

Abstract
In our previous studies, DAZAP2 gene expression was down-regulated in untreated patients of multiple myeloma (MM). For better studying the structure and function of DAZAP2, a full-length cDNA was isolated from mononuclear cells of a normal human bone marrow, sequenced and deposited to Genbank (AY430097). This sequence has an identical ORF (open reading frame) as the NM_014764 from human testis and the D31767 from human cell line KG-1. Phylogenetic analysis and structure prediction reveal that DAZAP2 homologues are highly conserved throughout evolution and share a polyproline region and several potential SH2/SH3 binding sites. DAZAP2 occurs as a single-copy gene with a four-exon organization. We further noticed that the functional DAZAP2 gene is located on Chromosome 12 and its pseudogene gene is on Chromosome 2 with electronic location of human chromosome in Genbank, though no genetic abnormalities of MM have been reported on Chromosome 12. The ORF of human DAZAP2 encodes a 17-kDa protein, which is highly similar to mouse Prtb. The DAZAP2 protein is mainly localized in cytoplasm with a discrete pattern of punctuated distribution. DAZAP2 may associate with carcinogenesis of MM and participate in yet-to-be identified signaling pathways to regulate proliferation and differentiation of plasma cells.
Key words: multiple myeloma, DAZAP2, SH2/SH3, polyproline
Introduction
Given that there is a large amount of data on the sequence of new genes whose functions are yet unknown in public databases, and hence a great challenge to researchers in the functional study, bioinformatics has become a powerful tool for predicting gene’s function. Gene expression patterns in diseases (including cancer) in combination with special analytical software do give scientists directions for further research in acquiring functional information of new genes. Multiple myeloma (MM) is a disorder characterized by the uncontrolled proliferation and accumulation of malignant plasma cells in the bone marrow (1). To identify the MM-associated genes and delineate their pathogenic correlation, we used cDNA profiling to assess the expression of 1,999 genes in bone marrow mononuclear cells from MM patients and normal subjects. The levels of the most significantly down-regulated gene DAZAP2 (deleted in azoospermia associated protein 2; ref. 2) were verified by Western blotting analysis of MM samples (7/11 or 63.6%), but all normal subjects (control) expressed DAZAP2 (3). DAZAP2 expression at both the mRNA and protein levels was negatively correlated with the pathogenesis of MM. The DAZAP2 protein had originally been identified as an interacting protein of germ-cell-specific RNA-binding proteins DAZ (deleted in azoospermia; ref. 4). The published DAZAP2 cDNA comes from a human testis cDNA library, located on Chromosome 2 with unknown function (4). To explore the functional significance of the down-regulated DAZAP2 gene in MM, we isolated this gene, characterized its product from human bone marrow mononuclear cells of a normal subject, and searched the available databases. By suitable software, we identified its homologues and characterized its molecular phylogenies. Since the positions of functional and/or structural importance tend to be more conserved in evolution, we may find important functional region of DAZAP2 by amino acid sequence alignment of 15 vertebrates’ DAZAP2 proteins. Furthermore, the expression pattern of DAZAP2 may also provide hints of its function. All these analyses tried to shed new light on the function of the DAZAP2 gene.
Results
Sequence analysis, molecular phylogeny and genomic organization of DAZAP2
A full-length DAZAP2 cDNA was isolated from mononuclear cells of normal human bone marrow, sequenced and deposited to Genbank (AY430097). Figure 1 shows its nucleotide and predicted amino acid sequence. The DAZAP2 gene contains a short 5’ UTR, an ORF (open reading frame) from Nucleotides 84–590, and a relatively long 3’ UTR plus poly(A) tail. Its ORF is predicted to encode a 17-kDa protein with 168 amino acids and is the same as those of the DAZAP2 gene (NM_014764) from human testis (4) and D31767 (KIAA0058) from human cell line KG-1 (5). Human DAZAP2 is highly similar to mouse Prtb (proline codon-rich transcript, brain expressed; ref. 6). Further sequence analysis reveals that DAZAP2 contains a proline-rich region and several potential SH2 (YxxΨ) and SH3 (PxΨP) domain-binding motifs.
Fig. 1.

