

Abstract
In a resource management perspective, the understanding of the relative influence of the physical factors on species connectivity remains a major challenge and is also of great ecological and conservation biology interest. Despite the overfishing threat on the wedge clam Donax trunculus in Europe, relatively little information is known about its population genetic structure and connectivity and their consequences on conservation policies. We employed 16 microsatellite loci to characterise the genetic diversity and population structure of D. trunculus. A total of 514 samples from seven different localities along the Atlantic-Mediterranean transition, from the Atlantic (Gulf of Cádiz) to the north-western Mediterranean were genotyped. The analysis of the population genetic structure displayed a clear distinction along the Atlantic-Mediterranean transition with different clusters in the Atlantic Ocean, the Alboran Sea and the northwestern Mediterranean. Consequently, we recommend that these three areas should be considered as different management units. We showed that all populations seem to be at high long-term risk of extinction with the exception of the protected Doñana National Park population which still seems to have evolutionary potential. Therefore, our results emphasized the necessity of protection of this economic resource and the validity of molecular tools to evaluate the population dynamics.
Many soft and hard faunal barriers to dispersal have been identified in marine environments throughout the world, including, for instance, the Isthmus of Panama1,2,3 and the Terminal Tethyan Event4,5 as hard barriers, as well as the East Pacific Barrier6,7 and the Benguela upwelling8 as soft barriers. Some potential physical factors such as ocean currents, habitats discontinuities or temperature gradients are recognised as driving the biogeographic barriers structuring these regions. Thus, in a resource management perspective, the understanding of the relative influence of the physical factors on species connectivity remains a major challenge and is also of great ecological and conservation biology interest.
Genetic studies have demonstrated that gene flow between conspecifics either side of barrier is often limited for marine9,10,11 and terrestrial fauna12,13,14. Among the major marine biogeographic boundaries, the Strait of Gibraltar that separates the Mediterranean Sea from the Atlantic Ocean and the Almeria Oran Front, which is a semi-permanent dynamic oceanographic front connecting the main jet of incoming Atlantic water and the Mediterranean Sea, have been confirmed by several studies as effective barriers to dispersal11,15,16. The complexity of historical and contemporary physical factors may explain the causes of the reduced dispersal encountered in this region. It is also well established that the Mediterranean Sea was strongly affected by sea level fluctuations during the Messinian Salinity Crisis in the Miocene and subsequently during the Pleistocene glaciations, isolating it several times from the Atlantic15. However, although the actual reopening of the Gibraltar Strait allowed free migration across this area, genetic patterns of some marine species reveal reduced gene flow levels between populations at either side, confirming that it represents a major biogeographic barrier for many taxa. Strong genetic differentiation between Atlantic and Mediterranean populations has been shown in fish as well as in shellfish species with contrasting life histories10,17,18. To explain this phenomenon, two mains factors have been proposed: 1) the one-way surface current of Atlantic water flowing through the Strait of Gibraltar into the Mediterranean19 and, 2) the presence of a front of surface waters between Almeria in Southeast of Spain and Oran in Algeria, the so-called Almeria-Oran front (AOF)16. These factors could represent a barrier to gene flow between Atlantic and Mediterranean populations and be responsible for the reduced migration observed.
The wedge clam, Donax trunculus (Bivalvia: Donacidae), is an Atlanto-Mediterranean warm-temperate species found in the Black Sea, the Mediterranean Sea20 and from Senegal to the northern Atlantic coast of France21. D. trunculus constitutes a very important fishing resource due to its high economic value. Only in Europe, the recorded landings over the last 12 years equal 11,202 tons, with a maximum yield of 1,355 tons in 2005 followed by a steady decline reaching only 516 tons in 201122. Although landing data are only available since 1972 and fishermen were not obliged to declare their catches until 199823, the species has been intensively exploited over the last decades using primarily either artisanal hand-operated dredges in shallow water or commercial boat-operated dredges from subtidal to deeper waters24.
Until recently25,26 and despite the overexploitation threat faced by this shellfish resource, D. trunculus has not been studied from a conservation biology or genetics perspective. The few studies that have paid any attention to its genetic makeup have focused on its genome structure27 or its mitochondrial DNA inheritance25,28 only, but not with the aim to characterise its diversity. However, the availability of highly polymorphic codominant genetic markers, such as microsatellites, allows essential research on the population structure, its effective population size, contemporary phylogeography and conservation genetics of wedge clamps. Nanton et al.26 and Rico et al. (GenBank Accession Numbers HG792255 to HG792275) recently characterised respectively the first nineteen and sixteen polymorphic microsatellite markers in D. trunculus, providing a first battery of genetic tools for the study of the genetic structure of the species to allow the delineation of conservation units essential in fisheries management29.
Understanding connectivity among populations through genetic structure has important conservation implications because genetic assessment is a central criterion in determining the appropriate units and spatial scale for conservation and management30,31. Thus, in a resource management perspective, the aim of this study was to assess the genetic diversity as well as the genetic structure of population of the wedge clam Donax trunculus. For that, 514 individuals from 7 locations along the Atlantic and Mediterranean coasts of Spain were genotyped using 16 microsatellite markers. After evaluating the genetic diversity, the importance of the two well-known characterised biogeographic boundaries (i.e. the Strait of Gibraltar and the Almeria-Oran Front) on the population genetic structure was assessed. Finally, we discuss these findings in a perspective of biodiversity conservation and resource management.
Results
Genetic diversity
A total of 514 individuals from seven different localities were genotyped at 16 microsatellite loci. The genetic diversity found in each locus across samples is summarised in Table 1. Highly significant deviations from Hardy-Weinberg equilibrium were found in all sampling sites and for each locus. Exact tests for genotypic linkage disequilibrium confirmed the absence of linkage among most loci (91.6%; P < 0.05 after Bonferroni correction). Genotyping was consistent across the 514 samples as revealed by mis-scoring rates from two independent scorers below 2%. The number of alleles (NA) per locus ranged from 12 to 33 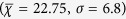 . Observed (HO) and expected (HE) heterozygosity and polymorphic information content ranged from 0.207 to 0.768
. Observed (HO) and expected (HE) heterozygosity and polymorphic information content ranged from 0.207 to 0.768 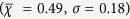 , 0.557 to 0.947
, 0.557 to 0.947 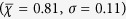 and 0.534 to 0.942
and 0.534 to 0.942 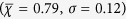 , respectively. The software Microchecker32 depicted the occurrence of null alleles in all loci and the presence of large alleles dropout for some loci. Thus, to better estimate the frequency of null alleles, we relied on the software Cervus33 (Table 1). The null allele frequencies results from Cervus revealed an estimate for each locus ranging from 6.6% to 55.3%
, respectively. The software Microchecker32 depicted the occurrence of null alleles in all loci and the presence of large alleles dropout for some loci. Thus, to better estimate the frequency of null alleles, we relied on the software Cervus33 (Table 1). The null allele frequencies results from Cervus revealed an estimate for each locus ranging from 6.6% to 55.3% 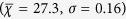 . and all loci, but two (Dtru 16 and Dtru 23), had a null alleles frequency above 10% (Table 1). Table 2 summarises the genetic diversity found for each sampling site. Observed and expected heterozygosity ranged from 0.423 to 0.533
. and all loci, but two (Dtru 16 and Dtru 23), had a null alleles frequency above 10% (Table 1). Table 2 summarises the genetic diversity found for each sampling site. Observed and expected heterozygosity ranged from 0.423 to 0.533 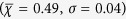 and from 0.756 to 0.826
and from 0.756 to 0.826 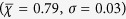 , respectively. Despite the strong departure from HWE observed, we estimated the effective population size, using NeEstimator to explore current demographic patterns in localities exposed to different fishing pressure. The Ne estimates ranged from 210 for Isla Canela to 1191.7 for Doñana (95% confidence interval of the parametric test ranging from 165.1 to infinite; Table 2).
