or concepts | Type | Definition |
| SNP | Biomolecule | Single nucleotide polymorphism |
| mRNA | Biomolecule | messenger RNA: RNA coding-protein |
| microRNA | Biomolecule | microRNA: non-coding RNA known to control mRNA expression. |
| lncRNA | Biomolecule | Long non-coding RNA: non-coding RNA |
| GO | Gene Ontology | Annotation of molecular function and biological process of genes and gene products |
| GWAS | Association study | Genome Wide Association Study: study assessing a correlation between SNPs and disease occurrence within a given population |
| PheWAS | Association study | Phenome Wide Association Study: study assessing phenotypes associated to genotypes |
| C. Methods | ||
| Abbreviations or concepts | Type | Definition |
| MIN | Network | Molecular Interaction Network: representation of biomolecules (nodes) and their physical interactions or functional annotations (edges) |
| PPIN | Network | Protein-Protein Interaction (PPI) Network: representation of proteins (nodes) and their physical interactions (edges) |
| Disease network | Network | Disease Network: representation of diseases (nodes) and their similarity degrees (edges) |
| Topological feature | Network | Identification of structural and functional distances by which biomolecules or diseases are arranged and connected in the network, such as shortest distance between every two proteins in PPIN |
| Long range interaction | Network | Indirect interactions or relationships between two biomolecules in their associated networks |
| Genetic or Disease Profile | Statistics | Set of scores computed SNPs associated to diseases and vice versa |
| Clustering | Machine Learning | Method for grouping elements into sets based on their relative similarities |
| TF-IDF | Information Retrieval | Term Frequency–Inverse Document Frequency: weighing method to adjust the frequency of occurrences of variables |
| D. Similarity Metrics | ||
| Abbreviations and concepts | Type | Definition |
| Shared loci | Loci | Number of genetic loci, SNPs, or genes and their relationships shared between diseases |
| Shared Function | Function | Biomolecules that are bioproducts of genetic loci such as RNAs, proteins, metabolites, etc. with assigned biological function and that are shared between diseases. The biological function is derived from gene ontology database, PPI, protein complex, and biological, molecular and metabolic pathways, etc. |
| Shared Phenotype | Phenotype | Any relationship that might be inferred between phenotypes, such as overlap, association, correlation, or co-occurrence. |
| Shared Function and Topology (FT) | Function, Topology | Disease relationships based both on functional and topological features of biomolecules such as structure of feedback loop, indirect PPI, protein subnetworks, or the whole MINs or PPIN |
| Shared Function, Topology, and Phenotype (FTT) | Function, Topology, Phenotype | Disease relationships based both on functional and topological features of biomolecules in addition to phenotypic attributes |
3 Results
To present and highlight previous, current, and future research directions underpinning the biology of comorbidity, we first sought to evaluate 59 studies and approaches that (i) integrated phenotypic and molecular information, (ii) used different computational and statistical methods, and (iii) uncovered disease similarity with and without a comorbidity context (Fig 1).
3.1 Survey
In the last decade, among these 59 studies that incorporated both phenotypic and molecular information, 44% generated or validated disease similarity metrics in a context of comorbidity. Interestingly, during the last two years, there has been a surge in studies assessing comorbidity, counting for 60% of our selected publications (Fig 2A). Looking across the spectrum of biomolecules, gene and protein information were predominantly used as input, counting respectively for 38% and 35% of surveyed publications, with at least a third being assessed in the context of disease comorbidity (Fig 2B). However, among the publications that analyzed SNP (14%) and RNA (13%), only 2% or none have evaluated disease comorbidity, respectively (Fig 2B). In the context of methods, studies mainly used network (34%) and statistics (28%) based methods followed by overlap (18%), machine learning (12%) and information retrieval (8%). With the exception of machine learning, a third or half of the publications using these methods analyzed shared molecular mechanisms between diseases including those that are comorbid (Fig 2B). Finally, among the 59 surveyed publications, the five disease similarity metrics were comparably represented (15-24%) with at least a third or more were generated to understand the biology of comorbidity with the exception of integrative disease similarity metrics based shared function, topology and phenotype (2% out of 17%). In light of these results, we focused our efforts on pioneer and emerging studies highlighting the importance and applicability of one-level molecular and phenotypic scales as well as integrative data based disease similarity metrics to decipher the biological underpinning of comorbidity.
3.2 Methodology Used to Generate Disease Similarity Metrics
Several reviews have extensively described and evaluated the performance of varying methods for generating disease similarity metrics in the context of data integration strategies and modeling [35-41]. Here, we focused on five categories of methods based on network, statistics, machine learning, information retrieval and overlap (Fig 2-3, and Table 1). We evaluated the distribution of these methods based on the number and type of biomolecules relatively to the number of phenotypes analyzed (Fig 3). Distribution of the types of methods widely changes according to the number of biomolecules and phenotypic/disease inputs. We found that predominantly network based methods utilize proteins as inputs and very few methods employ RNA. We also found that big datasets with ~104 biomolecules and as many phenotypes largely rely on proteins information and to a less extent on Loci (SNP and gene). It is worth noting that the SNPs analyzed are intragenic (Fig 3).
3.3 Uncovering Disease Similarity and its Impact in the Biological Underpinning of Comorbidity
In this section we outline, describe, and highlight pioneering and emerging studies that utilize different disease similarity metrics to understand and reveal shared biology and etiology between multiple diseases including those that mutually or gradually occur in a patient’s lifetime (Fig 4).
3.3.1 Disease Similarity Metrics Based on Shared Phenotype
The use of very fine-grained phenotypes, such as disease-associated traits, phenotypic ontologies, clinical synopsis, electronic health records, laboratory tests, and billing information, increases the likelihood of uncovering disease similarity associated molecular mechanisms [19, 42-47]. Following the work of Swanson et al., [48] who highlighted the usefulness of Medline for knowledge discovery, several groups have utilized text-mining strategies to extract and integrate detailed phenotypes from medical datasets with underlined molecular mechanisms [48-51]. For instance, Butte et al. generated a network connecting 7,466 genes to 281 biomedical concepts via 64,003 relationships, highlighting a high connectivity of gene-phenotype-environment interactions [52]. Similarly, van Driel et al. extracted specific disease-associated phenotypes from the full text and clinical synopses of the OMIM database to build a human phenome network made of 5,000 human diseases in which the strengths of connection between disease pairs correlates with their phenotypic similarities [53].