The nucleotide and predicted amino acid sequence of DAZAP2. The cDNA was cloned from total RNA of mononuclear cells of normal bone marrow. The polyproline region is shown by a rectangle. Potential SH2 (YxxΨ) and SH3 (PxΨP) domain-binding motifs are underlined. The initiation and stop codons are in bold. Triangles denote exon boundaries. The 3’-polyadenylation signals are underlined.
As shown in Figure 2A, human DAZAP2 has a highly related gene in two distant species, Apis mellifera (B1515614) and Ciona intestinalis (AY490098). This gene is highly conserved in vertebrates from zebrafish to human being whose homologues show a slow evolution. Comparison of 15 vertebrates’ DAZAP2 (Genbank IDs: AU297060, CB553719, XM_217038, NM_011873, BM967886, CD051977, CD467315, CD728885, AY490096, BC042215, BX078169, CB516510, AY490097, and BC059628) reveals that the polyproline region is a conserved structural motif (Figure 2B). Notably, its C-terminus is more conserved than its N-terminus.
Fig. 2.

Phylogenetic analysis and sequence alignment of DAZAP2. A. The tree topologies. The branch lengths, estimated by ML methods, denote nucleotide substitutions per nucleotide sites. The numbers on nodes are bootstrap test values. Left: ML at 1,000 puzzling steps for protein sequences under HKY85 model with five categories of Gamma substitution rate plus invariable sites (5G+I); Middle: ML at 1,000 puzzling steps for nucleotide sequences under JTT model with 5G+I; Right: MP at 200 replicates for protein sequences under default parameters. GenBank accession numbers are given along the tree. B. Sequence alignment of vertebrate DAZAP2 proteins. Dashes denote gaps or missing residues. The bold amino acids at the bottom represent 90% consensus in the alignment. The 17 prolines are largely conserved and the N-terminus is less conserved than the C-terminus. The single α-helix (underlined) present in the C-terminal end is flanked by potential extended β-sheets (not shown).
Genomic analysis by database searching (www.ncbi.nlm.nih.gov) showed that DAZAP2 occurs as a single-copy gene with a four-exon organization conserved throughout vertebrate evolution (Figure 3A). By electronic location of human chromosome in Genbank, we further noticed that the functional DAZAP2 gene is located on human Chromosome 12 rather than Chromosome 2 that had been reported (4). By comparing the DAZAP2 with its homologue sequence on Chromosome 2, we identified that the latter is a pseudogene that contains no introns but processed poly(A) tails and nonsense mutation that inactivates its protein expression (Figure 3B).
Fig. 3.

Genomic organization of DAZAP2 and its nucleotide sequence alignment with the pseudogene. A. The structure of four exons in human and pufferfish and five exons in sea squirt. The ATG initiation codon and stop codon are illustrated. The lengths of every exon (open bars) and intron (straight lines) are shown by the amount of base pairs. Note that rat, mouse and frog have the same gene organization as fish and human. B. The alignment between human DAZAP2 and pseudogene (φ). Nucleotide changes are spelled out. Dots denote identity and dashes denote deletion. For the wild-type gene, the initiation (M) codon and stop signals are in bold. For the pseudogene, the nonsense mutation TAG is bold, and the 311-bp and 321-bp insertion sequences are omitted.
DAZAP2 expression in human tissues
To define the pattern of DAZAP2 expression in various tissues, a human multiple tissue Northern blot was performed. As shown in Figure 4, DAZAP2 is expressed as a 1.9-Kb transcript in seven normal human tissues, including stomach, thyroid, spinal cord, lymph node, trachea, adrenal gland, and bone marrow. The results indicate a ubiquitous expression of the DAZAP2 gene in vivo and suggest an important functional role of the DAZAP2 gene in multiple tissues.
Fig. 4.

Expression of DAZAP2 in human tissues. A human multiple tissue Northern blot was hybridized with a DAZAP2 cDNA probe. Lane M: size markers. Lanes 1-7: stomach, thyroid, spinal cord, lymph node, trachea, adrenal gland, and bone marrow. A single and abundant transcript of about 1.9 Kb is seen in all tissues. The intensity of β-actin mRNA bands was relatively constant (not shown).
