

Abstract
Here we describe a patient who presented with a history of congenital diaphragmatic hernia, inguinal hernia, and recurrent umbilical hernia. She also has joint laxity, hypotonia, and dysmorphic features. A unifying diagnosis was not identified based on her clinical phenotype. As part of her evaluation through the Undiagnosed Diseases Network, trio whole-exome sequencing was performed. Pathogenic variants in FBN1 and TRPS1 were identified as causing two distinct autosomal dominant conditions, each with de novo inheritance. Fibrillin 1 (FBN1) mutations are associated with Marfan syndrome and a spectrum of similar phenotypes. TRPS1 mutations are associated with trichorhinophalangeal syndrome types I and III. Features of both conditions are evident in the patient reported here. Discrepant features of the conditions (e.g., stature) and the young age of the patient may have made a clinical diagnosis more difficult in the absence of exome-wide genetic testing.
Keywords: central hypotonia, congenital diaphragmatic hernia, fourth toe clinodactyly, hammertoe, inguinal hernia, joint laxity, long philtrum, malar flattening, pes planus, protruding ear, smooth philtrum, sparse anterior scalp hair, sparse lateral eyebrow, superior pectus carinatum, thin nail, thin upper lip vermilion, thoracic scoliosis, umbilical hernia
INTRODUCTION
There are more than 200 genetic disorders of connective tissue, which may affect skin, eyes and musculoskeletal, cardiovascular, and pulmonary systems. They can present with common features including joint laxity, hypotonia, short stature, and hernias (Jobling et al. 2014). Some common inherited disorders of the connective tissue include Ehlers–Danlos syndrome, osteogenesis imperfecta, Marfan syndrome, and cutis laxa. Diagnosis of these syndromes can be difficult because of the variability and overlapping phenotypes.