Integrative analysis of the phenome (22) and PPI networks facilitated the discovery of novel disease-associated genes [54, 55]. Recently, Zhou et al. utilized the similarity between clinical manifestations of diseases queried from PubMed to construct a Human Symptoms Disease Network (HSDN) made of 322 symptoms, such as pain and diarrhea, and 4,219 diseases [56]. This integrative analysis of the HSDN with disease networks based on shared genes and PPIs uncovered a positive correlation of clinical presentations of diverse diseases with that of their underlying molecular interactions. Despite the discoveries that made use of phenotypic data buried in OMIM or PubMed, these databases do not provide direct evidence for measuring the comorbidity based on patient-centered data.
Electronic Health Records (EHR) are important sources of unbiased and detailed phenotypic data. Text-mining strategies [49, 57, 58] are often utilized to mine the electronic health records and extract rich phenotypes to stratify the patients and find comorbidities [59], analyze disease progression to uncover directional comorbidity [60], or to derive, in combination with genotypic data, pleiotropic genetic loci [61]. For instance, Razhetsky et al., analyzed the EHR by developing a statistical model in which the overlapping of the latent genetic susceptibilities across multifactorial diseases determines if they are positively or negatively correlated [62]. They showed that some diseases are inversely comorbid such as the negative correlations found between breast cancer and both schizophrenia and bipolar disorder. While, Hidalgo et al. adopted a network-based approach showing positive correlations between comorbidities and mortality rates dependent on age, gender, and disease progression, such as the higher prevalence of hypercholesterolemia in white males [63]. The landmark study of Blair et al. analyzed 110 million EHRs and uncovered striking comorbidities between 95 Mendelian and 150 complex diseases. They found that each of the complex diseases is attributable to a set of combinations of Mendelian-associated loci [64]. Similarly, Melamed et al. found a comorbidity between Mendelian disorders and common cancers by evaluating shared genes and pathways [65]. They showed significant co-expression of genetically-altered genes associated with cancers with those of a set of Mendelian diseases. Altogether, the analysis of EHR uncovers novel phenotypic relationships among diseases stemming from common molecular and cellular mechanisms and that in part could explain their comorbidities [24, 66].
3.3.2 Disease Similarity Metrics Based on Shared Loci
During the last decade, many studies showed that genetic variants impact the prevalence of disease risk [67, 68]. Given the reported pleiotropic effects of such genetic variants, many studies assessed disease similarity based on their associated genetics (e.g. SNPs or genes) [69, 70]. Denny et al. analyzed the associations between 3,144 SNPs (single nucleotide variant) conferring risk of various diseases and traits with 1,358 phenotypes from 13,835 EHR in a combined phenome-genome wide association study [71]. They uncovered 63 SNPs with pleiotropic effects that mechanistically link different phenotypes together such as actinic keratosis, seborrheic keratosis, and non-melanoma skin cancer. Other studies developed methods that make use of GWAS p-values of SNPs to build the association profiles for SNPs or diseases. Sirota et al. calculated, for each disease-SNP, a variant score in which its associated odds ratio and p-value account for its allelic directionality and the disease association strength, respectively [72]. They segregated six common autoimmune diseases into two classes based on their genetic profiles with variant scores of 573 SNPs. The allele directionality analysis prioritized SNPs conferring risk to one disease class while protecting against another one, underlying the inverse occurrence of each of the disease classes. For example, the minor allele of rs11752919 increases the risk of developing multiple sclerosis (MS) and autoimmune thyroid disease (ATD), while decreasing that of rheumatoid arthritis (RA) and ankylosing spondylitis (AS). In contrast to traditional meta-analysis, Cotsapas et al. developed a novel statistical method that evaluates concurrent associations of a SNP to several phenotypes [73]. They applied these statistics to GWAS data of seven autoimmune diseases and found 47 SNPs associated with multiple diseases, where clustering the SNP associated disease profiles identified four distinctive groups with each one being associated with one or multiple diseases, indicating shared genetic structure.
Recently, some studies developed other gene-based statistics to construct and compare the genetic profiles of diseases. Chang et al. presented a method in which they first construct a gene-based score using SNPs associated GWAS p-values, and measure how strongly each gene is associated with different diseases through randomization [74]. They applied a principal component analysis (PCA) to a gene disease interaction matrix of 31 distinctive diseases for which they identified shared pathogenic genetic profiles. On the other hand, Li et al. built a disease-trait network with 1,439 genes connecting 69 disease and 85 traits together [75]. Specifically, they considered a disease–trait pair similar if the cosine of their genetic profiles, based on their gene associated frequency-inverse document frequency (TF-IDF) weighted scores, are statistically significant. The time of occurrence between traits and their associated diseases, derived from the EHR, segregated the inferred associations into three different categories: risk factors, diagnostics, and complications. This is of value to the prognosis of potential comorbidity, such as the evaluation of decreased platelet counts before the diagnosis of alcohol dependence syndrome.
3.3.3 Disease Similarity Metrics Based on Shared Function
Multiple studies developed function-based disease similarity metrics beyond a simple assessment of shared genetics [76] using functional activity of biomolecules such as the expression of microRNAs [77, 78] and long non-coding RNA [79], interactions between proteins (e.g. PPI) [80], and gene ontology of biological pathways and processes [26, 81-83]. In this context, Sam et al. used enrichment analysis to identify significant disease similarities by taking into account the number of direct interactions between proteins belonging to different disease nodes [80]. Similarly, Yang et al. developed a metric based on differentially co-expressed molecular pathways [83]. On the other hand, Li and colleagues developed a novel disease similarity metric based on shared gene function derived from gene ontology (GO) [81] using information theoretic measure [84]. They showed that disease similarity based on GO metrics correlate with the shortest distances of their associated protein in the PPI network.