, respectively. Despite the strong departure from HWE observed, we estimated the effective population size, using NeEstimator to explore current demographic patterns in localities exposed to different fishing pressure. The Ne estimates ranged from 210 for Isla Canela to 1191.7 for Doñana (95% confidence interval of the parametric test ranging from 165.1 to infinite; Table 2).
Table 1. Details of the 16 microsatellite markers developed and optimized.
| Locus ID | NA | Range | HO | HE | PIC | NAF | GenBank Acc. Nº |
|---|---|---|---|---|---|---|---|
| D.tru2 | 18 | 97–148 | 0.590 | 0.830 | 0.809 | 0.133 | HG792255 |
| D.tru4 | 14 | 151–179 | 0.641 | 0.750 | 0.716 | 0.123 | HG792256 |
| D.tru6 | 29 | 69–133 | 0.408 | 0.796 | 0.777 | 0.327 | HG792257 |
| D.tru8 | 14 | 127–169 | 0.670 | 0.823 | 0.796 | 0.109 | HG792258 |
| D.tru11 | 26 | 97–153 | 0.469 | 0.715 | 0.700 | 0.211 | HG792259 |
| D.tru14 | 19 | 75–135 | 0.207 | 0.557 | 0.534 | 0.540 | HG792260 |
| D.tru15 | 33 | 215–279 | 0.592 | 0.947 | 0.942 | 0.218 | HG792261 |
| D.tru16 | 26 | 70–124 | 0.768 | 0.912 | 0.903 | 0.093 | HG792262 |
| D.tru19 | 28 | 126–184 | 0.542 | 0.935 | 0.929 | 0.223 | HG792263 |
| D.tru22 | 12 | 215–249 | 0.280 | 0.689 | 0.639 | 0.410 | HG792264 |
| D.tru23 | 33 | 101–167 | 0.738 | 0.835 | 0.825 | 0.066 | HG792265 |
| D.tru26 | 28 | 55–119 | 0.590 | 0.924 | 0.917 | 0.223 | HG792266 |
| D.tru29 | 21 | 262–322 | 0.261 | 0.826 | 0.808 | 0.487 | HG792268 |
| D.tru32 | 25 | 196–284 | 0.323 | 0.674 | 0.659 | 0.425 | HG792271 |
| D.tru40 | 23 | 119–167 | 0.578 | 0.889 | 0.878 | 0.229 | HG792273 |
| D.tru49 | 15 | 144–182 | 0.238 | 0.801 | 0.771 | 0.553 | HG792275 |
514 individuals from 7 sampling sites were used for these analyses. Locus identity, (NA) number of alleles found, (Range) range of allele sizes, (HO) observed and (HE) expected heterozygosity, (PIC) polymorphism information content, (NAF) null-allele frequency and GenBank accession numbers.
Table 2. Characteristics of the sampling sites including the number of clams genotyped (N), observed (HO) and expected (HE) heterozygosity and the genetic effective population size (Ne estimate) with the lower (lower CI) and the higher (higher CI) confidence intervals.
| N | Ho | He | Ar | Ne estimate | lower CI | higher CI | Situation* | |
|---|---|---|---|---|---|---|---|---|
| Isla Canela | 85 | 0.533 | 0.799 | 14.327 | 210 | 165.1 | 238.6 | at high long-term risk of extinction |
| Doñana | 86 | 0.517 | 0.817 | 14.689 | 1191.7 | 526.9 | ∞ | at evolutionary potential |
| Caleta de Vélez | 56 | 0.516 | 0.773 | 13.109 | 782.5 | 320 | ∞ | at high long-term risk of extinction |
| Cabo de Gata | 86 | 0.463 | 0.826 | 13.573 | 357.2 | 245 | 635.6 | at high long-term risk of extinction |
| Gandía | 53 | 0.468 | 0.769 | 12.577 | 332.5 | 203.3 | 845.1 | at high long-term risk of extinction |
| Sant Carles de la Ràpita | 62 | 0.423 | 0.758 | 11.136 | 602.7 | 282.9 | ∞ | at high long-term risk of extinction |
| Roses | 86 | 0.486 | 0.756 | 15.202 | 600.3 | 340.4 | 2210.8 | at high long-term risk of extinction |
The use of ∞ indicates inestimable upper confidence limits* 56.
Genetic structure and gene flow
The analysis of molecular variance (AMOVA) revealed that the majority (98.6%) of the observed variation was explained by differences within the sampling sites, however a small but significant portion of the variation (1.1%) occurred among sampling sites between groups (Table 3). Results of the FST analysis revealed a significant genetic differentiation (P < 0.05) between the Gulf of Cádiz (Southern Atlantic Spanish coast), the Alboran Sea (Southwestern Mediterranean coast) and the Northwestern Mediterranean coast. This corroborates the presence of two well characterised biogeographical barriers, the Strait of Gibraltar34 and the AOF (P > 0.05; Table 4).
Table 3. Results of an analysis of molecular variance (AMOVA) showing the distribution of the genetic variation among groups, among populations within groups and within populations based on 16 microsatellites.
| Source of variation | Sum of squares | Variance components | Percentage variation | |
|---|---|---|---|---|
| Among groups | 21.734 | 0.023 | 1.08* | FCT = 0.011* |
| Among populations within groups | 12.101 | 0.007 | 0.31 | FSC = 0.003 |
| Within populations | 2127.061 | 2.083 | 98.60* | FST = 0.014* |
| Total | 2160.896 | 2.113 |
Gulf of Cádiz (Atlantic coast): Isla Canela and Doñana. Alboran Sea (southwestern Mediterranean coast): Caleta de Vélez and Cabo de Gata. Northwestern Mediterranean coast: Gandía, Sant Carles de la Ràpita and Roses.