We report the case of a female child suspected of having a connective tissue disorder based on findings of multiple hernias, hypotonia, and failure to thrive.
RESULTS
Clinical Presentation
A Caucasian female, age 3 yr, was referred to the Stanford Center for Undiagnosed Diseases with a suspected connective tissue disorder. The pregnancy and family history were unremarkable. Her father was 31 yr of age at the time of her birth, and her mother was 28 yr. She had been referred for genetic evaluation at 6 mo because of dysmorphic features, failure to thrive, and clinodactyly of her fourth toes bilaterally (Fig. 1; Supplemental Figs. S3A–H). Macrocephaly (head circumference 52.2 cm, 97th percentile), pes planus, pectus carinatum, scoliosis, and hypotonia were noted. Chest X-ray and abdominal magnetic resonance imaging (MRI) at 15 mo due to a history of gastroesophageal reflux disease (GERD) and poor weight gain revealed a right-sided diaphragmatic hernia, which was surgically repaired. Umbilical hernia was noted at 20 mo, and bilateral inguinal hernias were diagnosed at 2 yr.
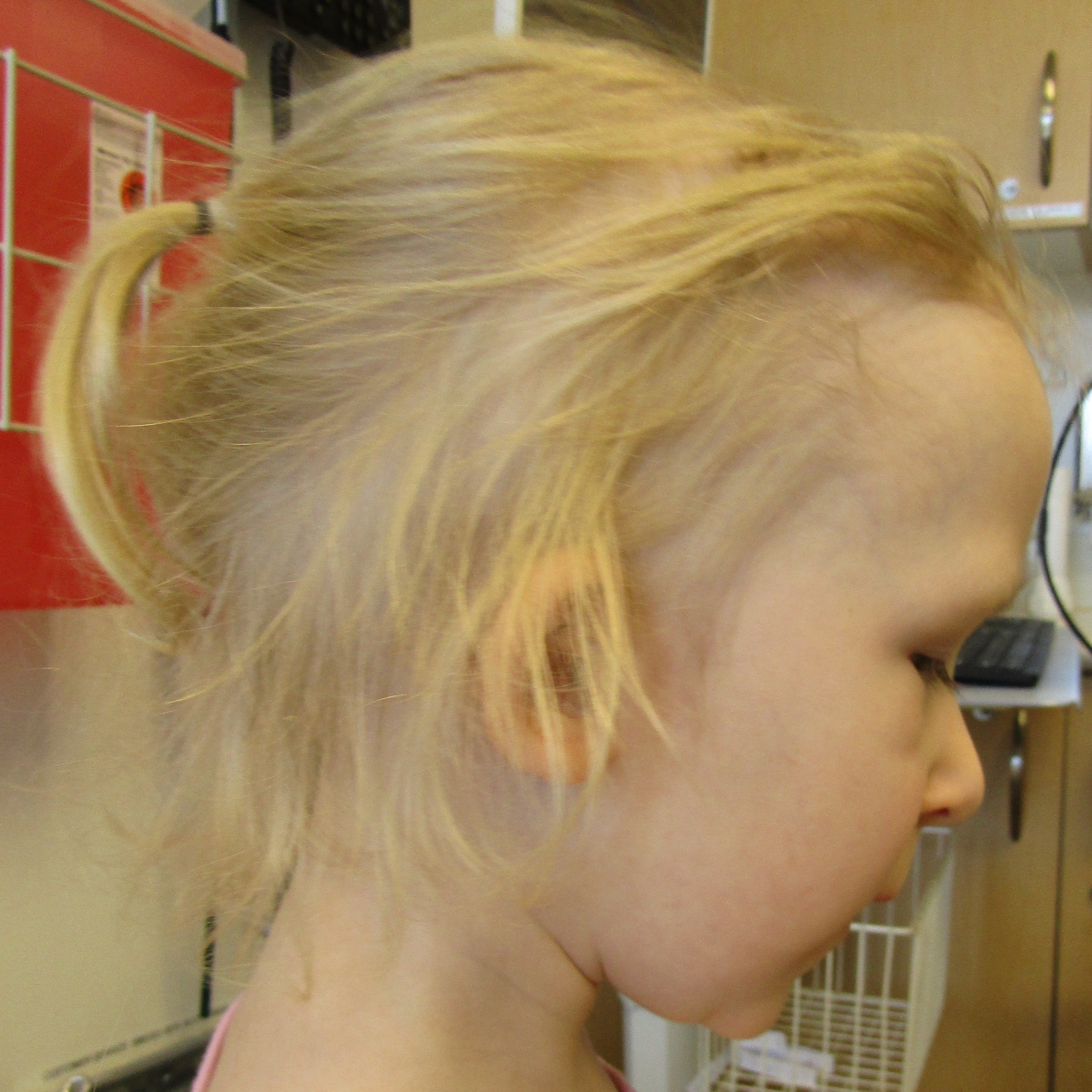
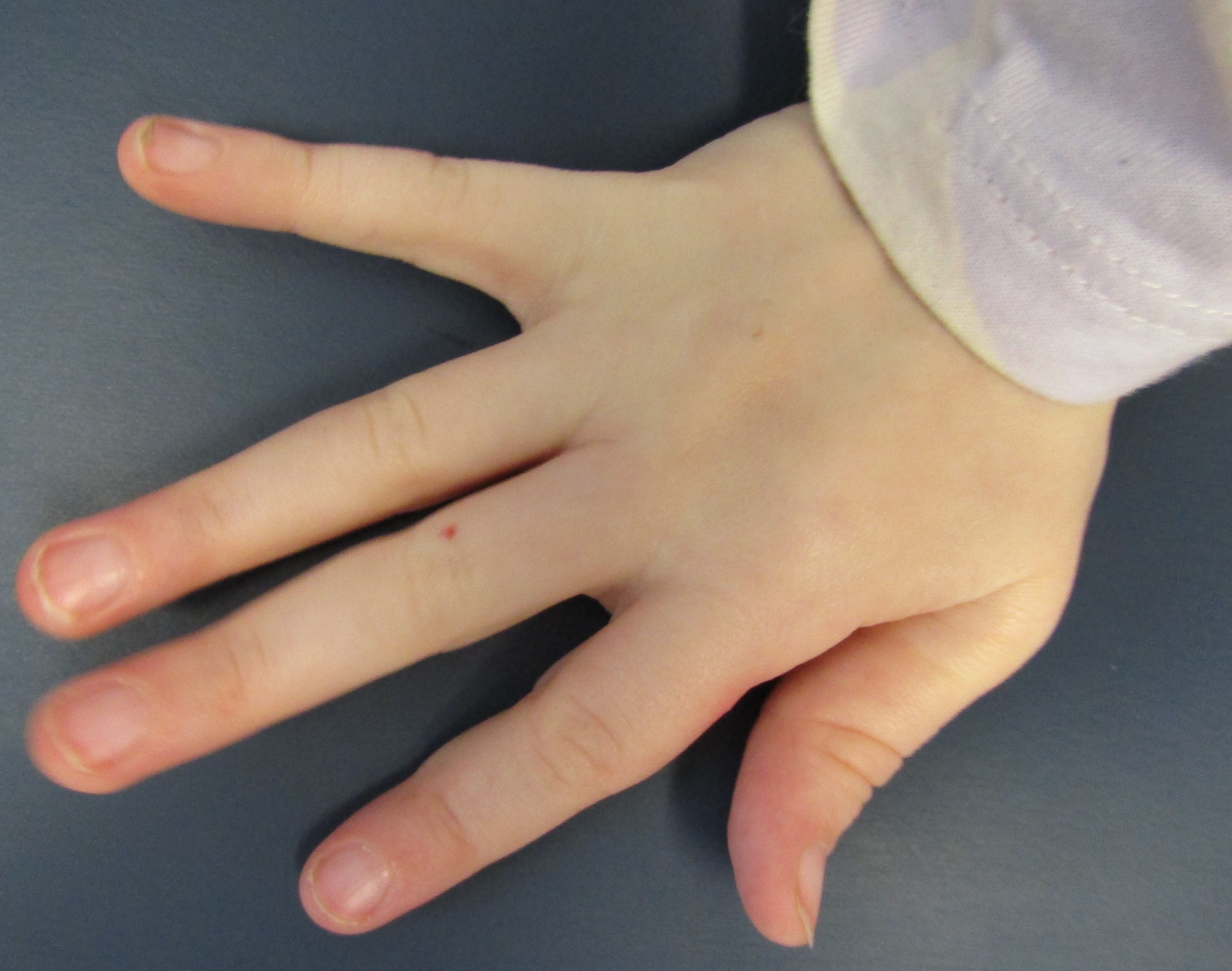
Figure 1.

Clinical features. (A) Sparse scalp hair with receding frontotemporal hairline and thin eyebrows. (B) Protruding ears, thin upper lip, long flat philtrum, and horizontal groove on chin. (C) Clinodactyly and hammer toes. (D) Scoliosis radiograph.
She had a normal ophthalmology exam at 16 mo of age, ruling out myopia and ectopia lentis. An echocardiogram done at 17 mo of age was reported as normal, ruling out mitral valve prolapse and aortic dilatation (aortic root Z-score = 2.44) (Fig. 2). Because she had features that were not specific to one genetic condition, a broad molecular genetic analysis was desired.
Figure 2.