Multiple lines of evidence showed that comorbidity might stem from shared functional properties and domains such as from PPI or co-expressed genes [24]; however, simple metrics based on the number of the shared genes are not always sufficient to capture comorbid diseases with no shared genes [27, 76, 85]. For example, Lee et al. showed the observed comorbidity between metabolic diseases, not reflected from the disease network of shared genes but from shared metabolic fluxes [27]. Using enrichment analysis, Zhernakova et al. identified three major immune pathways (e.g. T-cell differentiation, immune-cell signaling and the innate immune response) that explain similar pathogenesis among 11 immune-related diseases with significant comorbidities [26]. Wang et al. utilized a similarity metric based on the number of shared protein complexes between diseases showing a high connectivity between diseases associated with different classes such as linking the glycolipid metabolic diseases with multiple types of cancers [25]. They also observed a two-fold increase in the odds of comorbidity between diseases sharing protein complexes.
3.3.4 Disease Similarity Based on Shared Function and Topology
Leveraging the topological features of biomolecules, in addition to their functional attributes, has the benefit to retain and uncover relevant structural properties of the molecular relationships and interactions underpinning disease similarity [86]. Using topological properties of protein interactions and k-nearest neighbor’s classifier algorithm, Xu et al. showed that hereditary disease genes tend to cluster together in PPI networks curated from the literature rather than those predicted from high-throughput yeast two-hybrid mapping approach [87]. On the other hand, Kohler et al. utilized a random walk algorithm and PPI networks to reveal disease gene candidates and infer disease associated sub-networks [88]. They showed that similarity measures of long-range distance interactions outperform those of direct neighbors of disease genes. Recently, Hamenh et al. made use of an information flow algorithm for long-range interactions in the PPI networks to assign score relevance to all the proteins in the entire network for each disease [89]. These relevance score vectors were correlated to assess the degree of similarity between each disease. Other studies utilized long-range interaction based similarities in the context of long non-coding RNAs [90, 91]. For example, Yang et al. formulated the lncRNA-disease bi-partite graph with a resource-allocation process and applied a propagation algorithm to assess the relevance of each lncRNA and its associated protein-coding genes to various diseases [90]. Similarly, Chen X et al. showed, via machine learning methods, how disease similarity can be captured with the Gaussian kernel of interaction profiles of diseases with their associated lncRNAs [91].
Functionally related comorbidities can be identified through the analysis of similarity derived from the topological features of their associated PPI networks [66]. Duc-Hau et al. showed through Boolean simulation that diseases sharing shorter feedback loops in cell signaling network, particularly those positive, are more prone to be comorbid. This result suggests that amplified cell signals underpin the comorbidity [92]. Similarly, Paik et al. used network-based methods to study the shortest distance paths connecting the protein subnetworks and identified the overlapping degrees of subnetworks of the diseases and their associated traits [93]. They found disease overlap associated with PPI subnetworks reflective of their comorbidity with their associated traits. Importantly, Menche et al. assessed disease similarity using a novel network based distance measuring the overlapping degree between disease modules mapped to PPI networks [94]. With over 30 million EHR, they showed that the overlap of disease modules captured phenotypically different diseases with high comorbidity. Interestingly, this metric showed that diseases with high comorbidity and disease module overlap, such as lymphoma and myocardial infarction (RR=2.1), do not necessarily share similar genes. Following this study, Ghiassian et al developed an algorithm that utilized hypergeometric distribution and the connectivity patterns of disease associated proteins for detecting disease modules in the PPI [95].
3.3.5 Disease Similarity Based on Shared Function, Topology, and Phenotype
Modular nature of diseases reflects that sharing molecular mechanisms between diseases underlies their phenotypic similarities [53]. The natural extension of the use of long-range interactions is to incorporate the phenotypic similarity between diseases. By doing so, it is possible to infer the pathogenesis of diseases with unknown molecular mechanisms or to infer novel mechanisms for expanding known pathogenic disease basis [23]. There are several combined phenome-genome metrics that have been primarily devised to prioritize novel disease genes [96-100]. However, they make it possible to derive gene relevance profiles for each disease, using the propagation-based algorithm [96] or graph Laplacian [98], and optimizing the information flow in the genome-phenome network [97]. Some groups showed the adaptability of their gene prioritization metrics for assessing the disease similarity, for example, Li et al. used a coranking method to apply a random walk with restart algorithm to the bi-partite phenome-genome network [101]. They uncovered 122 associations between diseases sharing no obvious phenotype. On the other hand, Wu and colleagues [55] adapted a network alignment technique, which had been previously developed for finding conserved protein networks across different species [102]. They applied this approach to the genome-phenome networks and identified 39 bi-modules with diseases within each module belong to the same disease category. However, Hwang et al. addressed the genome-phenome integration problem with the use of regularized, non-negative matrix factorization and simultaneously identified the association between the dense clusters of similar phenotypes and those of genes [103]. While the diseases within the same class share similar phenotypic annotations, the ones from different classes also showed a similar pattern, suggesting similar molecular mechanisms could underpin different phenotypes.
Other groups also defined the disease gene similarity profiles. Wu et al. defined a score that includes topological distances between the genes on the PPI network in order to evaluate the correlation of the variation of the phenotypic profile of a disease relative to the other diseases with that of its associated genes’ profile relative to those of the other disease genes’ profiles [54]. The higher the score, the more similar the diseases are. Applying a bi-clustering method to the gene-disease similarity matrix identifies bi-modules with biological significance. Similarly, Zhao et al. developed a method based on gene closeness index, which makes an index by incorporating the relative closeness of a drug and target disease genes in the PPI network. Through the use of this index, the authors compute the gene closeness profile, which is subsequently partitioned to gene modules that importantly capture the diseases comorbidity [104].