*Significant P-value (<0.05).
Table 4. Pairwise F ST (above diagonal) and associated P-value (below diagonal) among pooled sampling sites.
| Gulf of Cádiz | Alboran Sea | Northwestern Mediterranean coast | |
|---|---|---|---|
| Gulf of Cádiz | – | 0.018 | 0.032 |
| Alboran Sea | 0.017* | – | 0.028 |
| Northwestern Mediterranean coast | 0.017* | 0.017* | – |
Gulf of Cadiz (Atlantic coast): Isla Canela and Doñana. Alboran Sea (southwestern Mediterranean coast): Caleta de Vélez and Cabo de Gata.
Northwestern Mediterranean Coast: Gandía, Sant Carles de la Ràpita and Roses. *Significant P-value.
STRUCTURE analyses using the RECESSIVEALLELES and LOCPRIOR functions further substantiated the existence of three genetically differentiated clusters: one in the Gulf of Cádiz; a second one for the samples collected east of the Strait of Gibraltar, in the Alboran Sea, but west of AOF in the Mediterranean Sea; and a third one for the samples collected northwest of the AOF in the Northwestern Mediterranean coast. A clear discrimination was obtained between these three clusters (Fig. 1). The assigned q-value for each pooled sampling site were of 0.806 ± 0.080 (range from 0.462 to 0.920) for the Gulf of Cadiz, 0.542 ± 0.084 (range from 0.357 to 0.736) for the Alboran Sea, and 0.845 ± 0.100 (range from 0.524 to 0.959) for the Northwestern Mediterranean Coast. Some individuals were not included into the mean individual admixture proportions of each pooled sampling site because they clustered into a different group: one individual from Isla Canela (q-value = 0.589) clustered into the Gulf of Cadiz and, two individuals from Caleta de Vélez (q-value = 0.438 and q-value = 0.355) and two from Cabo de Gata (q-value = 0.444 and q-value = 0.458) into the Northwestern Mediterranean Coast.
Figure 1. Assignment of individuals clams to each pooled sampling site in a conventional bar plot (results obtained from STRUCTURE).

The red cluster identifies the population of the Gulf of Cádiz, the green cluster the population of the Alboran Sea and the blue cluster the population of the Northwestern Mediterranean Coast.
Analyses using BayesAss suggest a consistent gene flow among the groups of populations (Table 5). The average migration rate in all pairwise comparisons ranged from 0.003 to 0.034. Each value represents the proportion of individuals that is derived from a corresponding source population for each generation. Altogether, the average migration rate was very low between samples from the Alboran Sea (i.e. source) and the two other groups of populations (i.e. sink), 0.034 and 0.017, respectively for the Gulf of Cadiz and the Northwestern Mediterranean Coast and were higher than any other pairwise comparison.
Table 5. Estimates of migration rates (proportion of individuals).
| Source | |||
|---|---|---|---|
| Destination | Gulf of Cadiz | Alboran Sea | Southwestern Mediterrannean coast |
| Gulf of Cadiz | 0.985 (0.007) | 0.009 (0.005) | 0.006 (0.004) |
| Alboran Sea | 0.032 (0.014) | 0.952 (0.016) | 0.017 (0.009) |
| Southwestern Mediterrannean coast | 0.003 (0.003) | 0.012 (0.006) | 0.985 (0.006) |
Values shown are the means of the posterior distributions of m, the migration rate into each group of populations, and their respective 95% confidence intervals in parentheses. Values in bold are the proportion of individuals derived from the source population each generation. Column headings are the source population, and row headings are the destination populations. Gulf of Cádiz (Atlantic coast): Isla Canela and Doñana. Alboran Sea (Southwestern Mediterranean Coast): Caleta de Vélez and Cabo de Gata. Northwestern Mediterranean Coast: Gandía, Sant Carles de la Ràpita and Roses.
Discussion
Several recent studies have clearly shown that the degree of connectivity among marine taxa separated by biogeographic barriers varies from extensive isolation to complete panmixia and thus the actual influence of these marine transitions remains controversial35. Furthermore, they have also shown that the differences observed are largely dependent on early life history traits, such as pelagic larval duration of the species concerned11,36. In fact, the number of studies that have utilised different molecular markers to unravel the contrasting results of the influence of biogeographical barriers to dispersal in marine environments is so vast that a new synthesis is required (e.g.37). The failure to account for differences in heterozygosity among markers and species may explain to some extent for the irreconcilable patterns observed across ecological and evolutionary timescales38. For example, a recent study on European sea bass Dicentrarchus labrax confirmed that the Atlantic and Mediterranean basins harbour two distinct lineages39. Similarly, Luttikhuizen et al.10 showed a very clear phylogeographic differentiation among the common shrimp (Crangon crangon) populations from the Atlantic Ocean and the Mediterranean Sea. Very high levels of genetic differentiation between Atlantic and Mediterranean populations (pairwise θB values range from 0.395 to 0.510) were also found in the Lusitanian sea star (Asterina gibbosa)18. On the other hand, the analysis of the genetic structure among samples from Atlantic Ocean and Mediterranean Sea of the European spiny lobster (Palinurus elephas) showed small but significant differences (FST for pairwise comparisons within basins = 0.003 ± 0.004 and FST among basins = 0.011 ± 0.005) indicating the presence of two partially-overlapping groups (overlapping of some localities from the Atlantic and Mediterranean basins)40. In contrast, a recent study on the genetic structure the holoplanktonic jellyfish, Pelagia noctiluca, across the Atlantic Ocean and Mediterranean Sea indicated a high degree of connectivity with little evidence of population subdivision between basins41. A very small but significant differentiation (FST = 0.005) among Atlantic and Mediterranean stocks was detected when using microsatellite allele frequencies but no evidence of differentiation was observed using mtDNA haplotype diversity. The magnitude of genetic differentiation observed between the different pooled sampling sites in the wedge clam can be considered as large values for marine taxa (from 2 to 3%).