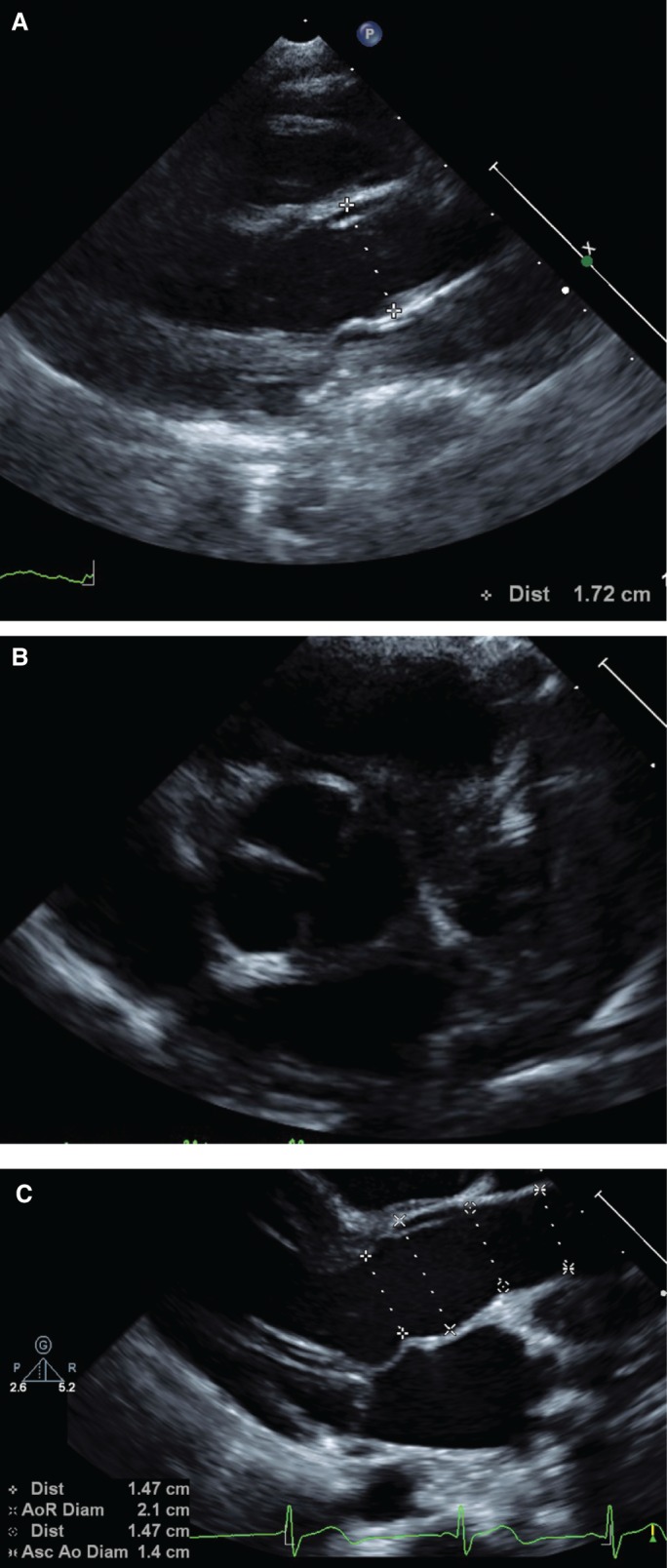
Echocardiogram images. (A) Aortic root at 17 mo, before Undiagnosed Diseases Network (UDN) evaluation. (B) Trileaflet aortic valve at 17 mo. (C) Aortic root at 4 yr and 3 mo.
Genomic Analyses
Previous genetic testing for this patient included a normal array comparative genomic hybridization ([arr(1-22,X)x2]) as well as negative Sotos syndrome (NSD1 gene, OMIM [Online Mendelian Inheritance in Man] 606681) sequencing and deletion/duplication analysis. Insurance authorization for whole-exome sequencing (WES) was denied.
The patient applied to the Undiagnosed Diseases Network (UDN) and was enrolled at the Stanford clinical site. The UDN is a National Institutes of Health (NIH) sponsored clinical research network, the goal of which is to diagnose disease in patients who do not have a clinical or molecular diagnosis (Gahl et al. 2015, 2016). As part of the research evaluation, whole-exome or genome sequencing is often performed in addition to an on-site clinical visit with multiple subspecialists to refine understanding of the phenotype. After her enrollment in the UDN, blood samples were obtained from the patient, her mother, and her father. DNA was extracted and sent to the UDN exome-sequencing core at Baylor Miraca Genetics Laboratories for trio WES. A summary of the exome-sequencing statistics for each family member is presented in Table 1. A tiered annotated output of all sequence variant calls in the patient is included in Supplemental Table 1, as given by the Sequence to Medical Phenotypes pipeline (Dewey et al. 2015). Alignment and variant filtering identified two high-quality de novo pathogenic variants in disease genes related to the clinical phenotype (Table 2).
Table 1.
Exome-sequencing metrics
| Proband | Mother | Father | |
|---|---|---|---|
| Bases covered at ≥20× | 35,296,438 | 35,322,751 | 35,239,444 |
| Average coverage | 124× | 134× | 110× |
| Number of reads | 126,028,542 | 131,764,296 | 111,657,880 |
| Percent of reads mapped | 94.48 | 95.24 | 94.21 |
Table 2.
Pathogenic variants identified by trio whole-exome sequencing
| Gene | FBN1 | TRPS1 |
|---|---|---|
| Chromosome position | 15:48758017 | 8:116616566 |
| HGVS DNA reference | NM_000138: c.4786C>T | NM_014112: c.1630C>T |
| HGVS protein reference | p.R1596X | p.R544X |
| Variant type | Nonsense | Nonsense |
| Predicted effect | Stop codon | Stop codon |
| dbSNP/dbVar ID | rs113871094 | N/A |
| Genotype | Heterozygous | Heterozygous |
| ClinVar ID | 36082 | N/A |
| Parent of origin | De novo | De novo |
| ExAC allele frequency | Not present | Not present |
| Gene loss-of-function intolerance (pLI) | 1.00 | 0.99 |
HGVS, Human Genome Variation Society; dbSNP, Database for Short Genetic Variations; dbVar, Database of Genomic Structural Variation; N/A, not available; ExAC, Exome Aggregation Consortium; pLI, probability of loss-of-function intolerance.
A heterozygous stop-gain variant c.4786C>T (p.R1596X) in the fibrillin 1 (FBN1; OMIM 134797) gene was detected. Pathogenic variants in the FBN1 gene lead to Marfan syndrome (MFS; OMIM 154700) and similar disorders (MASS phenotype, mitral valve prolapse syndrome, ectopia lentis syndrome). This variant has been previously reported as disease causing (Loeys et al. 2001) for MFS, a well-recognized autosomal dominant condition involving the connective tissue. Primary characteristics affect the ocular, skeletal, and cardiovascular systems (Dietz 2014). The principal diagnostic clinical features include aortic root enlargement and ectopia lentis (Loeys et al. 2010). A 29-yr-old patient with the p.R1596X variant has a reported diagnosis of classic MFS according to the original Ghent nosology, with major involvement of the ocular and cardiovascular systems and the dura (Loeys et al. 2001). Thoracic aortic aneurysms and aortic dissections (TAADs) have also been associated with this variant in one ClinVar submission (SCV000233837.2).
