

Abstract
Thymidylate synthase (TSase) catalyzes a hydride transfer in the last step of the de novo biosynthesis of the DNA nucleotide thymine. We compared two isozymes, namely, TSase from Escherichia coli (ecTSase) and TSase from Bacillus subtilis (bsTSase) that represent a case of divergent evolution. Interestingly, a highly conserved histidine (H147 of ecTSase) was proposed to serve a critical role in catalysis, but in bsTSase it is naturally substituted by valine (Val). Yet, bsTSase is more active than ecTSase, and the intrinsic kinetic isotope effects (KIEs) of both are temperature-independent, suggesting a similarly well-organized transition state (TS) for the catalyzed hydride transfer. To examine the role of that histidine (His) in TSase catalysis, we examined the kinetics of H147V ecTSase, which “bridges” between these two TSases. In contrast to both wild-type TSases, the single mutation results in deficient catalysis. The mutation leads to intrinsic KIEs that are temperature-dependent, indicating a substantial imperfection in its TS. The findings reveal two important features: a direct role of H147 in the hydride transfer step catalyzed by the ecTSase and the evolutionary compensation for its deficiency in bsTSase via extensive polymorphism across the protein. Very different active site residues are observed for these evolutionarily divergent isozymes, which result in a well-organized TS for both. It is suggested that evolutionary pressure compensated for the H to V substitution at the active site of bsTSase by polymorphism leading to a well-organized TS in both enzymes.
Keywords: thymidylate synthase, hydride transfer, isotope effect, marcus models, evolution
INTRODUCTION
Enzymes are key molecules required to carry out chemical reactions in all living organisms. Their ability to catalyze complex chemical transformations with exquisite specificity, in aqueous solutions at biological temperatures, makes them an essential focus of biochemical, bioengineering and clinical research. The determination of the roles of different functional groups in a protein, and their roles in its catalytic mechanism, may aid in the design of enzyme-targeting drugs and the engineering of biomimetic catalysts. In the studies described below, several aspects of enzyme mechanisms and the complex role of active site residues are examined, as are evolutionary aspects of enzymology using temperature dependence of kinetic isotope effect (KIE).
Thymidylate synthase (TSase, EC 2.1.1.45) is one of the most highly-conserved proteins found in nature [1-3]. This is not surprising, as it is involved in the last step of de novo synthesis of a precursor of DNA, 2’-deoxythymidine-5’-monophosphate (dTMP), using (6R)-N5,N10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) as a cofactor and 2’-deoxyuridine-5’-monophosphate (dUMP) as the substrate [1,3-6]. TSase is a homodimer with two identical active sites at the dimer interface. Previous studies [2] indicated a bi-bi ordered reaction mechanism for the wild-type (WT) TSase with the binding of dUMP followed by the cofactor, CH2H4folate.Owing to its essential role in cellular metabolism, TSase has served as a chemotherapeutic drug target for several decades (e.g., 5-fluorouracil and raltitrexed) [7, 8]. Detailed knowledge of the chemical mechanism and the role of active site residues of TSase may help in the development of new anti-cancer drugs with lower toxicity.
Despite the large collection of structural and kinetic data of TSase from several organisms, however, many aspects of its catalytic mechanism remain unresolved. Many studies have been carried out to elucidate the catalytic mechanism of TSase [1-3, 9]. The currently proposed mechanism(s) (Scheme 1) start with a nucleophilic activation of C6 of dUMP by the strictly conserved Cys146 (step 1), which may or may not occur in concert with a C5 enolate attack on the iminium methylene on CH2H4folate (step 2) [8], forming a ternary complex (C). This step is followed by the C5 proton abstraction and elimination of (6S)-H4folate, generating an exocyclic methylene intermediate (D). Finally, a hydride transfer from (6S)-H4folate to the exocyclic methylene forms the product dTMP, releasing the enzymatic cysteine (steps 5’ or 6 in Scheme 1). Two different mechanisms have been proposed for the C-H→C hydride transfer step catalyzed by this enzyme (steps 5 [ 10] vs. 5’ [1] in Scheme 1). This hydride transfer to C7 and the thiol departure from C6 of the exocyclic methylene intermediate could occur either in two steps, forming an intermediate enol at the C4 of enzyme-bound pyrimidine (step 5) [10], or in one concerted step as suggested by our recent hybrid quantum mechanics/molecular mechanics (QM/MM) studies (step 5’) [11]. A recent study examined secondary (2°) KIEs for the WT ecTSase, and strongly supported the concerted mechanism [12]. If the irreversible hydride transfer would have occurred prior to the C-S cleavage (where C6 is changing form sp3 to sp2), no 2° KIE would have been measured, as reported in ref [12]. This said, as the hydride transfer involves tunneling and is much faster than the C-S cleavage [13], it is possible that some negative charge will accumulate on carbonyl 4 of the exocyclic intermediate (D in scheme 1) during that process. Thus, the actual mechanism could be a combination of the two extreme cases drawn in scheme 1 as 5 and 5’.