In this study, the level of genetic differentiation observed between the main oceanographic discontinuities along the Gulf of Cádiz to the Western Mediterranean Sea (the Gibraltar Strait and the AOF) confirms that these biogeographic barriers to dispersal effectively restrict gene flow between D. trunculus populations as supported by our Bayesass analysis and also as reported in many other taxa11. Before the entry of the Atlantic waters throughout the Gibraltar Strait a branch of these waters recirculates near the Strait, in front of the Cape of Trafalgar, towards the northwest along the coast of Cádiz. This area is also influenced by the intense tidal-current regime of the Strait of Gibraltar and the strong topographic interaction between the swift along-shore tidal flow and a submerged ridge running perpendicular to the shoreline15,42. These processes originate persistently a patch of cold water that can affect the connectivity between populations at both sides of the Gibraltar Strait11,43. On the other hand, the AOF is a semi-permanent dynamic oceanographic front connecting the main jet of incoming Atlantic water and the Mediterranean Sea and it is situated at approximately 400 km east of the Strait of Gibraltar16. This front is formed by the convergence between two different water masses (of Atlantic and Mediterranean origin) present in the upper 300 m, and it is known to act as a barrier to gene flow in numerous species11,15. This phenomenon is clearly demonstrated through our results (Table 4 and Fig. 1), which displayed a significant genetic differentiation between the populations from the Gulf of Cádiz, the Alboran Sea and the Northwestern Mediterranean Coast. As such, these three areas should be considered as different management units. However, the levels of genetic differentiation obtained in this study might represent a conservative estimate as levels of heterozygosity may be artificially decreased due to the presence of null alleles. However, despite the heterozygous deficits of the loci used in this study, they were all sufficient for clearly differentiating the populations between the Atlantic Ocean and the Southeast and Northwest Mediterranean Coast of Spain.
The high incidence of null alleles found in this study is consistent with many reports in natural populations of bivalve species analysed with DNA markers such as microsatellites26,44,45. In a previous study on D. trunculus, Nanton et al.26 showed the presence of null alleles (range from 0.109 to 0.277) in 10 loci out of 19. Another study aiming to examine the genetic variability and relationships between two shellfish (Ruditapes philippinarum) populations in Korea also reported the occurrence of null alleles in 7 loci out of 1046. The presence of null alleles has also been reported in a study that assessed the genetic diversity of four wild and six hatchery stocks of the hard clam (Mercenaria mercenaria) in Florida. Null alleles were present in all loci (range from 0.015 to 0.296), but for 4 loci out of 7, the frequency of null alleles was particularly high (>0.146)47. Although the causes of these heterozygote deficits remain uncertain, several hypotheses have been suggested to explain them including the Wahlund effect, selection, inbreeding and the presence of null alleles48. Inbreeding seems unlikely given the large population size of bivalves. Moreover, we cannot demonstrate from these results, selection against heterozygotes. Indeed, although microsatellite markers are supposed to be neutral, it may be possible that one or more loci are linked to genes under selection49 or are under selective constrains due to DNA structure50. Another reason that may explain the presence of heterozygote deficits would be scoring errors. However, we can confidently discard scoring errors as a cause of heterozygous deficits because the robustness of the genotyping was thoroughly assessed by double checking independently all genotypes by two experienced scorers and secondly by using the software Microchecker (unpublished data).
Alternatively and certainly a far more interesting result would be that the deviations from HWE are due to the known severe exploitation that the species has been subjected over the last decades. For example, Hoarau et al.51 used DNA from archived otoliths collected between 1924 and 1972 together with 2002 juvenile’s tissue to estimate effective population size (Ne) in plaice (Pleuronectes platessa). They found that populations examined between 1924 and 1960 were in Hardy-Weinberg equilibrium, whereas populations examined after approximately 1970 were not. They performed extensive testing to rule out that these deviations for HWE were not due to genotyping artefacts and Wahlund effects. Subsequently, the authors were able to attribute the significant heterozygote deficiencies found from 1970 onward to inbreeding and overexploitation. Unfortunately, we do not have access to archived samples to verify if the populations where in HWE in the past. However, we attempted to rule out genotyping errors (see above) and PCR artefacts using the following approaches. First, we redesigned primers whenever the flanking sequences where adequate (D.tru2, D.tru8, D.tru14, D.tru28, and D.tru32). Secondly, we re-amplified 10 loci including the loci for which we redesigned primers and 5 more which produced codominant amplicons for a subsample of individuals (approx. 200) under relaxed annealing temperatures (4 °C lower) to verify that additional alleles had not been missed. None of these strategies resulted in a reduction of the HWE deviations and thus, it was concluded that PCR artefacts were minimal and insufficient to explain the large heterozygous deficits observed. While it is impossible with the current dataset and with the funding that as available for the project to demonstrate that overexploitation may play and important part in the lack of conformance with HWE, this explanation remains plausible and interesting.
In this study, the microsatellite loci characterized for D. trunculus showed variable levels of genetic variation than those previously found26. The allele number of these polymorphic markers ranged from 12 to 33, which is higher than previous results on D. trunculus26 as well as the most part of the studies on other bivalve species such as Sinonovacula constricta52 and Mactra chinensis53. A heterozygosity deficit has been observed for each locus and all sampling sites (Table 1; Table 2). Although these results are higher than those reported by Nanton et al.26, and are consistent with the levels of variability found in both for D. trunculus and other clam species54.
The effective population size (Ne) is an important parameter of numerically small populations, which is positively correlated with their ability to persist in a changing environment and to evolve. The 50/500 rule often cited by conservation practitioners55 has been recently revised to ≥100/1000 by Frankham et al.56. They estimated that a minimum of 100 for Ne was necessary to avoid immediate risk of extinction due to inbreeding depression. Populations with Ne less than 500–1000 were at high long-term risk of extinction due to the loss of ecologically relevant genetic diversity by drift and populations with Ne above 1000 were considered as being able to retain their evolutionary potential. D. trunculus populations have been intensively exploited over the last decades, and particularly in Europe24, and thus, all populations seem to be at high long-term risk of extinction except in Doñana, a managed area located in a National Park where effective population sizes appear to be larger (Table 2)57. However, although we used the Pcrit value recommended for microsatellite markers (Pcrit = 0.02;58), these results should be taken with caution given the presence of null alleles, and as a consequence of the heterozygote deficits. Interestingly however, Isla Canela suffers from severe overexploitation from both, registered and licensed fishermen and from Illegal, Unregulated and Unreported (IUU) extraction and has the lowest Ne estimate. This result supports to some extent the robustness of the analyses57.