A heterozygous nonsense variant c.1630C>T (p.R544X) in the TRPS1 gene (OMIM 604386) was also detected in the patient. The TRPS1 gene is a zinc finger transcription factor, and is associated with trichorhinophalangeal syndrome, types I (TRPS1; OMIM 190350) and III. Trichorhinophalangeal syndrome type I (OMIM 190350) is an autosomal dominant condition that also affects the skeletal system, with prominent dysmorphic craniofacial features. Dysmorphic findings include sparse hair, bulbous tip of the nose, long flat philtrum, thin upper vermilion border, and protruding ears (Momeni et al. 2000). Missense mutations in the GATA zinc finger domain (exon 6) have a more severe phenotype of TRPS type III, whereas mutations leading to haploinsufficiency (frameshift, nonsense, or splicing) cause TRPS type I (Ludecke et al. 2001). TRPS type II is due to a contiguous gene deletion encompassing the TRPS1 and EXT1 genes (Ludecke et al. 1995). The p.R544X variant has been previously reported and associated with TRPS1 in one patient (Ito et al. 2013). Allele frequency of this variant is assumed to be very low, as it is absent from the 1000 Genomes Project, the Exome Variant Server, National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Project (ESP), and Exome Aggregation Consortium (ExAC) databases (http://evs.gs.washington.edu/EVS/) (The 1000 Genomes Project Consortium 2015; Lek et al. 2016). The TRPS1 gene encodes a 1281-amino acid protein with seven exons. Important motifs include exon 6, which encodes a presumptive GATA DNA-binding zinc finger, and an IKAROS-like zinc-finger motif in the carboxyl terminus, which enables protein–protein interactions (Momeni et al. 2000; Ludecke et al. 2001). R544X is in exon 4 of the TRPS1 gene and is predicted to result in a stop codon. Because of the location of this variant in the TRPS1 protein and the nonsense effect of the variant, nonsense-mediated decay of the resulting mutant protein is likely (Richards et al. 2015). The probability of loss-of-function intolerance (pLI) score for TRPS1 is 0.99. Reported germline TRPS1 mutations number at least 55, not including gross deletions and rearrangements typically associated with trichorhinophalangeal syndrome type II (Human Gene Mutation Database [HGMD], ClinVar). Nonsense variants in TRPS1 have been reported as pathogenic (17/55 in HGMD, 5/14 in ClinVar). Therefore, the TRPS1 p.R544X variant was classified as pathogenic based on the nonsense/null variant, de novo inheritance, low allele frequency in the general population, and multiple lines of computational evidence supporting a deleterious effect (Richards et al. 2015).
Whole-exome sequencing and Sanger sequencing of the parental samples did not detect the c.4786C>T FBN1 variant or the c.1630C>T TRPS1 variant. De novo mutations are assumed, although the possibility of cryptic somatic or germline mosaicism in either parent cannot be excluded. These variants were confirmed in the proband and absent in parents by Sanger sequencing (Supplemental Fig. S1A–F), whereas no variant reads were mapped in either parent for either mutation as visualized in the Integrative Genomics Viewer (Supplemental Fig. S2A–F).
DISCUSSION
We report on a female with a suspected connective tissue disorder who was found to have two pathogenic de novo variants by trio WES causing two autosomal dominant syndromes: Marfan syndrome and TRPS1. Both pathogenic variants have been previously reported to be associated with these conditions, but to our knowledge this is the first report of these conditions appearing in the same individual.
The TRPS1 R544X variant found in our patient was previously reported in a 5-yr-old female who presented with hair growth retardation (Ito et al. 2013). Upon examination she also had a pear-shaped nose, thin upper lip, thin toenails, and brachydactyly of the big toes. These features are similar to that of our patient (Table 3). Immunohistochemical staining of a skin sample from the previously reported patient with the TRPS1 R544X variant showed low immunoreactivity with anti-TRPS1 antibody, suggesting haploinsufficiency (Ito et al. 2013). This is consistent with genotype–phenotype correlations for TRPS1 missense mutations leading to haploinsufficiency (Ludecke et al. 2001). A characteristic skeletal finding of TRPS1 is cone-shaped epiphyses of the phalanges, which typically present after 2 yr. These were seen in the previously reported patient, but were not investigated in our patient. Without whole-exome sequencing, this subtle dysmorphic diagnosis might not have been appreciated.
Table 3.
Correlation of clinical phenotype using Human Phenotype Ontology (HPO)
| System | Patient | Marfan syndrome | Trichorhinophalangeal syndrome type 1 |
|---|---|---|---|
| Skeletal | Joint laxitya | Joint laxity | |
| Pectus carinatumb | Pectus excavatumc/carinatum | Pectus carinatum | |
| Thumb sign | Thumb sign | ||
| Scoliosisd | Scoliosis | Scoliosis | |
| Pes planuse | Pes planus | Pes planus | |
| Height = 97.0 cm (31st percentile by CDC, 17th percentile by WHO) | Bone overgrowth/tall staturef | Short statureg | |
| Hammertoeh Toe clinodactylyi |
|||
| Hammertoe arachnodactylyj | Short fingersk | ||
| Unknown | Cone-shaped epiphysisl (>2 yr) | ||
| Ocular | Negative exam at 3 yr | Ectopia lentism, myopian | |
| Cardiovascular | Aortic root dilated (Z = 3.24 at 4 yr 3 mo) | Aortic dilatationo | |
| Normal mitral and tricuspid valves at 4 yr 3 mo | Mitralp/tricuspid valve prolapseq | ||
| Connective tissue | Congenital diaphragmaticr, umbilicals, and inguinal herniast | Herniasu | |
| Hair, nails | Sparse scalp hairv Sparse eyebrowsw |
Sparse scalp hair Sparse eyebrows |
|
| Thin nailsx | Thin nails | ||
| Craniofacial | Full nasal tip | Bulbous nosey | |
| Long,z flataa philtrum | Long, flat philtrum | ||
| Thin upper lip vermilionbb | Thin upper lip vermilion | ||
| Protruding earscc | Protruding ears | ||
| Malar hypoplasiadd | Malar hypoplasia | ||
| Muscular | Hypotoniaee | Hypotonia |
CDC, Centers for Disease Control and Prevention; WHO, World Health Organization.