Scheme. 1.
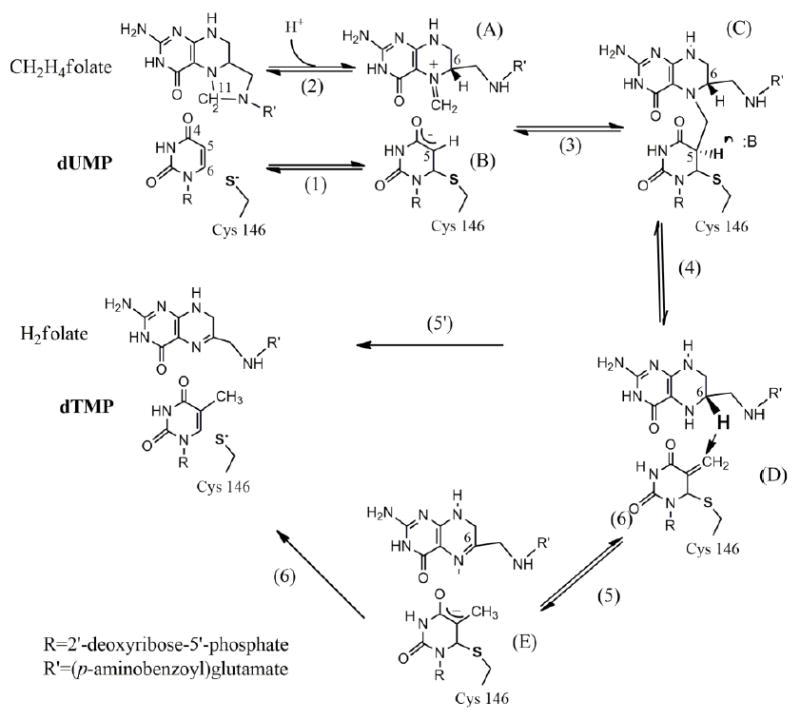
The proposed chemical mechanism for TSase.
Finer-Moore et al. suggested that a histidine residue (H147 of Escherichia coli TSase, ecTSase) is part of a network of H-bonds that includes carbonyl 4 of dUMP [3]. That residue may act as the general acid/base in the protonation/deprotonation of the C4 carbonyl during the catalytic cycle. Consequently, the mutational studies of H147V described here address the two possible mechanisms as they alter the H-bonding network of the C4 carbonyl. This histidine is highly conserved; however, an isozyme that is among the most active TSases [14], and provides the majority of TSase activity in Bacillus subtilis (bsTSase-A), has a hydrophobic valine at the equivalent position (V162). The other TSase from B. subtilis, bsTSase-B, has 64.8% sequence identity (81.8% homology) with ecTSase. bsTSase-A is an interesting case: thermostable and phylogenetically very close to TSase from phage Φ3T (97% sequence identity), it shares only 37.4% sequence identity (60.3% homology, fig.1) with the TSase from E. coli [15]. bsTSase-A has been incorporated most likely from the phage [15]. Consequently, ecTSase and bsTSase-A represent two genetically and structurally unrelated enzymes that are both highly evolved and catalyze the same complex reaction, which is likely a result of divergent evolution (fig. 2 A) [15, 16]. In spite of the low homology, the overall subunit topology and active site locations are similar in both crystal structures of ecTSase and bsTSase-A (addressed as bsTSase hereafter) as shown in fig. 2 B. The resemblance between the overlays in fig. 2 B is similar to that for glycolytic enzymes from Archea domain and E. coli, which are also thought to have resulted by divergent evolution [17]. If H147 plays an important role in ecTSase catalysis, studying the ways in which bsTSase compensates for the valine substitution can provide much-needed insights into the roles of the active site residues in TSase catalysis and how evolution preserves needed aspects of catalysis in diverged active sites.