Both populations, Isla Canela and Doñana, are geographically very close to each other (85 km) and share a similar habitat structure: long sandy beaches close to a river mouth (Guadiana and Guadalquivir Rivers, respectively) (Fig. 2). However, they also present clear differences in respect to the management of the Donax trunculus fishery. The Doñana beach belongs to the Doñana National Park, and since 1996 has a regulation of the D. trunculus fishery: only 160 shellfish collectors are allowed (have a license). These shellfish collectors have to work using only an artisanal rake with a long net attached to the end of the drag (fishing from boats is forbidden) on the intertidal zone. During five days a week, they can collect a maximum of 25 kg of D. trunculus individuals per day and these must be larger than 25 mm. Furthermore, they have to respect the breeding season (two months a year) without fishing, and they cannot collect when toxic algae are detected in the wedge clams. In the case of Isla Canela, in addition to the shellfish collectors with license that work in the D. trunculus fishery, there are numerous illegal collectors who operate in the area throughout the year and sell their catches in the streets of Huelva and Seville and other smaller villages in both Provinces of Andalusia and, in general, visitors to the beach also harvest clams mostly during the summer months (CR & JAC personal observations). Fishing from boats is also allowed, so the area of collection is wider, covering both the subtidal and intertidal zones. Moreover, there is neither a strict and respected reproductive period without fishery, nor a strict control over the D. trunculus minimum sizes. All these differences in the exploitation of D. trunculus fishery are reflected in the size structure of both populations. For example, in July of 2011, 2012 and 2014, although significant differences in D. trunculus density between the two beaches were only observed in 2014, individuals from Doñana were always significantly larger than those from Isla Canela (Supplementary Methods and Results; Figs 1 and 2), possibly reflecting different age structures resulting from over-harvesting in Isla Canela. This observation is also in line with the observed differences in effective population size between these locations.
Figure 2. Geographical location of sampling sites on the East Atlantic Ocean and Western Mediterranean Sea Coast in Spain (Google Earth, 2016, version 7.1.5.1557, https://www.earth.google.com).

In conclusion, for the first time, the genetic diversity and population structure of Donax trunculus is described along the Atlantic-Mediterranean transition. Using 16 microsatellite markers, we identified the presence of a large proportion of null alleles in this species. However, the presence of null alleles in the loci appears to have no effect on the population genetic structure uncovered. Our results showed that all populations seem to be at high long-term risk of extinction with the exception of the Doñana National Park population which still seems to have evolutionary potential. Interestingly, this population is rigorously managed by the authorities of the National Park with restrictions in the number of fishing licences, the amount of allowed harvest and the use of hand operated dredges. This work represents a contribution for the understanding of the genetic diversity and population structure of this species, and de facto in terms of biodiversity conservation and resource management.
Methods
Sample collection
With the assistance of a licensed fisherman and using both artisanal hand- and boat-operated dredges, Donax trunculus individuals were obtained in seven different locations in the Month of July 2011 in Spain (Fig. 2): Gulf of Cádiz/Atlantic coast (Isla Canela and Doñana National Park), Alboran Sea/Southwestern Mediterranean Coast (Caleta de Vélez and Cabo de Gata) and Northwestern Mediterranean Coast (Gandía, Sant Carles de la Ràpita and Roses). All the samples used in this study belong to species targeted by the commercial fishing industry and are neither protected, nor require approval of any animal ethics committee since they are invertebrates. Permit of sampling in the Doñana National Park was granted for the study.
DNA extraction
DNA was extracted from different wedge clam’s tissues using a slightly modified version of Aljanabi and Martinez59 salting out method. Namely, after addition of the saline solution, the mixture was centrifuged for 30 min at 10,000 g. Also, DNA precipitation was achieved by incubation at −20 °C with 600 μL isopropanol for 30 min. then, after washing out the pellet with 70% ethanol, we centrifuged at 10,000 g for 10 min.
Microsatellite analysis
A total of 514 of Donax trunculus individuals were genotyped with 16 microsatellite markers (D.tru2, D.tru4, D.tru6, D.tru8, D.tru11, D.tru14, D.tru15, D.tru16, D.tru19, D.tru22, D.tru23, D.tru26, D.tru29, D.tru32, D.tru40, D.tru49). The PCR amplifications were done on an Applied Biosystems 9700 DNA thermal cycler. The PCR products were then separated on a capillary sequencer (ABI 3130x Genetic Analyzer, Applied Biosystems, USA) using GeneScan™ 500 LIZ® Size Standard. Allele sizes were determined with the Gene Mapper®V4.0 program (Applied Biosystems, USA).
Genetic diversity analyses
Deviation from Hardy-Weinberg equilibrium and linkage disequilibrium were tested using Genepop (version 4.060 for each marker. We used the software MicroChecker32 to test the presence of genotyping errors due to stuttering, allele drop-out or null alleles. Results showed the presence of null alleles for almost each marker, as well as stuttering and large allele drop-out for some of them (results not shown), but no genotyping errors from allele dropouts or stuttering affected allele scoring. Additionally, a deficit of heterozygosity for almost each marker was observed. Consequently, all samples were re-amplified for each of the 10 loci under relaxed annealing temperature (4 °C lower) to verify that additional alleles had not been missed. After scoring all individuals, the same analyses were re-conducted for each marker. Furthermore, primers were redesigned for the loci where sufficient and adequate flanking regions were available. Primer sequences for this new primers are; D.tru2 F 5′FAMACCACCAATTCTCCTACGG3′, D.tru2 R 5′ATGTGGGCGATGATTTCCT3′; D.tru8 F 5′PET TAAAATTGCCATGCGTGCAG3′, D.tru8 R 5′GAAATATATTGCAGGCTGGTAGG3′; D.tru14 F 5′VIC TTTTTGTTCTTCTGAATAGTGCAA3′, D.tru14 R 5′TTTACAGCGTGTCGCCATCT3′; D.tru28 F 5′FAM CTGAGAAGTAATAAAACGTGAATTGTTG3′, D.tru28 R 5′CAACAGAGCCACTGATGACAA3′; D.tru32 F 5′NED TGGGTCCTGGAGGGTAAAAT3′, D.tru32 R 5′TTAGTCAGAACCTCAACTCTCAAA3′.
The total number of alleles (NA), the observed (HO) and expected (HE) heterozygosity, the polymorphism information content61 and the null-allele frequency (NAF) were also determined for each locus and each sampling site using the program CERVUS 3.033. Effective population size for all the populations were also estimated using the linkage disequilibrium method62 as implemented in NeEstimator (version 2.058).
Analyses of population genetic structure
In order to assess the significance of the level of genetic differentiation, an analysis of molecular variance (AMOVA) was implemented in Arlequin (version 3.5.2.1)63. Specifically, we evaluated the amount of variance attributable to groups of sampling sites (Gulf of Cádiz: Isla Canela and Doñana; Alboran Sea: Caleta de Vélez and Cabo de Gata; Northwestern Mediterranean coast: Gandía, San Carles de la Ràpita and Roses), sampling sites within the groups and within sampling sites. The extent of genetic differentiation among the group of sampling locations was assessed using pairwise FST estimates64 obtained in FSTAT version 2.9.365.
The individual admixture proportions (q-values) for each individual in each sampling site, considering each one like a population, was performed using STRUCTURE v.2.266. Because of the presence of null alleles, analyses were done incorporating RECESSIVEALLELES option. In this case, the program assumes that the recessive allele is never observed in homozygous state but it might be present (e.g. when there might be null alleles). Moreover, the genetic differentiation of population being weak, the LOCPRIOR model has been used. The program assumes an initially probability that each individual is a resident of its sampling locality calculating posterior probabilities that individuals belong to their sampled locality. The number of clusters for each analysis was therefore assessed using the ad hoc statistic ∆K67. An admixture model with correlated allele frequency was used for each analysis with 250,000 steps of the Markov-Chain preceded by a burnin-period of 100,000 steps.