HPO IDs: a0002761; b0000768; c0000767; d0002650; e0001763; f0000098; g0004322; h0001765; i0001863; j0001166; k0009381; l0010579; m0001083; n0000545 o0002616; p0001634; q0001704; r0000776; s0001537; t0000023; u0100790; v0002209; w0000535; x0001816; y0000414; z0000343; aa0000319; bb0000219; cc0000411; dd0000272; ee0008947.
Growth alterations leading to abnormal height is a significant feature in both TRPS1 and MFS. Short stature in TRPS1 is progressive with age and is therefore more pronounced in adulthood (Ludecke et al. 2001). In contradistinction, long bone overgrowth in patients with Marfan syndrome often leads to absolute or relative increased height as well as arachnodactyly. In TRPS1, growth retardation is due to premature closure of the growth plates. TRPS1 is involved in regulation of chondrocyte and perichondrium development by repressing expression of RUNX2 (Napierala et al. 2008). Studies of FBN1 in the skeletal pathway suggest involvement of the TGFβ and BMP pathways (Quarto et al. 2012a,b). Typically for children with MFS, tall stature (height ≥ 97th percentile, or >3 SD above the mean) is a common presenting feature (Lipscomb et al. 1997; Faivre et al. 2009). TGF-β is up-regulated in MFS, which recruits and proliferates osteoprogenitors, but down-regulates BMP; this inhibits osteoblast proliferation, differentiation, and mineralization. The height of our patient (97.0 cm: 31st percentile by the Centers for Disease Control and Prevention [CDC]; 17th percentile by the World Health Organization [WHO]) may reflect epistasis between FBN1, promoting TGF-β-related chondrogenesis, and TRPS1 haploinsufficiency, leading to growth retardation through premature closure of epiphyses. This perhaps confused the potential clinical/phenotypic diagnosis of Marfan syndrome before genetic testing was initiated.
The FBN1 R1596X variant was reported in a family with phenotypic features similar to our patient (De Backer et al. 2007). A 4-yr-old male presented with inguinal hernia at age 2 yr and with flat feet. He had pectus excavatum, arachnodactyly, and tall stature (103 cm, 97th percentile), and an echocardiogram showed dilation of the proximal aorta (Z-score = 3.2). Six other members of the family were subsequently found to have the same FBN1 variant, with limited expression of the disease (3/7 fulfilled the Ghent criteria). RNA expression studies in this family were done on two sisters: one satisfied the Ghent criteria (proband's mother), and the other did not. The authors found that they had similar expression and concluded that was not the cause for the intrafamilial variation.
Clinical diagnosis of Marfan syndrome in children has been noted to be more difficult as compared to adults because of the delay in onset of clinical features (De Backer et al. 2007). This is especially true in the absence of ocular or cardiovascular features. Revised Ghent diagnostic criteria for MFS combine family history, medical history, physical exam, and molecular FBN1 analysis (Loeys et al. 2010). The skeletal findings, such as bone overgrowth and joint laxity, are also clinically assessed in the revised Ghent criteria as part of the Systemic Score, where features are assigned a value and a total value score greater than or equal to 7 can be included as diagnostic criteria (Loeys et al. 2010). In the absence of a family history, the diagnosis is made when aortic root enlargement or ectopia lentis is present in addition to other findings. Our patient had a normal ocular exam and presented with no identified cardiovascular features. Using the systemic score for MFS, our patient presented with the thumb sign, pectus carinatum, pes planus, and scoliosis; this gave a systemic score of 5. Therefore, she did not overtly meet clinical criteria for MFS upon presentation to the UDN. Echocardiogram performed during the UDN evaluation, but after WES results were available, revealed mild aortic root dilation (Z-score = 3.24), aortic annulus (Z-score = 4.13), and sinotubular junction (Z-score = 3.03) (The Marfan Foundation; https://www.marfan.org/dx/zscore). These findings combined with the known pathogenic FBN1 variant allowed the diagnosis of MFS to be established. FBN1 variants and MFS have been reported in association with congenital diaphragmatic hernia (CDH), which was present in our patient (Petersons et al. 2003; Faivre et al. 2009; Jetley et al. 2009; Beck et al. 2015). However, this characteristic is not part of the revised Ghent diagnostic criteria or systemic score. When the diagnosis of MFS in people under the age of 20 yr is suspected, a suggestion is to use “nonspecific connective tissue disorder” or “potential MFS” until aortic root dilation is present (Z-score ≥ 3.0) (Dietz 2014). Also, the Kid-Short Marfan Score (SMS) diagnostic tool can be used, but this also relies heavily on cardiac, ocular, and family history features (Mueller et al. 2013). Patients similar to ours, with pathogenic de novo FBN1 variants and a milder skeletal phenotype, will likely remain undiagnosed in childhood based on clinical features alone. This is problematic, as the first diagnostic manifestation is skeletal for a quarter of children with FBN1 mutations (Faivre et al. 2009). Importantly, the clinical diagnosis of MFS has implications for ongoing surveillance and treatment to avoid aortic catastrophe.
There was no family history for our patient of Marfan syndrome or specific phenotypic findings suggesting TRPS1. This of course is consistent with the de novo nature of the variants. Approximately 25% of patients with MFS have a de novo variant (Dietz 2014). Because of the small number of reported pathogenic TRPS1 variants (approximately 55), the frequency of de novo variants has yet to be determined. Although the p.R544X was assumed to be de novo in the previous report (Ito et al. 2013), those parents did not have molecular analysis to confirm. Half of the TRPS1 variants reported by Momeni et al. (2000) were sporadic (3/6). Thirty TRPS1 cases studied by Ludecke et al. (2001) were isolated, but causative TRPS1 variants were found in only 24 of those cases, and only 15 cases had mother and father available for analysis. Though rare, multiple prior reports have found more than one pathogenic de novo variant on WES (Tunovic et al. 2014; Yoo et al. 2015; Gahl et al. 2016). With the increase in clinical WES, multiple de novo pathogenic variants are likely to be more common than previously appreciated.