Fig. 1.

Sequence alignment of WT ecTSase (ec) and WT bsTSase-A (bs). The + sign indicates homology and identical residues are specified between the two sequences.
Fig. 2.

A. Phylogenetic relationship among selected thymidylate synthases as calculated by GeneWorks and described in details in ref [15]. The lengths of the horizontal lines in this figure are proportional to the estimated genetic distance between the sequences. The L. casei TSase is indicated as it is considered to be very close to the E. coli enzyme [3],and the human and T4 phage are indicated as these organisms are of general interest. B. Superimposed ribbon diagrams of the ternary complexes of ecTSase (green; PDB ID 1TSN) and bsTSase (cyan; PDB ID 1B02), both with 5-fluoro-2’-deoxyuridine-5’-monophosphate (FdUMP) in magenta and yellow and CH2H4folate (red and gray).
In the current study we compare two genetically unrelated TSases,ecTSase and bsTSase-A, that seem to result from divergent evolution and a mutant of ecTSase whose mutation makes it more like bsTSase at an important residue (H147V). Both steady state rates and KIEs were used to assess the differences between these enzymes on the TSase-catalyzed hydride transfer step (5 or 5’in scheme 1).
MATERIALS AND METHODS
Materials
[2-14C] dUMP (specific radioactivity 53 Ci/mol) and [5-3H] dUMP (specific radioactivity 13.6 Ci/mmol) were purchased from Moravek Biochemicals. [3H]NaBH4 (specific radioactivity 15 Ci/mmol) was purchased from American Radiolabeled Chemicals. [2H]NaBH4 (> 99.5% D) was purchased from Cambridge Isotopes. Dihydrofolate (H2-folate) was synthesized by following the procedure of Blakely [18]. Unlabeled CH2H4folate was a generous gift from Merck and Cie (Switzerland). [2-3H]iPrOH and [2-2H]iPrOH were prepared by the reduction of acetone with [3H]NaBH4 and [2H]NaBH4, respectively, as described in previous publications [19]. The WT bsTSase enzyme was produced and purified by following a published procedure [20]. Site-directed mutagenesis of the ecthyA gene was carried out using the Quick-Change site-directed mutagenesis kit from Stratagene. The mutant protein was purified following the same procedure for the WT. Ultima Gold liquid scintillation cocktails were purchased from Packard Bioscience. Liquid scintillation vials were purchased from Research Products International Corp. All other materials were purchased from Sigma. The steady-state experiments were performed using a Hewlett-Packard Model 8452A diode-array spectrophotometer equipped with a temperature-controlled cuvette assembly. All the purifications and analytical separations were performed using an Agilent Technologies model 1100 HPLC (High Pressure Liquid Chromatography) system, with a column from Supelco (C18, 250 × 4.6 mm, 5 μm, Discovery®). The radioactive samples were analyzed using a Liquid Scintillation Counter (LSC, from Perkin Elmer Biosciences).
Synthesis of [6R-xH]CH2H4folate for KIE experiments on hydride transfer
The [6R-xH]CH2H4folate was synthesized following the published procedure [21]. In brief, the synthesis is a one-pot preparation that utilizes two enzymes and a single chemical reaction: The reduction of NADP+ by [2-xH]iPrOH to produce [4R-xH]NADPH was catalyzed by alcohol dehydrogenase from Thermoanarobium brockii (tbADH) and the reduction of H2folate by [4R-xH]NADPH to produce [6S-xH]H2folate was catalyzed by dihydrofolate reductase.[6S-xH]H2folate was trapped by adding formaldehyde, forming [6R-xH]CH2H4folate. Strict anaerobic conditions were maintained by a glucose/glucose oxidase in situ oxygen scavenging system. The synthesized [6R-xH]CH2H4folate was purified by reverse phase HPLC (RP HPLC), lyophilized, and stored at -80 °C prior to use.