4 Discussion
This literature review shows how the use of phenome enhances our knowledge about shared pathogenesis of diseases. Mining the phenotypic data from databases, such as OMIM and PubMed, helped uncover the correlation between phenotypic similarity with shared molecular mechanisms and extensive interrelationships between phenome, genome, and environment. In addition, we showed the joint analysis of the rich phenotypic information buried in EHR with molecular layers would give rise to discovering cellular mechanisms underpinning the interaction between diseases, such as inferring the pleiotropic loci [71], prioritizing diseases associated traits as risk factors [75], and also explaining the genetics of comorbidities [64]. It was through the use of EHR that Rzhetsly et al. [62] in their premier work linked the comorbidity with shared genetic susceptibility mechanisms, and later by Park et al. [24] and Lee et al. [27] for explaining comorbidity in disease modularity framework. The landmark work by Blaire et al. [64] highlights the immediate practicality of EHR for geneticists to systematically screen different combinations of rare or common phenotypes that might be genetically linked or vice versa, which would be otherwise discovered with lengthy and costly epidemiological studies. Importantly, several ongoing national efforts will facilitate studying the gene-phenotype-environment interactions, such as i2b2 [105], Vanderbilt BioVu [106], and the Electronic Medical Records and Genomics (eMERGE) consortium [107-109]. The future of genomic research depends on the effective use of EHR with sophisticated text mining methods to extract the informative phenotypic data from all of their different tiers, namely structured, codified and narrative [110-112].
There are unique limitations for each type of “omics” dataset that differentially impact data mining. Advanced technologies, such as microarrays or next generation sequencing (NGS), have enabled cost-effective, high-throughput measures of the whole genome biological activity. However, they present sequence assembly and accuracy challenges affecting each of the produced “omics” datasets. These limitations introduce noise and biases. For example, in the context of NGS, DNAseq and RNAseq introduce gene-specific biases [113] and can produce low mappability reads, causing base mis-calls and mis-alignments often being filtered out, which leads to missing data [114]. This particularly plagues pseudogenes and their coding paralogs or orthologs, as well as genomic duplication blocs, repetitive regions, and multi-gene clusters such as cytochromes [113, 115, 116]. Highly polymorphic regions, such as HLA or regions often subject to DNA rearrangements, also suffer from mappability issues [117], leading to a mismatch to the reference scaffold. In addition, computational tools [118], library preparation [119], and sequencing platforms [120] frequently introduce biases.
In contrast to RNAseq, batch effect issues are recurrent in gene expression microarrays and often require cross-batch and cross-sample normalization methods that don’t necessarily correct for technical variations, while the identification of differentially expressed genes is highly dependent on these analytical methods [122] and are notoriously platform (or even probe) specific [121]. Moreover, microarrays often present probe specificity issues [121]- due to hybridization steps and/or the lack of appropriate probe design taking into account isoforms. Nanostring technology can also be rate limiting and highly expensive to measure the entire transcriptome in addition to requiring distinct analyses from those used in expression arrays or RNAseq [118]. Lastly, protein-protein interaction datasets are incomplete and limited to more studied cases [123]. Data mining can be improved when explicitly modeled with the specific biases present in different datasets. For example, integrating expression data from high coverage RNAseq provides the opportunity to better evaluate a larger dynamic range of mRNA expression and their associated alternative splicing isoforms than expression microarrays.
This review unveiled a major opportunity to use more machine learning methods, as they have only been utilized in a limited number of manuscripts to date. Further, none of the reviewed methods addressed intergenic SNPs located far from the protein coding loci. Statistical modeling predominantly employed protein omics datasets as inputs, while straightforward overlap of nodes was the predominant method for locus assignment. Network modeling appears more versatile as it was applied equally to all types of omics datasets. Published network models always employed a larger number of biomolecules than statistical methods, suggesting a lack of predictive power of the former on smaller datasets. Here, we provide more details on similarity methods. We visualized the distribution of biologically informed similarity metrics according to multiple variables, such as the computational methods and the types and counts of biomolecules or diseases. These results demonstrate the efficacy of naïve as well as complex similarity metrics between biomolecular functions. The common theme among these metrics is the integration of various interaction types of data capturing the cellular variation, such as genetic polymorphisms, GO, protein complexes, and long-range interactions among biomolecules. Interestingly, devising biologically informed metrics or solely employing computational approaches could converge to similar findings. For example, Lee et al. highlighted the role of biologically inspired metrics thorough finding that metabolic diseases rarely share genes; however, they interact through metabolic fluxes and their similarities reflect their comorbidities [27]. Without making this assumption, Li and Patra reached the same conclusion by applying a random walk algorithm to the phenome-genome network. We also showed how the different computational methods could integrate cellular and phenotypic data [101]. The performances of the computational methods have been primarily assessed in the context of gene prioritization. Relative to the algorithms strictly relying on the shortest topological distances, random walk and diffusion-based ones perform better, as they evaluate the global topology of the network while profiling the similarity of a disease and its known associated genes relative to other genes [124]. Additionally, some studies showed integration of the disease phenotypic similarities enhancing their performances [96, 125] since the primary premise implies that the genes associated with a disease also underlie the pathogenesis of those phenotypically similar. Different frameworks and methods and their relative performances for integrative data analysis have been covered in previous reviews [35-41]. Despite the significant contribution of a disease similarity framework to uncover the underpinning molecular mechanisms of comorbidity, its current formulation needs the integration of other types of data to explain how the interaction between different cellular domains underpins comorbidity. Omics data provide functional and physical interactions between various molecular tiers inferred from biochemical measures rather than functional associations based on knowledge base and correlations studies. The integration of omics data with different degrees of specificity and sensitivity [126-128] enhances the performance of modeling [129-131] and optimizes accuracy and recovery of more comprehensive disease mechanism similarities. For example, the use of eQTL data (expression quantitative trait loci) [132, 133] linking mRNA expression to SNPs enabled the recovery of not only shared mRNAs among disease-associated SNPs but also their shared downstream biological pathways and their relative interactions [134]. Other studies showed that disease similarity stemmed from shared regulatory mechanisms through the mapping of disease-SNPs with transcription factor (TF) binding sites or genomic regulatory elements such as enhancers (e.g. ChIP-seq, DNAseq) along with RNA gene expression (RNAseq) and long-range chromatin interaction regions (ChIA-PET) [135-138]. Cell type-specific interaction networks have also been also used to uncover disease similarity [139, 140] [141-144]. Moreover, the use of other regulatory molecular players, such as microRNAs and lncRNAs, improves the formulation of the interaction between diseases for better mechanistic explanation of comorbidity.