Expression and intracellular localization of DAZAP2 protein
To verify the expression of DAZAP2 protein in eukaryotic cells, pEGFP-N1-DAZAP2 was transfected into COS7 cells. Western-blotting analysis detected a 44-kDa protein in cells transfected with pEGFP-N1-DAZAP2 and a 27-kDa protein in cells transfected with pEGFP-N1 alone by using an anti-GFP antibody. Thus, the DAZAP2 protein expressed in COS7 cells was about 17-kDa (Figure 5), a result that is consistent with ORF prediction and Western blot of native DAZAP2 protein expressed in the bone marrow (data not shown).
Fig. 5.
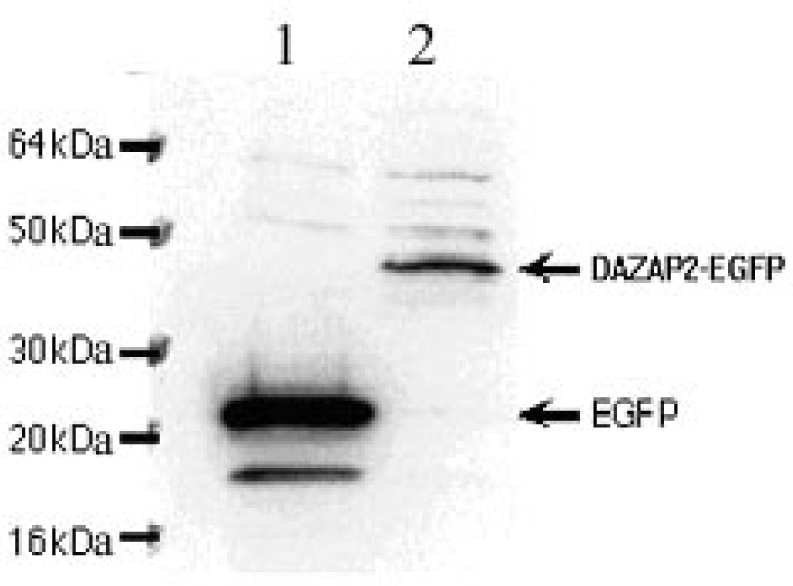
Immunoblotting analysis of the fusion protein DAZAP2-EGFP expression. Lane 1: EGFP protein; the MW (molecular weight) of EGFP protein is about 27 kDa. Lane 2: DAZAP2-EGFP fusion protein; the MW of DAZAP2-EGFP fusion protein is about 44 kDa.
The intracellular localization of DAZAP2 protein was next examined by immunofluorescent microscopy. In control cells expressing GFP alone, the green fluorescence was distributed evenly in the cytosol and in the nucleus (Figure 6A). By contrast, COS7 cells expressing DAZAP2-GFP showed a largely cytosolic location with small distinctive punctated staining (Figure 6B, green color). The green fluorescence of DAZAP2-GFP fusion protein showed no apparent overlap with the staining of Golgi (Figure 6B, red color), indicating that DAZAP2 protein mainly resides in the cytoplasm with a discrete pattern of punctated structures.
Fig. 6.

Intracellular localization of DAZAP2 protein. COS7 cells were transfected with either pEGFP vector or DAZAP2-GFP plasmid and visualized under confocal microscopy. A. Expression of GFP in control cells. Note the green fluorescence of GFP is evenly distributed in the cytosol and the nucleus. B. The DAZAP2-GFP fusion protein (green) shows largely a cytosolic distribution with small discrete punctuated staining; its green fluorescence appears not to overlap with the staining of Golgi (red).