BAYESASS version 3.068 was used to estimate direction and rates of migration that has occurred more recently (i.e., within the last several generations) between the different groups of populations (Gulf of Cadiz, Alboran Sea and Northwestern Mediterrannean coast). Analysis were performed pooling locations as recommended by Meirmans69 (i.e. few populations with many individuals). This method allows for deviations from HWE but assumes linkage disequilibrium and constant migration rates for two generations prior to sampling. Migrants are defined by hybrid genotypes and the program determines a migration rate m, which is the proportion of first generation migrants. Parameters were as follows: number of iterations was 3,000,000, of which 106 are burn in, and the sampling frequency was 2,000.
Additional Information
How to cite this article: Marie, A. D. et al. Implications for management and conservation of the population genetic structure of the wedge clam Donax trunculus across two biogeographic boundaries. Sci. Rep. 6, 39152; doi: 10.1038/srep39152 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
C.R. gratefully acknowledges the Spanish Ministry of Education (Grant № PR2010–0601) and the Spanish Scientific Research Council for the financial support received during his stay at L.B. laboratory. C.R. also acknowledges F. Hiraldo Cano, J.J. Negro Balmaseda, Eric Normandeau and Guillaume Côté for their support during his stay in Canada. This research was financially supported by Organismo Autónomo de Parques Nacionales Grant OAPN122_2010 to C.R., E.M., J.A.C. and P.D. We are also grateful for the support given, throughout the project, by the Coordination Office of Doñana National Park and the Consejería de Agricultura, Pesca y Desarrollo Rural of the Andalusian Government. We also thank Dr Serrão who handled the MS as EBM and two anonymous referees for their constructive comments on a previous version.
Footnotes
Author Contributions C.R., E.M., J.A.C. and P.D. designed the project. P.D. and J.C. organised and sampled all specimens. C.R. conducted the laboratory work, the microsatellite development and the first round of genotyping. E.K. conducted the second round of genotyping and all the genotyping of the second round of amplifications. C.L. contributed with microsatellite experimental design and analyses. A.D.M. analysed the results. A.D.M and C.R. wrote the manuscript. L.B. provided advice in the experimental design, execution of the project and laboratory facilities. All authors reviewed and contribute to the writing of the manuscript.
References
- Farris D. W. et al. Fracturing of the Panamanian Isthmus during initial collision with South America. Geology 39, 1007–1010, doi: 10.1130/g32237.1 (2011). [DOI] [Google Scholar]
- O’Dea A. et al. Environmental change preceded Caribbean extinction by 2 million years. PNAS 104, 5501–5506, doi: 10.1073/pnas.0610947104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton N. & Weigt L. A. New dates and new rates for divergence across the Isthmus of Panama. Philos. Trans. R. Soc. B-Biol. Sci. 265, 2257–2263, doi: 10.1098/rspb.1998.0568 (1998). [DOI] [Google Scholar]
- Rögl F. Palaeogeographic Considerations for Mediterranean and Paratethys Seaways (Oligocene to Miocene). Annalen des Naturhistorischen Museums in Wien. Serie A, Für Mineralogie und Petrographie, Geologie und Paläontologie, Anthropologie und Prähistorie 99, 279–310 (1997). [Google Scholar]
- Cowman P. F., Bellwood D. R. & Rocha L. A. The historical biogeography of coral reef fishes: global patterns of origination and dispersal. J. Biogeogr. 40, 209–224, doi: 10.1111/jbi.12003 (2013). [DOI] [Google Scholar]
- Lessios H. A., Kessing B. D. & Robertson D. R. Massive gene flow across the world’s most potent marine biogeographic barrier. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 265, 583–588, doi: 10.1098/rspb.1998.0334 (1998). [DOI] [Google Scholar]
- Lessios H. A. & Robertson D. R. Crossing the impassable: genetic connections in 20 reef fishes across the eastern Pacific barrier. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 273, 2201–2208, doi: 10.1098/rspb.2006.3543 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons M. J., Buecher E., Thibault-Botha D. & Helm R. R. Patterns in marine hydrozoan richness and biogeography around southern Africa: implications of life cycle strategy. J. Biogeogr. 37, 606–616, doi: 10.1111/j.1365-2699.2009.02237.x (2010). [DOI] [Google Scholar]
- Quesada H., Zapata C. & Alvarez G. A multilocus allozyme discontinuity in the mussel Mytilus galloprovincialis - the interaction of ecological and life-history factors. Mar. Ecol.-Prog. Ser. 116, 99–115, doi: 10.3354/meps116099 (1995). [DOI] [Google Scholar]
- Luttikhuizen P. C., Campos J., Bleijswijk J. v., Peijnenburg K. T. C. A. & van der Veer H. W. Phylogeography of the common shrimp, Crangon crangon (L.) across its distribution range. Mol. Phylogenet. Evol. 46, 1015–1030, doi: 10.1016/j.ympev.2007.11.011 (2008). [DOI] [PubMed] [Google Scholar]
- Galarza J. A., Turner G. F., Macpherson E. & Rico C. Patterns of genetic differentiation between two co-occurring demersal species: the red mullet (Mullus barbatus) and the striped red mullet (Mullus surmuletus). Can. J. Fish. Aqua. Sci. 66, 1478–1490, doi: 10.1139/f09-098 (2009). [DOI] [Google Scholar]
- Gómez-Díaz E., González-Solís J., Peinado M. A. & Page R. D. M. Phylogeography of the Calonectris shearwaters using molecular and morphometric data. Mol. Phylogenet. Evol. 41, 322–332, doi: 10.1016/j.ympev.2006.05.006 (2006). [DOI] [PubMed] [Google Scholar]
- Jaramillo-Correa J. P. et al. The Strait of Gibraltar as a major biogeographic barrier in Mediterranean conifers: a comparative phylogeographic survey. Mol. Ecol. 19, 5452–5468, doi: 10.1111/j.1365-294X.2010.04912.x (2010). [DOI] [PubMed] [Google Scholar]
- Mata V. A., da Silva L. P., Lopes R. J. & Drovetski S. V. The Strait of Gibraltar poses an effective barrier to host-specialised but not to host-generalised lineages of avian Haemosporidia. Int. J. Parasit. 45, 711–719, doi: 10.1016/j.ijpara.2015.04.006 (2015). [DOI] [PubMed] [Google Scholar]
- Patarnello T., Volckaert F. A. & Castilho R. Pillars of Hercules: is the Atlantic–Mediterranean transition a phylogeographical break? Mol. Ecol. 16, 4426–4444, doi: 10.1111/j.1365-294X.2007.03477.x (2007). [DOI] [PubMed] [Google Scholar]
- Tintore J., La Violette P. E., Blade I. & Cruzado A. A Study of an Intense Density Front in the Eastern Alboran Sea: The Almeria–Oran Front. J. Phys. Oceanogr. 18, 1384–1397, doi: (1988). [DOI] [Google Scholar]
- Lowe C. D., Martin L. E., Montagnes D. J. S. & Watts P. C. A legacy of contrasting spatial genetic structure on either side of the Atlantic–Mediterranean transition zone in a marine protist. PNAS 109, 20998–21003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baus E., Darrock D. J. & Bruford M. W. Gene-flow patterns in Atlantic and Mediterranean populations of the Lusitanian sea star Asterina gibbosa. Mol. Ecol. 14, 3373–3382, doi: 10.1111/j.1365-294X.2005.02681.x (2005). [DOI] [PubMed] [Google Scholar]
- Bryden H. L., Candela J. & Kinder T. H. Exchange through the Strait of Gibraltar. Prog. Oceanogr. 33, 201–248, doi: 10.1016/0079-6611(94)90028-0 (1994). [DOI] [Google Scholar]
- Bayed A. & Guillou J. A contribution to the study of populations of the Donax genus - The population of Donax trunculus L (Mollusca, Bivalvia). Annales De L Institut Oceanographique 61, 139–147 (1985). [Google Scholar]
- Tebble N. British bivalve shells. A handbook for identification (1966). [Google Scholar]
- FAO-FIGIS. In Fisheries Global Information System (http://www.fao.org/fishery/statistics/global-capture-production/query/en, 2013).