This review is limited to recent scientific publications that met stringent inclusion criteria of a minimum of five analyzed diseases. There is an overabundance of manuscripts using rate-limiting biological or curation approaches to identify similarity of mechanisms between two diseases, but most of these would not scale up affordably to a large number of disease comorbidities. This review is therefore steered by design towards computational methods.
5 Conclusion
The premise of biomodularity of diseases implies that diseases should no longer be treated as simple traits due to their complex phenotypic presentation and genetic pleiotropy. This review highlights the significance of improving our understanding disease’s underpinnings using similarity metrics that uncover shared molecular mechanisms. Future studies shall benefit from the rapid increase of large cohorts and personal omics data to infer biological activity and function, especially using previously overlooked intergenic loci. It is also important that future studies model genetic and biomolecular interactions as non-linear interdependencies may modulate the severity of comorbidities. Finally, the use of detailed phenotypes and availability of clinical databases provide new opportunities to develop more robust and comprehensive diseases similarity metrics that account for the spatio-temporal epidemiology of disease comorbidity.
Acknowledgments
Dr. Lussier is supported in part by The National Library of Medicine (K22 LM00830804), The University of Arizona Cancer Center (P30CA023074), the Center for Biomedical Informatics and Biostatistics of the University of Arizona Health Sciences, and the BIO5 Institute.
References
- 1.National Insitute on Aging, National Insitute of Health, World Health Organization, Global Health and Aging. NIH Publication; Oct. 2011. p. 11-7737. [Google Scholar]
- 2.Dye C. After 2015: infectious diseases in a new era of health and development. Philos Trans R Soc Lond B Biol Sci 2014;369(1645): 20130426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valderas JM, Starfield B, Sibbald B, Salisbury C, Roland M. Defining comorbidity: implications for understanding health and health services. Ann Fam Med 2009;7(4):357-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Global status report on noncommunicable diseases 2014; 2014. [Google Scholar]
- 5.Hunter DJ, Reddy KS. Noncommunicable diseases. N Engl J Med 2013;369(14):1336-43. [DOI] [PubMed] [Google Scholar]
- 6.Jakovljevic M, Ostojic L. Comorbidity and multimorbidity in medicine today: challenges and opportunities for bringing separated branches of medicine closer to each other. Psychiatr Danub 2013;25(Suppl 1):18-28. [PubMed] [Google Scholar]
- 7.Barnett K., et al. , Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet, 2012. 380(9836): p. 37-43. [DOI] [PubMed] [Google Scholar]
- 8.Pantalone KM, Hobbs TM, Wells BJ, Kong SX, Kattan MW, Bouchard J, et al. Clinical characteristics, complications, comorbidities and treatment patterns among patients with type 2 diabetes mellitus in a large integrated health system. BMJ Open Diabetes Res Care 2015;3(1):e000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demetrius LA, Magistretti PJ, Pellerin L. Alzheimer’s disease: the amyloid hypothesis and the Inverse Warburg effect. Front Physiol 2014;5:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabares-Seisdedos R, Rubenstein JL. Inverse cancer comorbidity: a serendipitous opportunity to gain insight into CNS disorders. Nat Rev Neurosci 2013;14(4):293-304. [DOI] [PubMed] [Google Scholar]
- 11.Tabares-Seisdedos R, Dumont N, Baudot A, Valderas JM, Climent J, Valencia A, et al. No paradox, no progress: inverse cancer comorbidity in people with other complex diseases. Lancet Oncol 2011;12(6): 604-8. [DOI] [PubMed] [Google Scholar]
- 12.Robinson D, Jr., Hackett M, Wong J, Kimball AB, Cohen R, Bala M, et al. Co-occurrence and comorbidities in patients with immune-mediated inflammatory disorders: an exploration using US healthcare claims data, 2001-2002. Curr Med Res Opin 2006;22(5):989-1000. [DOI] [PubMed] [Google Scholar]
- 13.Bonavita V, De Simone R. Towards a definition of comorbidity in the light of clinical complexity. Neurol Sci. 2008;29 Suppl 1:S99-102. [DOI] [PubMed] [Google Scholar]
- 14.van Weel C, Schellevis FG. Comorbidity and guidelines: conflicting interests. Lancet 2006;367(9510):550-1. [DOI] [PubMed] [Google Scholar]
- 15.Beckles MA, Spiro SG, Colice GL, Rudd RM, American College of Chest Physicians The physiologic evaluation of patients with lung cancer being considered for resectional surgery. Chest 2003;123(1 Suppl):105S-114S. [DOI] [PubMed] [Google Scholar]
- 16.Lee L, Cheung WY, Atkinson E, Krzyzanowska MK. Impact of comorbidity on chemotherapy use and outcomes in solid tumors: a systematic review. J Clin Oncol 2011;29(1):106-17. [DOI] [PubMed] [Google Scholar]
- 17.Sogaard M, Thomsen RW, Bossen KS, Sørensen HT, Nørgaard M. The impact of comorbidity on cancer survival: a review. Clin Epidemiol 2013;5(Suppl 1):3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolff JL, Starfield B, Anderson G. Prevalence, expenditures, and complications of multiple chronic conditions in the elderly. Arch Intern Med 2002;162(20):2269-76. [DOI] [PubMed] [Google Scholar]
- 19.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12(1):56-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freudenberg J, Propping P. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics. 2002;18 Suppl 2:S110-5. [DOI] [PubMed] [Google Scholar]
- 21.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL. The human disease network. Proc Natl Acad Sci U S A 2007;104(21):8685-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature 2001;409(6822):853-5. [DOI] [PubMed] [Google Scholar]
- 23.Oti M, Brunner HG. The modular nature of genetic diseases. Clin Genet 2007;71(1):1-11. [DOI] [PubMed] [Google Scholar]
- 24.Park J, Lee DS, Christakis NA, Barabási AL. The impact of cellular networks on disease comorbidity. Mol Syst Biol 2009;5:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, Liu W, Ning S, Ye J, Huang T, Li Y, Wang P, et al. Community of protein complexes impacts disease association. Eur J Hum Genet 2012;20(11):1162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet 2009;10(1):43-55. [DOI] [PubMed] [Google Scholar]