Discussion
Multiple myeloma is a malignant disorder of plasma cells for which the molecular etiology is still unknown (1). The fact that the down-regulation of DAZAP2 occurred at both the mRNA (2) and protein levels (3) provides a strong indication for its specific association with MM. A full-length 1,913 bp DAZAP2 cDNA (AY430097) isolated from human bone marrow mononuclear cells has an identical ORF as the published DAZAP2 from human testis library and KG-1 cell line. To explore the sequence features and functional significance of DAZAP2, we searched available databases and used three program packages and methods to identify its homologues and characterize its molecular phylogenies. A more detailed picture of DAZAP2 molecular evolution was obtained when we isolated three closely related cDNA fragments of DAZAP2 from three distant species (AY490096, AY490097, AY490098). The three program packages and methods we employed are TREE-PUZZLE, MEGA2 and T-coffee. TREE-PUZZLE is a quartet-based maximum-likelihood phylogenetic analysis tool, including a broad variety of evolutionary models (7), for example, models for DNA sequences (TN, ref. 8), models for protein sequences (JTT and VT, ref. 9., 10.). MEGA2 is a set of software that can compute evolutionary distances based on the observed differences in amino acid and nucleotide sequences (11). The bootstrap test and the interior branch length tests can examine the reliability of the inferred phylogenies 12., 13.. Most of the bootstrap test values on nodes of the tree topologies have a high score (Figure 2A), which elucidate the accuracy of the phylogenetic tree (14). From the result of the phylogenetic tree, in which human DAZAP2 has a highly related gene in two distant species, Apis mellifera and Ciona intestinalis, we concluded that DAZAP2 has an early origin. This suggests that DAZAP2 might have vital cellular functions. T-coffee is a method for multiple alignment that provides a dramatic improvement in accuracy, and its resulting alignments are significantly more reliable as compared to the most commonly used alternatives (15). By this reliable method, we showed that DAZAP2 was conserved throughout evolution, particularly in the C-terminus of the protein we defined a potentially important function domain for DAZAP2. Its vertebrate homologues virtually carried the same genomic organization and structural features (four exons and three introns; the others are 5’ and 3’ untranslated regions). Moreover, by nucleotide sequence alignment, we found that human has a functional gene on Chromosome 12 and its pseudogene on Chromosome 2, yet Chromosome 12 has not been implicated to harbor MM-associated gene abnormalities 1., 16.. The pseudogene protein expression was inactivated by a nonsense mutation (substitution TAG for TAC). Structure prediction revealed multiple sequence motifs in DAZAP2 including several potential SH2 and SH3 binding sites, which also conserved throughout evolution. Protein-protein interactions mediated by SH2 and SH3 domains have been shown to play a central role in multiple signaling pathways that regulate cell proliferation and differentiation (17). For instance, the SH3-SH2-SH3 Grb2 adapter links the receptors to the Ras pathway (18); Nck/Dock, composed of three SH3 domains and one SH2 domain, links cell surface receptors to the actin cytoskeleton (19). Hence, it is of great interest to uncover the role of DAZAP2 in such signaling events in regulating the proliferation of tumor cells in MM, should the disruption of these interactions result in loss-of-function phenotypes.
As shown here and elsewhere 4., 5., DAZAP2 was broadly expressed in normal tissues and cell lines. Given this ubiquitous occurrence of DAZAP2 and testis specificity of DAZ, their interaction is unlikely to play a critical role in the pathogenesis of MM, although the expression of DAZ was not assessed. It is of further interest to note the mouse homologue Prtb, which was first cloned from mouse brain (6) and located in the cytoplasm (20). Its expression is increased in osteoclast cell line (MC3P3-E1) upon adhesion to polystyrene with or without surface-adsorbed serum proteins, suggesting that Prtb is involved in cell adhesion-mediated function (20). The human 17-kDa DAZAP2 protein was also mainly localized in cytoplasm as mouse Prtb protein. Nevertheless, whether human DAZAP2 and mouse or rat Prtb genes are functionally equivalent in the setting of MM pathogenesis and progression remains to be elucidated.
Materials and Methods
Bone marrow sample
Normal control bone marrow samples were obtained from the Department of Hematology of Xiangya Hospital, Central South University, China.
Total RNA preparation
Mononuclear cells were isolated from bone marrow samples by the lymphocyte isolation solution. Total RNA was prepared from the bone marrow mononuclear cells using Trizol kit (Invitrogen, Carlsberg, USA). RNA concentration was determined on a UV spectrophotometer. The quality of RNA was assessed by electrophoresis on 1.2% agarose gel.