- Gaspar M. B., Ferreira R. & Monteiro C. C. Growth and reproductive cycle of Donax trunculus L., (Mollusca: Bivalvia) off Faro, southern Portugal. Fish Res. 41, 309–316, doi: 10.1016/s0165-7836(99)00017-x (1999). [DOI] [Google Scholar]
- Gaspar M. B. et al. Depth segregation phenomenon in Donax trunculus (Bivalvia: Donacidae) populations of the Algarve coast (southern Portugal). Sci. Mar. 66, 111–121, doi: 10.3989/scimar.2002.66n2111 (2002). [DOI] [Google Scholar]
- Nantón A., Freire R., Arias-Pérez A., Gaspar M. B. & Méndez J. Identification of four Donax species by PCR–RFLP analysis of cytochrome c oxidase subunit I (COI). Eur. Food Res. Technol. 240, 1129–1133, doi: 10.1007/s00217-015-2416-z (2015). [DOI] [Google Scholar]
- Nantón A., Arias-Pérez A., Méndez J. & Freire R. Characterization of nineteen microsatellite markers and development of multiplex PCRs for the wedge clam Donax trunculus (Mollusca: Bivalvia). Mol. Biol. Rep. 41, 5351–5357, doi: 10.1007/s11033-014-3406-0 (2014). [DOI] [PubMed] [Google Scholar]
- Plohl M. & Cornudella L. Characterization of Interrelated Sequence Motifs in Four Satellite DNAs and Their Distribution in the Genome of the Mollusc Donax trunculus. J. Mol. Evol. 44, 189–198, doi: 10.1007/pl00006135 (1997). [DOI] [PubMed] [Google Scholar]
- Theologidis I., Fodelianakis S., Gaspar M. B. & Zouros E. Doubly uniparental inheritance (DUI) of mitochondrial dna in Donax trunculus (Bivalvia: Donacidae) and the problem of its sporadic detection in Bivalvia. Evolution 62, 959–970, doi: 10.1111/j.1558-5646.2008.00329.x (2008). [DOI] [PubMed] [Google Scholar]
- Hauser L., Waples R. S. & Carvalho G. R. Special Issue: Advances in Marine Fish and Fisheries Genetics. Fish. Fish. 9, 331–332, doi: 10.1111/j.1467-2979.2008.00307.x (2008). [DOI] [Google Scholar]
- Waples R. S. & Gaggiotti O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15, 1419–1439, doi: 10.1111/j.1365-294X.2006.02890.x (2006). [DOI] [PubMed] [Google Scholar]
- Funk W. C., McKay J. K., Hohenlohe P. A. & Allendorf F. W. Harnessing genomics for delineating conservation units. Trends Ecol Evol 27, 489–496, doi: 10.1016/j.tree.2012.05.012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosterhout C., Hutchinson W. F., Wills P. M. & Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538, doi: 10.1111/j.1471-8286.2004.00684.x (2004). [DOI] [Google Scholar]
- Kalinowski S. T., Taper M. L. & Marshall T. C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16, 1099–1106, doi: 10.1111/j.1365-294X.2007.03089.x (2007). [DOI] [PubMed] [Google Scholar]
- Riesgo A. et al. Comparative description of ten transcriptomes of newly sequenced invertebrates and efficiency estimation of genomic sampling in non-model taxa. Front. Zool. 9, 33, doi: 10.1186/1742-9994-9-33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cisneros A., Palacín C., Ben Khadra Y. & Pérez-Portela R. Low genetic diversity and recent demographic expansion in the red starfish Echinaster sepositus (Retzius 1816). Sci. Rep. 6, doi: 10.1038/srep33269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangensteen O. S., Turon X., Pérez-Portela R. & Palacín C. Natural or Naturalized? Phylogeography Suggests That the Abundant Sea Urchin Arbacia lixula Is a Recent Colonizer of the Mediterranean. PLoS One 7, doi: 10.1371/journal.pone.0045067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen B. W. et al. Comparative phylogeography of the ocean planet. PNAS 113, 7962–7969, doi: 10.1073/pnas.1602404113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellberg M. E. Gene Flow and Isolation among Populations of Marine Animals. Annu. Rev. Ecol. Evol. Syst. 40, 291–310, doi: 10.1146/annurev.ecolsys.110308.120223 (2009). [DOI] [Google Scholar]
- Souche E. L. et al. Range-wide population structure of European sea bass Dicentrarchus labrax. Biol. J. Linnean Soc. 116, 86–105, doi: 10.1111/bij.12572 (2015). [DOI] [Google Scholar]
- Palero F., Abellό P., Macpherson E., Beaumont M. & Pascual M. Effect of oceanographic barriers and overfishing on the population genetic structure of the European spiny lobster (Palinurus elephas). Biol. J. Linnean Soc. 104, 407–418, doi: 10.1111/j.1095-8312.2011.01728.x (2011). [DOI] [Google Scholar]
- Glynn F. et al. High-resolution genetic analysis reveals extensive gene flow within the jellyfish Pelagia noctiluca (Scyphozoa) in the North Atlantic and Mediterranean Sea. Biol. J. Linnean Soc. 117, 252–263, doi: 10.1111/bij.12654 (2016). [DOI] [Google Scholar]
- García Lafuente J. & Ruiz J. The Gulf of Cádiz pelagic ecosystem: A review. Prog. Oceanogr. 74, 228–251, doi: 10.1016/j.pocean.2007.04.001 (2007). [DOI] [Google Scholar]
- Sala-Bozano M., Ketmaier V. & Mariani S. Contrasting signals from multiple markers illuminate population connectivity in a marine fish. Mol. Ecol. 18, 4811–4826, doi: 10.1111/j.1365-294X.2009.04404.x (2009). [DOI] [PubMed] [Google Scholar]
- Launey S., Ledu C., Boudry P., Bonhomme F. & Naciri-Graven Y. Geographic Structure in the European Flat Oyster (Ostrea edulis L.) as Revealed by Microsatellite Polymorphism. J. Hered. 93, 331–351, doi: 10.1093/jhered/93.5.331 (2002). [DOI] [PubMed] [Google Scholar]