Genome sequencing has allowed more accurate analysis of the frequency of de novo mutations (Conrad et al. 2011; Kong et al. 2012; Francioli et al. 2015). Paternal age is a known factor for de novo mutations during DNA replication in spermatogenesis and has been confirmed with these recent genome investigations; however, the father of our patient was 31 yr at the time of her birth. Clinical guidelines suggest that paternal age of >40 yr reaches significance for increased risk of pathogenic de novo variants to offspring in autosomal dominant genetic conditions (Toriello and Meck 2008). The annual mutation rate is estimated to increase by one to two mutations per year with paternal age (Kong et al. 2012; Francioli et al. 2015). The overall mutation rates based on whole-genome studies are estimated at 0.97–1.20 × 10−8 per nucleotide per generation (Conrad et al. 2011; Kong et al. 2012). Most de novo mutations will take place in noncoding regions; Francioli et al. found that only 1.22% of de novo mutations in studied genomes were in exonic regions. The estimated prevalence of MFS is 1/5000–1/10,000 (Dietz 2014). The prevalence of clinically recognized TRPS1 is low; approximately 100 cases have been published (Orphanet Report Series 2016). The prevalence of TRPS1 is estimated to be 1/1,000,000. Therefore, the likelihood of a co-occurrence of MFS and TRPS1 is 1/5,000,000,000–1/10,000,000,000, indicating our patient may be unique among the world's population (7.4 billion) (World Population Clock; http://www.worldometers.info/world-population/, accessed July 7, 2016); the likelihood of de novo mutations causing these specific phenotypes would be even lower. WES presents a unique advantage over single gene sequencing and gene panels to interrogate the exome for these rare de novo variants responsible for clinical disease.
This is the first report, to our knowledge, of MFS and TRPS type 1 appearing in the same individual, along with molecular discovery of FBN1 and TRPS1 de novo pathogenic variants by WES. The expected phenotypes of Marfan syndrome and trichorhinophalangeal syndrome type 1 partially overlap (e.g., scoliosis, pectus carinatum, and pes planus are common to both) yet are distinct in others (e.g., skeletal and craniofacial features), which made it difficult to arrive at a clinical diagnosis prior to WES. Because of the overlapping or subtle characteristics of these conditions in our patient (height, multiple and recurrent hernias, joint laxity, hypotonia, and dysmorphic features), clinical diagnoses were only possible after genetic evaluation through WES. Additionally, if targeted testing for connective tissue disease had been done, then the diagnosis of TRPS1 may not have been appreciated. This case highlights the importance and utility of WES for undiagnosed genetic disease in thoroughly identifying multiple conditions and allowing management of life-threatening sequellae.
METHODS
Whole-Exome Sequencing
For the paired-end precapture library procedure, genome DNA was fragmented by sonication and ligated to the Illumina multiplexing paired-end (PE) adapters. The adapter-ligated DNA was the polymerase chain reaction (PCR) amplified using primers with sequencing barcodes (indexes). For target enrichment/exome capture procedure, the precapture library was enriched by hybridizing to biotin-labeled VCRome 2.1 in-solution exome probes (Bainbridge et al. 2011) for 64–72 h at 47°C. Additional probes for more than 2600 Mendelian disease genes were also included in the capture in order to improve the exome coverage. For massively parallel sequencing, the postcapture library DNA was subjected to sequence analysis on Illumina HiSeq platform for 100-bp paired-end reads. The following quality control metrics of the sequencing data are generally achieved: >70% of reads aligned to target, >95% target base covered at >20×, >85% target base covered at >40×, mean coverage of target bases >100×. Single-nucleotide polymorphism (SNP) concordance to genotype array: >99%. As a quality control measure, DNA is also analyzed by a SNP array (Illumina Human Exome-12v1 array). The SNP data are compared with the WES data to ensure correct sample identification and to assess sequencing quality.
Data Analysis and Interpretation by Mercury 1.0
The output data from Illumina HiSeq are converted from BCI files to FASTQ by Illumina CASAVA 1.8 software, and mapped by BWA (Li and Durbin 2009) to the reference haploid human genome sequence (Genome Reference Consortium human genome build 37, human genome 19). The variant calls are performed using Atlas-SNP and Atlas-indel developed in-house by the Baylor College of Medicine Human Genome Sequencing Center (BCM HGSC) (Yang et al. 2013). The variant annotations are performed using in-house developed software: HGSC-SNP-anno and HGSC-indel-anno. Synonymous variants, intronic variants not affecting splice site, and common benign variants are excluded from interpretation unless they were previously reported as pathogenic variants. The variants were interpreted according to American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015) and patient phenotypes. Variants related to patient phenotypes are confirmed by Sanger sequencing for patient and parents.
ADDITIONAL INFORMATION
Data Deposition and Access
Whole-exome sequencing data have been submitted to the Database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap) per NIH study protocol and patient consent (accession number phs001232.v1.p1). The FBN1 and TRPS1 variants have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000328213.1 and SCV000328214.1. Patient-derived biospecimens are available by contacting the corresponding author.
Ethics Statement
The clinical trial was approved by the Institutional Review Board of Stanford University and the NIH (NIH Study Reference Number 15-HG-0130), and written informed consent and assent was obtained from the patient and family members.