Formation of thiol-trapped Intermediates
The assessment of the formation of the thiol-trapped intermediate was carried out in the following reaction mixture: 100 μM dUMP, 200 μM CH2H4folate, 2 mM tris(2-carboxyethyl)phosphine (TCEP), 50 mM MgCl2 , ~0.25 Mdpm [2-14C]dUMP and 25 mM thiol (dithiotheritol (DTT) and β-mercaptoethanol (ß-ME)) in 100 mM tris(hydroxymethyl)aminomethane (Tris)/HCl buffer (pH 7.5), as described previously [22]. The reaction was incubated for 60 min at 25 °C prior to HPLC analysis after adding H147V ecTSase. The products were analyzed by HPLC separation followed by flow scintillation analysis.
Steady-state kinetics
The optical method [23], which measures the increase of absorbance at 340 nm upon conversion of CH2H4folate to H2folate (Δε340nm = 6.4 mM-1cm-1), was used here to determine the initial velocities for the H147V ecTSase mutant. The temperature dependence of initial rates of CH2H4folate was measured within a range of 5-35°C. These experiments were conducted, at a minimum, in triplicate in a reaction mixture containing TCEP.
To measure the Michaelis constant of dUMP ( ) we fixed the concentration of CH2H4folate at 50 μM, which is not inhibitory at 25 °C. A fixed dUMP concentration of 200 μM was used for the measurements of the Michaelis constant of CH2H4folate, ( ). The data at each temperature were fitted to the non-linear Michaelis-Menten equation with substrate inhibition (fig. 3 A and table 1) using the following equation [24]:
| (1) |
Fig. 3.
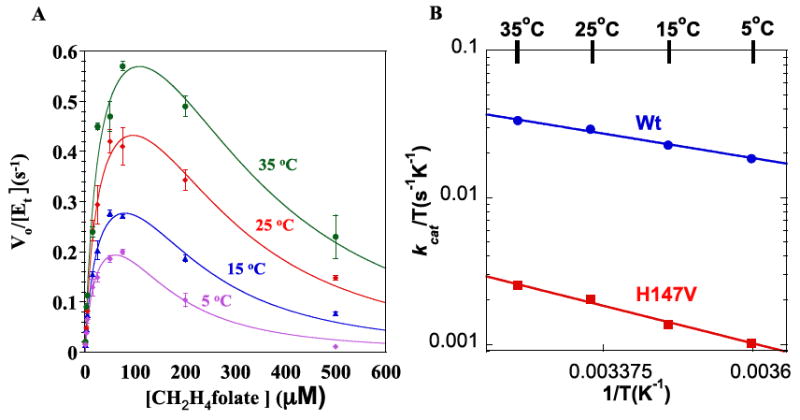
(A) Steady-state initial rates of H147V vs. varying concentrations of CH2H4folate with 200 μM dUMP. At each temperature, the data were fit to eq. 1 by least-squares nonlinear regression. (B) The Eyring plots for kcat of WT (blue) and H147V (red) ecTSase.
Table 1.
Steady-state kinetic parameters of the WT and H147V (at 25 °C) and WT bsTSase (at 30 °C).
where [E]t is the total enzyme concentration, [S] is the concentration of CH2H4folate and KI is the inhibition constant of CH2H4folate.
To further investigate the H147V mutational effect on ecTSase, we studied the temperature dependence of kcat and evaluated its activation parameters (fig. 3 B and table 2) entropy, enthalpy and free energy of activation (ΔS‡, ΔH‡ and ΔG‡), using a least squares nonlinear regression fitted to the Eyring equation.
| (2) |
Table 2.