Isolation and sequencing of DAZAP2 cDNA from bone marrow
Based on the sequence of DAZAP2, gene-specific primers (GSPs) in either sense (S) or antisense (A) were designed to isolate the DAZAP2 gene from mononuclear cells of bone marrow of normal control by the method of rapid amplification of cDNA ends (RACE). For the 5’ RACE, 2.5 μg of total RNA was reverse-transcribed with the 5’-end phosphorylated GSP (RT1): 5’-ACAACATGTGTCT-3’. The RNA strand of DNA-RNA duplex was digested and the single cDNA strand was circularized as described in the 5’-Full RACE kit (TaKaRa Bio Inc., Dalian, China) (21). The cDNA was then amplified sequentially with two different pairs of GSPs as follows: S1, 5’-GGTATGATGCAGGTGCCAG-3’/A1, 5’-GACCGACTGGATAAT-3’; and S2, 5’-CGTCCTCGTAACTCAGCGG-3’/A2, 5’-GATACAGAGAAACTACAGG-3’. For the 3’ RACE, 1.0 μg of total RNA was reverse-transcribed with supplied oligo dT-3 sites adapter primer, and then amplified once with adapter primer 5’-CTGATCTAGAGGTACCGGATCC-3’ and GSP (S3), 5’-GGTGGCTACACCATCTGGTG-3’, as described in the 3’-Full RACE kit (TaKaRa Bio Inc.). PCR products were analyzed on 1.5% agarose gel. The 5’ and 3’ RACE products were purified, subcloned into the pEGM-Teasy vector (Promega, San Luis Obispo, USA), and then sequenced on an automated ABI377 sequencer.
Sequence analysis, structure prediction and phylogenetic analysis
Sequence analysis and structural prediction were performed by searching public databases on the website servers of NCBI (http://www.ncbi.nlm.nih.gov/BLAST/), NetStar (http://www.cbs.dtu.dk/services) and Scan Prosite (http://us.expasy.org/cgi-bin/scanprosite). The tree topologies were estimated using maximum likelihood (ML; ref. 7) and maximum parsimony (MP; ref. 11). The sequence alignment was carried out using T-coffee (15) followed by manual inspection.
Analysis of DAZAP2 expression in human tissues
The human multiple tissue Northern blot with poly(A)+ RNA prepared from various tissues (Clontech, Palo Alto, USA) was hybridized with the 32P-labeled DAZAP2 cDNA probe. The probe spans the entire ORF of DAZAP2. Human β-actin cDNA was used as a control probe. The blot was hybridized and washed under highly stringent conditions.
Transfection, immunoblotting and immunolocalization
The recombinant pEGFP-DAZAP2 expression vector was constructed as follows. The primers for DAZAP2 amplification were 5’-CGGAATTCGCCGCCACCATGAACAGCAAAGGTC-3’ (sense) and 5’-GCGGATCCCGCCAGATGGTGTAGCCACCATC-3’ (antisense), which contain EcoR I and BamH I sites (in bold), respectively. The ORF of DAZAP2 was amplified by PCR using the cDNA template derived from mononuclear cell RNA of normal bone marrow. PCR products were digested with EcoR I and BamH I followed by ligation to the pEFGP-N1 vector (Clontech) and transformation into E. coli DH5α cells. The ORF and fusion with the GFP gene in the pEGFP-N1-DAZAP2 vector were verified by EcoR I and BamH I digestion followed by DNA sequencing.
COS7 cells (ATCC) were used as a host for DAZAP2-GFP fusion protein expression. Transfection was performed with LipofectAMINE-2000 using the manufacturer’s protocol (Invitrogen, Carlsbad, USA). For immunoblotting analysis, cells were seeded in a 3-cm dish, transfected with DAZAP2-GFP plasmid and cultured for 24 h. The cells were lysed and the total proteins (10 μg) were separated on a 4%-20% precast Tris-glycine gel, and were then electro-transferred to PVDF (polyvinylidene difluoride) membranes. The protein bands were visualized by using an ECL-plus kit (Amersham, Piscataway, USA). Immunostaining was done as above with the primary anti-GFP antibody (Clontech; ref. 22). Briefly, cells grown on coverslips were fixed with 3.7% paraformadelhyde for 30 min, permeated with 0.1% Triton X-100 for 10 min, and immunostained with the respective antibodies for 1 h after blocking with 5% BSA (bovine serum albumin) for 30 min. Cells were then stained with cy2- or cy3-labeled secondary antibodies (The Jackson Laboratory, Bar Harbor, USA). After several times of washing in PBS (phosphate-buffered saline), coverslips were mounted on slides with anti-fade buffer and observed under confocal microscopy.