Acknowledgments
Members of the Undiagnosed Diseases Network: Mercedes E. Alejandro, Carlos A. Bacino, Ashok Balasubramanyam, Lindsay C. Burrage, Gary D. Clark, William J. Craigen, Shweta U. Dhar, Lisa T. Emrick, Brett H. Graham, Neil A. Hanchard, Mahim Jain, Seema R. Lalani, Brendan H. Lee, Richard A. Lewis, Azamian S. Mashid, Paolo M. Moretti, Sarah K. Nicholas, Jordan S. Orange, Jennifer E. Posey, Lorraine Potocki, Jill A. Rosenfeld, Daryl A. Scott, Alyssa A. Tran, Hugo J. Bellen, Michael F. Wangler, Shinya Yamamoto, Christine M. Eng, Donna M. Muzny, Patricia A. Ward, Yaping Yang, Andrea L. Gropman, David B. Goldstein, Nicholas Stong, Yong-hui Jiang, Allyn McConkie-Rosell, Loren D.M. Pena, Kelly Schoch, Vandana Shashi, Rebecca C. Spillmann, Jennifer A. Sullivan, Nicole M. Walley, Alan H. Beggs, Lauren C. Briere, Cynthia M. Cooper, Laurel A. Donnell-Fink, Elizabeth L. Krieg, Joel B. Krier, Sharyn A. Lincoln, Joseph Loscalzo, Richard L. Maas, Calum A. MacRae, J. Carl Pallais, Lance H. Rodan, Edwin K. Silverman, Joan M. Stoler, David A. Sweetser, Chris A. Walsh, Cecilia Esteves, Ingrid A. Holm, Isaac S. Kohane, Paul Mazur, Alexa T. McCray, Matthew Might, Rachel B. Ramoni, Kimberly Splinter, David P. Bick, Camille L. Birch, Braden E. Boone, Donna M. Brown, Dan C. Dorset, Lori H. Handley, Howard J. Jacob, Angela L. Jones, Jozef Lazar, Shawn E. Levy, J. Scott Newberry, Molly C. Schroeder, Kimberly A. Strong, Elizabeth A. Worthey, Jyoti G. Dayal, David J. Eckstein, Sarah E. Gould, Ellen M. Howerton, Donna M. Krasnewich, Carson R. Loomis, Laura A. Mamounas, Teri A. Manolio, John J. Mulvihill, Anastasia L. Wise, Ariane G. Soldatos, Matthew Brush, Jean-Philippe F. Gourdine, Melissa Haendel, David M. Koeller, Jennifer E. Kyle, Thomas O. Metz, Katrina M. Waters, Bobbie-Jo M. Webb-Robertson, Euan A. Ashley, Jonathan A. Bernstein, Annika M. Dries, Paul G. Fisher, Jennefer N. Kohler, Daryl M. Waggott, Matthew T. Wheeler, Patricia A. Zornio, Patrick Allard, Hayk Barseghyan, Esteban C. Dell'Angelica, Katrina M. Dipple, Naghmeh Dorrani, Matthew R. Herzog, Hane Lee, Stan F. Nelson, Christina G.S. Palmer, Jeanette C. Papp, Janet S. Sinsheimer, Eric Vilain, Christopher J. Adams, Elizabeth A. Burke, Katherine R. Chao, Mariska Davids, David D. Draper, Tyra Estwick, Trevor S. Frisby, Kate Frost, Valerie Gartner, Rena A. Godfrey, Mitchell Goheen, Gretchen A. Golas, Mary “Gracie” G. Gordon, Catherine A. Groden, Mary E. Hackbarth, Isabel Hardee, Jean M. Johnston, Alanna E. Koehler, Lea Latham, Yvonne L. Latour, C. Christopher Lau, Denise J. Levy, Adam P. Liebendorder, Ellen F. Macnamara, Valerie V. Maduro, Thomas C. Markello, Alexandra J. McCarty, Jennifer L. Murphy, Michele E. Nehrebecky, Donna Novacic, Barbara N. Pusey, Sarah Sadozai, Katherine E. Schaffer, Prashant Sharma, Sara P. Thomas, Nathanial J. Tolman, Camilo Toro, Zaheer M. Valivullah, Colleen E. Wahl, Mike Warburton, Alec A. Weech, Guoyun Yu, David R. Adams, William A. Gahl, May Christine V. Malicdan, Cynthia J. Tifft, Lynne A. Wolfe, Paul R. Lee, John H. Postlethwait, Monte Westerfield, Anna Bican, Rizwan Hamid, John H. Newman, John A. Phillips III, Amy K Robertson, Joy D. Cogan.
Author Contributions
D.B.Z. and M.T.W. analyzed and interpreted the data and drafted and critically reviewed the manuscript. M.W. and C.M.E. generated, analyzed, and interpreted the sequencing data and drafted the manuscript. A.D. obtained consent from the patient and family and critically reviewed the manuscript. J.K. obtained images for the manuscript and critically reviewed the manuscript. L.F. critically reviewed the manuscript. D.W. analyzed and interpreted the sequencing data and critically reviewed the manuscript. P.A.Z. and J.A.B. obtained consent from the patient and family, collected the clinical data, and critically reviewed the manuscript. M.A.M. and E.F. collected and provided the clinical data and critically reviewed the manuscript. J.A.B., P.G.F., E.A.A., M.T.W., and members of the Undiagnosed Diseases Network conceived of the study and critically reviewed the manuscript.
Funding
This work was supported in part by the Intramural Research Program of the National Human Genome Research Institute and by the NIH Common Fund, through the Office of Strategic Coordination, Office of the NIH Director under Award Numbers U01 HG007708, U01 HG007709, U01 HG007703, U01 HG007530, U01 HG007942, U01 HG007690, U01 HG007674, U01 HG007672, U01 TR001395, U01 HG007943, and U54 NS093793. This work was also supported by the Stanford Clinical and Translational Science Award (CTSA) to Spectrum (UL1 TR001085). The CTSA program is led by the National Center for Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Competing Interest Statement
E.A.A. and M.T.W. report holding stock in Personalis, Inc.