- 27.Lee DS, Park J, Kay KA, Christakis NA, Oltvai ZN, Barabási AL. The implications of human metabolic network topology for disease comorbidity. Proc Natl Acad Sci U S A 2008;105(29):9880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan SY, Loscalzo J. The emerging paradigm of network medicine in the study of human disease. Circ Res 2012;111(3):359-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gustafsson M, Nestor CE, Zhang H, Barabási AL, Baranzini S, Brunak S, et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med 2014;6(10):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furlong LI. Human diseases through the lens of network biology. Trends Genet 2013;29(3):150-9. [DOI] [PubMed] [Google Scholar]
- 31.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005;122(6):957-68. [DOI] [PubMed] [Google Scholar]
- 32.Barrenas F, Chavali S, Alves AC, Coin L, Jarvelin MR, Jörnsten R, et al. Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms. Genome Biol 2012;13(6):R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gustafsson M, Edström M, Gawel D, Nestor CE, Wang H, Zhang H, et al. Integrated genomic and prospective clinical studies show the importance of modular pleiotropy for disease susceptibility, diagnosis and treatment. Genome Med 2014;6(2):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vidal M, Cusick ME, Barabasi AL. Interactome networks and human disease. Cell 2011;144(6):986-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gligorijevic V, Malod-Dognin N, Przulj N. Integrative methods for analysing big data in precision medicine. Proteomics 2015. [DOI] [PubMed] [Google Scholar]
- 36.Gligorijevic V, Przulj N. Methods for biological data integration: perspectives and challenges. J R Soc Interface 2015;12(112). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le D-H, Hoai NX, Kwon Y-K. A Comparative Study of Classification-Based Machine Learning Methods for Novel Disease Gene Prediction. Knowledge and Systems Engineering. Springer; 2015. p. 577-88. [Google Scholar]
- 38.Wang X, Gulbahce N, Yu H. Network-based methods for human disease gene prediction. Brief Funct Genomics 2011;10(5):280-93. [DOI] [PubMed] [Google Scholar]
- 39.Moreau Y, Tranchevent LC. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat Rev Genet 2012;13(8):523-36. [DOI] [PubMed] [Google Scholar]
- 40.Mitra K, Carvunis AR, Ramesh SK, Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet 2013;14(10):719-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Harazi O, Al Insaif S, Al-Ajlan MA, Kaya N, Dzimiri N, Colak D. Integrated Genomic and Network-Based Analyses of Complex Diseases and Human Disease Network. J Genet Genomics 2016. June 20;43(6):349-67. [DOI] [PubMed] [Google Scholar]
- 42.Freimer N, Sabatti C. The human phenome project. Nat Genet 2003;34(1):15-21. [DOI] [PubMed] [Google Scholar]
- 43.Houle D, Govindaraju DR, Omholt S. Phenomics: the next challenge. Nat Rev Genet 2010;11(12):855-66. [DOI] [PubMed] [Google Scholar]
- 44.Snyder M, Weissman S, Gerstein M. Personal phenotypes to go with personal genomes. Mol Syst Biol 2009;5:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oti M, Huynen MA, Brunner HG. The biological coherence of human phenome databases. Am J Hum Genet 2009;85(6):801-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pendergrass SA, Brown-Gentry K, Dudek SM, Torstenson ES, Ambite JL, Avery CL, et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet Epidemiol 2011;35(5):410-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Almasy L. The role of phenotype in gene discovery in the whole genome sequencing era. Hum Genet 2012;131(10):1533-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swanson DR. Fish oil, Raynaud’s syndrome, and undiscovered public knowledge. Perspect Biol Med 1986;30(1):7-18. [DOI] [PubMed] [Google Scholar]
- 49.Jensen LJ, Saric J, Bork P. Literature mining for the biologist: from information retrieval to biological discovery. Nat Rev Genet 2006;7(2):119-29. [DOI] [PubMed] [Google Scholar]
- 50.Agarwal P, Searls DB. Can literature analysis identify innovation drivers in drug discovery? Nat Rev Drug Discov 2009;8(11):865-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen AM, Hersh WR. A survey of current work in biomedical text mining. Brief Bioinform 2005;6(1):57-71. [DOI] [PubMed] [Google Scholar]
- 52.Butte AJ, Kohane IS. Creation and implications of a phenome-genome network. Nat Biotechnol 2006;24(1):55-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA. A text-mining analysis of the human phenome. Eur J Hum Genet 2006;14(5):535-42. [DOI] [PubMed] [Google Scholar]
- 54.Wu X, Jiang R, Zhang MQ, Li S. Network-based global inference of human disease genes. Mol Syst Biol 2008;4:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu X, Liu Q, Jiang R. Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics 2009;25(1):98-104. [DOI] [PubMed] [Google Scholar]
- 56.Zhou X, Menche J, Barabási AL, Sharma A. Human symptoms-disease network. Nat Commun 2014;5:4212. [DOI] [PubMed] [Google Scholar]
- 57.Wilke RA, Xu H, Denny JC, Roden DM, Krauss RM, McCarty CA, et al. The emerging role of electronic medical records in pharmacogenomics. Clin Pharmacol Ther 2011;89(3):379-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jensen PB, Jensen LJ, Brunak S. Mining electronic health records: towards better research applications and clinical care. Nat Rev Genet 2012;13(6):395-405. [DOI] [PubMed] [Google Scholar]
- 59.Roque FS, Jensen PB, Schmock H, Dalgaard M, Andreatta M, Hansen T, et al. , Using electronic patient records to discover disease correlations and stratify patient cohorts. PLoS Comput Biol 2011;7(8):e1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jensen AB, Moseley PL, Oprea TI, Ellesøe SG, Eriksson R, Schmock H, et al. Temporal disease trajectories condensed from population-wide registry data covering 6.2 million patients. Nat Commun 2014;5:4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010;26(9):1205-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci U S A 2007;104(28):11694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hidalgo C.A., et al. , A dynamic network approach for the study of human phenotypes. PLoS Comput Biol, 2009. 5(4): p. e1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blair DR, Lyttle CS, Mortensen JM, Bearden CF, Jensen AB, Khiabanian H, et al. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell 2013;155(1):70-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melamed RD, Emmett KJ, Madubata C, Rzhetsky A, Rabadan R. Genetic similarity between cancers and comorbid Mendelian diseases identifies candidate driver genes. Nat Commun 2015;6:7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun K, Gonçalves JP, Larminie C, Przulj N. Predicting disease associations via biological network analysis. BMC Bioinformatics 2014;15:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sivakumaran S, Agakov F, Theodoratou E, Prendergast JG, Zgaga L, Manolio T, et al. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet 2011;89(5):607-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet 2013;14(7):483-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hall MA, Verma A, Brown-Gentry KD, Goodloe R, Boston J, Wilson S, et al. Detection of pleiotropy through a Phenome-wide association study (PheWAS) of epidemiologic data as part of the Environmental Architecture for Genes Linked to Environment (EAGLE) study. PLoS Genet 2014;10(12):e1004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pendergrass SA, Brown-Gentry K, Dudek S, Frase A, Torstenson ES, Goodloe R, et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) Network. PLoS Genet 2013;9(1):e1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol 2013;31(12):1102-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sirota M, Schaub MA, Batzoglou S, Robinson WH, Butte AJ. Autoimmune disease classification by inverse association with SNP alleles. PLoS Genet 2009;5(12):e1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet 2011;7(8):e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang D, Keinan A. Principal component analysis characterizes shared pathogenetics from genome-wide association studies. PLoS Comput Biol 2014;10(9):e1003820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li L, Ruau DJ, Patel CJ, Weber SC, Chen R, Tatonetti NP, et al. Disease risk factors identified through shared genetic architecture and electronic medical records. Sci Transl Med 2014;6(234):234ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barrenas F, Chavali S, Holme P, Mobini R, Benson M. Network properties of complex human disease genes identified through genome-wide association studies. PLoS One 2009;4(11):e8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 2009;37(Database issue):D98-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, et al. An analysis of human microRNA and disease associations. PLoS One 2008;3(10):e3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, et al. LncRNADisease: a database for long-noncoding RNA-associated diseases. Nucleic Acids Res 2013;41(Database issue):D983-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sam L, Liu Y, Li J, Friedman C, Lussier YA. Discovery of protein interaction networks shared by diseases. Pac Symp Biocomput 2007:76-87. [PMC free article] [PubMed] [Google Scholar]
- 81.Li H, Lee Y, Chen JL, Rebman E, Li J, Lussier YA. Complex-disease networks of trait-associated single-nucleotide polymorphisms (SNPs) unveiled by information theory. J Am Med Inform Assoc 2012;19(2):295-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Y, Agarwal P. A pathway-based view of human diseases and disease relationships. PLoS One 2009;4(2):e4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang J, Wu SJ, Dai WT, Li YX, Li YY. The human disease network in terms of dysfunctional regulatory mechanisms. Biol Direct 2015;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tao Y, Sam L, Li J, Friedman C, Lussier YA. Information theory applied to the sparse gene ontology annotation network to predict novel gene function. Bioinformatics 2007;23(13):i529-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davis DA, Chawla N.V. Exploring and exploiting disease interactions from multi-relational gene and phenotype networks. PloS one, 2011. 6(7): p. e22670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ideker T, Sharan R. Protein networks in disease. Genome Res 2008;18(4):644-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu J, Li Y. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics 2006;22(22):2800-5. [DOI] [PubMed] [Google Scholar]
- 88.Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 2008;82(4):949-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamaneh M.B., Yu Y.K., Relating diseases by integrating gene associations and information flow through protein interaction network. PLoS One, 2014. 9(10): p. e110936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang X, Gao L, Guo X, Shi X, Wu H, Song F, et al. A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PLoS One 2014;9(1):e87797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen X, Yan GY. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013;29(20):2617-24. [DOI] [PubMed] [Google Scholar]
- 92.Le DH, Kwon YK. The effects of feedback loops on disease comorbidity in human signaling networks. Bioinformatics 2011;27(8):1113-20. [DOI] [PubMed] [Google Scholar]
- 93.Paik H, Heo HS, Ban HJ, Cho SB. Unraveling human protein interaction networks underlying co-occurrences of diseases and pathological conditions. J Transl Med 2014;12:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, et al. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015;347(6224):1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghiassian SD, Menche J, Barabasi AL. A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput Biol 2015;11(4):e1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol 2010;6(1):e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen Y, Jiang T, Jiang R. Uncover disease genes by maximizing information flow in the phenome-interactome network. Bioinformatics 2011;27(13):i167-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hwang T, Zhang W, Xie M, Liu J, Kuang R. Inferring disease and gene set associations with rank coherence in networks. Bioinformatics 2011;27(19):2692-9. [DOI] [PubMed] [Google Scholar]
- 99.Xie M, Xu Y, Zhang Y, Hwang T, Kuang R. Network-based Phenome-Genome Association Prediction by Bi-Random Walk. PLoS One 2015;10(5):e0125138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen H, Zhang Z. Similarity-based methods for potential human microRNA-disease association prediction. BMC Med Genomics 2013;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li Y, Patra JC. Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics 2010;26(9):1219-24. [DOI] [PubMed] [Google Scholar]
- 102.Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, et al. Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci U S A 2005;102(6):1974-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hwang T, Atluri G, Xie M, Dey S, Hong C, Kumar V, et al. Co-clustering phenome-genome for phenotype classification and disease gene discovery. Nucleic Acids Res 2012;40(19):e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao S, Li S. A co-module approach for elucidating drug-disease associations and revealing their molecular basis. Bioinformatics 2012;28(7):955-61. [DOI] [PubMed] [Google Scholar]