- Chiesa S. et al. Null alleles of microsatellites for Manila clam Ruditapes philippinarum. Anim. Genet. 47, 135–136, doi: 10.1111/age.12382 (2016). [DOI] [PubMed] [Google Scholar]
- Kim E. M. et al. New polymorphic microsatellite markers for the Korean manila clam (Ruditapes philippinarum) and their application to wild populations. Genet. Mol. Res. 13, 8163–8173, doi: 10.4238/2014.October.7.11 (2014). [DOI] [PubMed] [Google Scholar]
- Hargrove J. S., Sturmer L., Scarpa J. & Austin J. D. Assessment of Genetic Diversity in Wild and Aquaculture Stocks of Mercenaria mercenariain Florida. J. Shellfish Res. 34, 355–365, doi: 10.2983/035.034.0218 (2015). [DOI] [Google Scholar]
- Zouros E. & Foltz D. W. Possible explanations of the heterozygote deficiency in Bivalve mollusks. Malacologia 25, 583–591 (1984). [Google Scholar]
- Astanei I., Gosling E., Wilson J. I. M. & Powell E. Genetic variability and phylogeography of the invasive zebra mussel, Dreissena polymorpha (Pallas). Mol. Ecol. 14, 1655–1666, doi: 10.1111/j.1365-294X.2005.02530.x (2005). [DOI] [PubMed] [Google Scholar]
- Rico C., Rico I. & Hewitt G. 470 Million Years of Conservation of Microsatellite Loci among Fish Species. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 263, 549–557, doi: 10.1098/rspb.1996.0083 (1996). [DOI] [PubMed] [Google Scholar]
- Hoarau G. et al. Low effective population size and evidence for inbreeding in an overexploited flatfish, plaice (Pleuronectes platessa L.). Proc. R. Soc. Lond. Ser. B-Biol. Sci. 272, 497–503, doi: 10.1098/rspb.2004.2963 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H. T., Jiang H. B., Liu X. Q., Wu X. P. & Wei X. M. Polymorphic microsatellite loci for the razor clam, Sinonovacula constricta. Genet. Mol. Res. 14, 145–148, doi: 10.4238/2015.January.15.17 (2015). [DOI] [PubMed] [Google Scholar]
- Li H. J., Zhang J. J., Li H. B., Gao X. G. & He C. B. Development and characterization of 13 polymorphic microsatellite markers for the Chinese surf clam (Mactra chinensis) through Illumina paired-end sequencing. Conserv. Genet. Resour. 6, 877–879, doi: 10.1007/s12686-014-0229-1 (2014). [DOI] [Google Scholar]
- Pereira S., Arias A., Méndez J., Insua A. & Freire R. Isolation of twelve microsatellite markers in the pullet carpet shell Venerupis pullastra (Bivalvia: Veneridae). Conserv. Genet. Resour. 2, 201–203, doi: 10.1007/s12686-009-9134-4 (2010). [DOI] [Google Scholar]
- Jamieson I. G. & Allendorf F. W. How does the 50/500 rule apply to MVPs? Trends Ecol. Evol. 27, 578–584, doi: 10.1016/j.tree.2012.07.001 (2012). [DOI] [PubMed] [Google Scholar]
- Frankham R., Bradshaw C. J. A. & Brook B. W. Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 170, 56–63, doi: 10.1016/j.biocon.2013.12.036 (2014). [DOI] [Google Scholar]
- Rico C., Drake P., Macpherson E. & Cuesta J. A. Estimación de la diversidad genética y del tamaño efectivo de la población de coquina Donax trunculus del Parque Nacional de Doñana y su contribución a áreas no protegidas., (Internal Report. Organismo Autónomo Parques Nacionales, Ministerio de Agricultura, Alimentación y Medio Ambiente, Madrid Spain, 2015).
- Do C. et al. NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14, 209–214, doi: 10.1111/1755-0998.12157 (2014). [DOI] [PubMed] [Google Scholar]
- Aljanabi S. M. & Martinez I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 25, 4692–4693, doi: 10.1093/nar/25.22.4692 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F. Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Res. 8, 103–106, doi: 10.1111/j.1471-8286.2007.01931.x (2008). [DOI] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. & Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 4, 44–57, doi: 10.1038/nprot.2008.211 (2008). [DOI] [PubMed] [Google Scholar]
- Waples R. S. & Do C. H. I. Ldne: a program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Res. 8, 753–756, doi: 10.1111/j.1755-0998.2007.02061.x (2008). [DOI] [PubMed] [Google Scholar]
- Excoffier L. & Lischer H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 10, 564–567, doi: 10.1111/j.1755-0998.2010.02847.x (2010). [DOI] [PubMed] [Google Scholar]
- Weir B. S. & Cockerham C. C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 38, 1358–1370, doi: 10.2307/2408641 (1984). [DOI] [PubMed] [Google Scholar]
- Goudet J. F. S. T. A. T. (Version 1.2): A Computer Program to Calculate F-Statistics. J. Hered. 86, 485–486 (1995). [Google Scholar]
- Pritchard J. K., Stephens M. & Donnelly P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 155, 945–959, doi: 10.3410/f.1015548.197423 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G., Regnaut S. & Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620, doi: 10.1111/j.1365-294X.2005.02553.x (2005). [DOI] [PubMed] [Google Scholar]
- Wilson G. A. & Rannala B. Bayesian Inference of Recent Migration Rates Using Multilocus Genotypes. Genetics 163, 1177–1191, doi: 10.3410/f.1012487.188326 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirmans P. G. Nonconvergence in Bayesian estimation of migration rates. Mol. Ecol. Res. 14, 726–733, doi: 10.1111/1755-0998.12216 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.