Referees
Gholson J. Lyon
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, Albert TJ, Burgess DL, Gibbs RA. 2011. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol 12: R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck TF, Campeau PM, Jhangiani SN, Gambin T, Li AH, Abo-Zahrah R, Jordan VK, Hernandez-Garcia A, Wisniewski WK, Muzny D, et al. 2015. FBN1 contributing to familial congenital diaphragmatic hernia. Am J Med Genet A 167A: 831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Cassals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Backer J, Loeys B, Leroy B, Coucke P, Dietz H, De Paepe A. 2007. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin Genet 72: 188–198. [DOI] [PubMed] [Google Scholar]
- Dewey FE, Grove ME, Priest JR, Waggott D, Batra P, Miller C, Wheeler M, Zia A, Pan C, Karzcewski KJ, et al. 2015. Sequence to medical phenotypes: a framework for interpretation of human whole genome DNA sequence data. PLoS Genet 11: e1005496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz HC. 2014. Marfan Syndrome. GeneReviews. University of Washington, Seattle: http://www.ncbi.nlm.nih.gov/books/NBK1335/. [PubMed] [Google Scholar]
- Faivre L, Masurel-Paulet A, Collod-Béroud G, Callewaert BL, Child AH, Stheneur C, Binquet C, Gautier E, Chevallier B, Huet F, et al. 2009. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics 123: 391–398. [DOI] [PubMed] [Google Scholar]
- Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, Genome of the Netherlands Consortium, van Duijn CM, Swertz M, Wijmenga C, et al. 2015. Genome-wide patterns and properties of de novo mutations in humans. Nat Genet 47: 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Wise AL, Ashley EA. 2015. The undiagnosed diseases network of the National Institutes of Health: a national extension. JAMA 314: 1–2. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, Adams DR, Tifft CJ, for Members of the UDN. 2016. The NIH undiagnosed diseases program and network: applications to modern medicine. Mol Genet Metab 117: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Shimomura Y, Farooq M, Suzuki N, Sakabe J, Tokura Y. 2013. Trichorhinophalangeal syndrome with low expression of TRPS1 on epidermal and hair follicle epithelial cells. J Dermatol 40: 396–398. [DOI] [PubMed] [Google Scholar]
- Jetley NK, Al-Assiri AH, Al Awadi D. 2009. Congenital para esophageal hernia: a 10 year experience from Saudi Arabia. Indian J Pediatr 76: 489–493. [DOI] [PubMed] [Google Scholar]
- Jobling R, D'Souza R, Baker N, Lara-Corrales I, Mendoza-Londono R, Dupuis L, Savarirayan R, Ala-Kokko L, Kannu P. 2014. The collagenopathies: review of clinical phenotypes and molecular correlations. Curr Rheumatol Rep 16: 394. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O'Donnell-Luria A, Ware J, Hill A, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb KJ, Clayton-Smith J, Harris R. 1997. Evolving phenotype of Marfan's syndrome. Arch Dis Child 76: 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A. 2001. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med 161: 2447–2454. [DOI] [PubMed] [Google Scholar]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, et al. 2010. The revised Ghent nosology for the Marfan syndrome. J Med Genet 47: 476–485. [DOI] [PubMed] [Google Scholar]
- Ludecke HJ, Wagner MJ, Nardmann J, La Pillo B, Parrish JE, Willems PJ, Haan EA, Frydman M, Hamers GJH, Wells DE, et al. 1995. Molecular dissection of a contiguous gene syndrome: localization of the genes involved in the Langer–Giedion syndrome. Hum Mol Genet 4: 31–36. [DOI] [PubMed] [Google Scholar]
- Ludecke HJ, Schaper J, Meinecke P, Momeni P, Groß S, von Holtum D, Hirche H, Abramowicz MJ, Albrecht B, Apacik C, et al. 2001. Genotypic and phenotypic spectrum in Tricho-Rhino-Phalangeal syndrome types I and III. Am J Hum Genet 68: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, Hennekam R, Meinecke P, Zabel B, Rosenthal A, et al. 2000. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet 24: 71–74. [DOI] [PubMed] [Google Scholar]
- Mueller GC, Stark V, Steiner K, Weil J, Kodolitsch Y, Mir TS. 2013. The Kid-Short Marfan Score (Kid-SMS)—an easy executable risk score for suspected paediatric patients with Marfan syndrome. Acta Paediatr 102: e84–e89. [DOI] [PubMed] [Google Scholar]
- Napierala D, Sam K, Morello R, Zheng Q, Munivez E, Shivdasani RA, Lee B. 2008. Uncoupling of chondrocyte differentiation and perichondrial mineralization underlies the skeletal dysplasia in tricho-rhino-phalangeal syndrome. Hum Mol Genet 17: 2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanet Report Series. 2016. Prevalence of rare diseases: bibliographic data. Rare diseases collection, March 2016, Number 1: diseases listed in alphabetical order. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf. [Google Scholar]
- Petersons A, Liepina M, Spitz L. 2003. Neonatal intrathoracic stomach in Marfan's syndrome: report of two cases. J Pediatr Surg 38: 1663–1664. [DOI] [PubMed] [Google Scholar]
- Quarto N, Leonard B, Li S, Marchand M, Anderson E, Behr B, Francke U, Reijo-Pera R, Chiao E, Longaker M. 2012a. Skeletogenic phenotype of human Marfan embryonic stem cells faithfully phenocopied by patient-specific induced-pluripotent stem cells. Proc Natl Acad Sci 109: 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarto N, Li S, Renda A, Longaker MT. 2012b. Exogenous activation of BMP-2 signaling overcomes TGFβ-mediated inhibition of osteogenesis in Marfan embryonic stem cells and Marfan patient-specific induced pluripotent stem cells. Stem Cells 30: 2709–2719. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello HV, Meck JM. 2008. Statement on guidance for genetic counseling in advanced paternal age. Genet Med 10: 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunovic S, Barkovich J, Sherr EH, Slavotinek AM. 2014. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am J Med Genet A 164A: 1744–1749. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. 2013. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HJ, Kim K, Kim IH, Rho S, Park J, Lee KY, Kim SE, Choi BY, Kim N. 2015. Whole exome sequencing for a patient with Rubinstein–Taybi syndrome reveals de novo variants besides an overt CREBBP mutation. Int J Mol Sci 16: 5697–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Data Availability Statement
Whole-exome sequencing data have been submitted to the Database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap) per NIH study protocol and patient consent (accession number phs001232.v1.p1). The FBN1 and TRPS1 variants have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000328213.1 and SCV000328214.1. Patient-derived biospecimens are available by contacting the corresponding author.