- 105.Murphy S, Churchill S, Bry L, Chueh H, Weiss S, Lazarus R, et al. Instrumenting the health care enterprise for discovery research in the genomic era. Genome Res 2009;19(9):1675-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 2008;84(3):362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kho AN, Pacheco JA, Peissig PL, Rasmussen L, Newton KM, Weston N, et al. Electronic medical records for genetic research: results of the eMERGE consortium. Sci Transl Med 2011;3(79):79re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Manolio TA. Collaborative genome-wide association studies of diverse diseases: programs of the NHGRI’s office of population genomics. Pharmacogenomics 2009;10(2):235-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shah NH. Mining the ultimate phenome repository. Nat Biotechnol 2013;31(12):1095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sinnott JA, Dai W, Liao KP, Shaw SY, Ananthakrishnan AN, Gainer VS, et al. Improving the power of genetic association tests with imperfect phenotype derived from electronic medical records. Hum Genet 2014;133(11):1369-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu S, Liao KP, Shaw SY, Gainer VS, Churchill SE, Szolovits P, et al. Toward high-throughput phenotyping: unbiased automated feature extraction and selection from knowledge sources. J Am Med Inform Assoc 2015;22(5):993-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kohane IS. Using electronic health records to drive discovery in disease genomics. Nat Rev Genet 2011;12(6):417-28. [DOI] [PubMed] [Google Scholar]
- 113.SEQC/MAQC-III Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 2014;32(9):903-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 2014;15(2):121-32. [DOI] [PubMed] [Google Scholar]
- 115.Rouchka EC, Cha IE. Current trends in pseudogene detection and characterization. Current Bioinformatics 2009;4(2):112-9. [Google Scholar]
- 116.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Revi Genet 2012;13(1):36-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brandt DY, Aguiar VR, Bitarello BD, Nunes K, Goudet J, Meyer D. Mapping Bias Overestimates Reference Allele Frequencies at the HLA Genes in the 1000 Genomes Project Phase I Data. G3 (Bethesda) 2015;5(5):931-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Richard AC, Lyons PA, Peters JE, Biasci D, Flint SM, Lee JC, et al. Comparison of gene expression microarray data with count-based RNA measurements informs microarray interpretation. BMC Genomics 2014;15:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Toedling J, Servant N, Ciaudo C, Farinelli L, Voinnet O, Heard E, et al. Deep-sequencing protocols influence the results obtained in small-RNA sequencing. PLoS One 2012;7(2):e32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harismendy O, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol, 2009. 10(3): p. R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yauk CL, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, et al. Comprehensive comparison of six microarray technologies. Nucleic Acids Res 2004;32(15):e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qin LX, Zhou Q. MicroRNA array normalization: an evaluation using a randomized dataset as the benchmark. PLoS One 2014;9(6):e98879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gillis J, Ballouz S, Pavlidis P. Bias tradeoffs in the creation and analysis of protein-protein interaction networks. J Proteomics 2014;100:44-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Navlakha S, Kingsford C. The power of protein interaction networks for associating genes with diseases. Bioinformatics 2010;26(8):1057-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lage K, Karlberg EO, Størling ZM, Olason PI, Pedersen AG, Rigina O, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 2007;25(3):309-16. [DOI] [PubMed] [Google Scholar]
- 126.Tanay A, Sharan R, Kupiec M, Shamir R. Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc Natl Acad Sci U S A 2004;101(9)2981-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Troyanskaya OG, Dolinski K, Owen AB, Altman RB, Botstein D. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci U S A 2003;100(14)8348-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Joyce AR, Palsson BO. The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol 2006;7(3):198-210. [DOI] [PubMed] [Google Scholar]
- 129.Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, et al. Gene prioritization through genomic data fusion. Nat Biotechnol 2006;24(5):537-44. [DOI] [PubMed] [Google Scholar]
- 130.Zitnik M, Janjić V, Larminie C, Zupan B, Pržulj N. Discovering disease-disease associations by fusing systems-level molecular data. Sci Rep 2013;3:3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Linghu B, Snitkin ES, Hu Z, Xia Y, Delisi C. Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network. Genome Biol 2009;10(9):R91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jansen RC, Nap RP. Genetical genomics: the added value from segregation. Trends Genet 2001;17(7):388-91. [DOI] [PubMed] [Google Scholar]
- 133.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348(6235): 648-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li H, Achour I, Bastarache L, Berhout J, Gardeux V, Li J, et al. Integrative genomics analyses unveil downstream biological effectors of disease-specific polymorphisms buried in intergenic regions. NPJ Genomic Medicine 2016;1:16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. , Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337(6099):1190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karczewski KJ, Dudley JT, Kukurba KR, Chen R, Butte AJ, Montgomery SB, et al. Systematic functional regulatory assessment of disease-associated variants. Proc Natl Acad Sci USA 2013;110(23):9607-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sallari R, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 2014;24(1):1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155(4):934-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518(7539):337-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015;28(4):529-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cornish AJ, Filippis I, David A, Sternberg MJ. Exploring the cellular basis of human disease through a large-scale mapping of deleterious genes to cell types. Genome Med 2015;7(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Magger O, Waldman YY, Ruppin E, Sharan R. Enhancing the prioritization of disease-causing genes through tissue specific protein interaction networks. PLoS Comput Biol 2012;8(9):e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kotlyar M, Pastrello C, Sheahan N, Jurisica I. Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res 2016;44(D1):D536-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 2014;507(7493):462-70. [DOI] [PMC free article] [PubMed] [Google Scholar]