Acknowledgments
This work was supported by grants (No.39880021, 30270750, 30300406) from the National Natural Science Foundation of China. The accession numbers of the nucleotide sequences deposited to the GenBank are AY430097, AY490096, AY490097, and AY490098.
References
- 1.Bataille R., Harousseau J.L. Multiple myeloma. New Engl. J. Med. 1997;336:1657–1664. doi: 10.1056/NEJM199706053362307. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y.W. Gene expression profile changes in human multiple myeloma. Bull. Hunan Med. Univ. 2003;28:201–205. [PubMed] [Google Scholar]
- 3.Shi Y.W. Preparation of antibody of rabbit anti-human DAZAP2 and application in multiple myeloma research. Life Sci. Res. 2004 (Chinese). in press. [Google Scholar]
- 4.Tsui S. Identification of two novel proteins that interact with germ-cell-specific RNA-binding proteins DAZ and DAZL1. Genomics. 2000;65:266–273. doi: 10.1006/geno.2000.6169. [DOI] [PubMed] [Google Scholar]
- 5.Nomura N. Prediction of the coding sequences of unidentified human genes. II. The coding sequences of 40 new genes (KIAA0041- KIAA0080) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1994;1:223–229. doi: 10.1093/dnares/1.5.223. [DOI] [PubMed] [Google Scholar]
- 6.Yang W., Mansour S.L. Expression and genetic analysis of prtb, a gene that encodes a highly conserved proline-rich protein expressed in the brain. Dev. Dyn. 1999;215:108–116. doi: 10.1002/(SICI)1097-0177(199906)215:2<108::AID-DVDY3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt H.A.K. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 8.Tamura K., Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 9.Jones D.T. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 10.Müller T., Vingron M. Modeling amino acid replacement. J. Comput. Biol. 2000;7:761–776. doi: 10.1089/10665270050514918. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;12:1244–1245. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- 12.Nei M., Kumar S. Oxford University Press; New York, United States: 2000. Molecular Evolution and Phylogenetics. [Google Scholar]
- 13.Swofford D.L. Phylogenetic inference. In: Hillis D.M., editor. Molecular Systematics. 2nd ed. Sinauer Assoc., Inc.; Sunderland, United States: 1996. pp. 407–514. [Google Scholar]
- 14.Baxevanis A.D., Ouellette B.F., editors. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins. John Wiley & Sons; New York, United States: 1998. pp. 193–199. [Google Scholar]
- 15.Notredame C. T-Coffee: a novel method for multiple sequence alignments. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 16.Hallek M. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- 17.Sudol M. From Src homology domains to other signaling modules: proposal of the protein recognition code. Oncogene. 1998;17:1469–1474. doi: 10.1038/sj.onc.1202182. [DOI] [PubMed] [Google Scholar]
- 18.Li W., She H. The SH2 and SH3 adapter Nck: a two-gene family and a linker between tyrosine kinases and multiple signaling networks. Histol. Histopathol. 2000;15:947–955. doi: 10.14670/HH-15.947. [DOI] [PubMed] [Google Scholar]
- 19.Li W. Nck/Dock: an adapter between cell surface receptors and the actin cytoskeleton. Oncogene. 2001;20:6403–6417. doi: 10.1038/sj.onc.1204782. [DOI] [PubMed] [Google Scholar]
- 20.Ommerfeldt D.W. Proline-rich transcript of the brain (prtb) is a serum-responsive gene in osteoblasts and upregulated during adhesion. J. Cell. Biochem. 2002;84:301–308. doi: 10.1002/jcb.10018. [DOI] [PubMed] [Google Scholar]
- 21.Maruyama I.N. cRACE: a simple method for identification of the 5’ end of mRNAs. Nucleic Acids Res. 1995;23:3796–3797. doi: 10.1093/nar/23.18.3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z. The αvß1 integrin functions as a fibronectin receptor but does not support fibronectin matrix assembly and cell migration on fibronectin. J. Cell Biol. 1993;122:235–242. doi: 10.1083/jcb.122.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]