Activation parameters for kcat of WT and H147V ecTSases.
| TSase | ΔH‡ kcal/mol | TΔS‡ at 25 °C kcal/mol | ΔG‡ kcal/mol |
|---|---|---|---|
| WTa | 3.4 ± 0.2 | -12.8 ± 0.2 | 16.2 ± 0.4 |
| H147V | 5.2 ± 0.4 | -12.6 ± 0.4 | 17.8 ± 0.6 |
Data from ref [25]
Competitive KIEs on the hydride transfer (step 5 in scheme 1)
The competitive H/T and D/T method was used to measure the KIE on the hydride transfer step in the temperature range of 5-35 °C, using the same procedure published for the WT ecTSase enzyme [25]. The only change was that the reducing agent, β-mercaptoethanol (50 mM) (used for the WT bsTSase), was replaced by 2 mM TCEP to avoid formation of the thiol-trapped intermediate [22] in the H147V ecTSase mutant. In short all experiments were carried out in Tris HCl buffer (pH =7.5, adjusted at each temperature), with 2 mM TCEP, 1 mM EDTA (Ethylenediaminetetraacetic acid), 5 mM HCHO(Formaldehyde), 1.5 Mdpm [6R-T]CH2H4folate, [6R-xH]CH2H4folate (x = 1 for H/T and x = 2 for D/T experiments), 0.5 Mdpm [2-14C]dUMP, CH2H4folate and dUMP to enable 20% molar excess of [2-14C]dUMP. The reaction mixture was preincubated at each temperature and the reaction was initiated by the addition of H147V ecTSase. 100 μl aliquots were removed from the same reaction at five different time points and quenched with 30 μM of 5-fluoro-2’-deoxyuridine-5’-monophosphate (FdUMP). Three infinity time points (t∞) were obtained by adding concentrated WT ecTSase. Two to time points (reaction mixture with no enzymes added) were used as controls for the experiment. All quenched samples were analyzed by HPLC and LSC as described in previous publications [22, 25]. The observed competitive KIEs on the second order rate constant (e.g., T(V/K)H for H/T KIE) were calculated from equation 3 [25, 26]:
| (3) |
where Rt and R∞ are 3H /14C ratios in dTMP and f is the fractional conversion of the reaction. f was calculated by equation 4
| (4) |
Intrinsic KIEs were calculated using equation 5 by combining the Swain-Schadd relationship and Northrop’s method [27-29]:
| (5) |
where ki is the rate constant for the reaction involving isotope i, kH/kT is the intrinsic KIE, and and are the observed competitive KIE values on the second order rate constant. Although intrinsic KIE kH/kT is the only unknown in equation 5, it cannot be solved analytically. A program was developed to solve this equation numerically and is available on our web site: http://www.chem.uiowa.edu/kohen-research-group/tools
The intrinsic KIEs were calculated from their observed values by equation 5. All the observed and intrinsic H/T KIEs for the hydride transfers vs. the reciprocal of the absolute temperature are presented in fig. 4 and the intrinsic KIEs were fitted to the Arrhenius equation:
| (6) |
Fig. 4.

Arrhenius plots of intrinsic H/T KIE on log scale vs. the reciprocal of the absolute temperature (1/T) on the hydride transfers catalyzed by the WT ecTSase (blue) [25],WT bsTSase (green) and H147V ecTSase (red). The triangles represent the observed H/T KIEs, and squares represent the intrinsic H/T KIEs. The lines represent the least-squares nonlinear regression of the intrinsic KIEs fitted to equation 6.Error bars are always included but are sometimes smaller than symbols.
where k is the microscopic rate constant of the isotopically sensitive step (the hydride transfer step in this instance) and the subscripts L denote the light isotope (H or D) while the subscripts T denote the heavy isotope (T) of hydrogen; ΔEa is the difference in the energy of activation between the light and heavy isotopes (ΔEa = EaL - EaT); R is the gas constant, T is the absolute temperature, and AL/AT is the isotope effect on the pre-exponential factors.
RESULTS
Comparison of the hydride transfer step in bsTSase, ecTSase, and its H147V mutant
We compared the nature of the hydride transfer (step 5 in Scheme 1) in the WT bsTSase, H147V ecTSase and WT ecTSase by examination of the temperature dependence of their intrinsic KIEs, in the 5-40 °C temperature range using published methods [25]. This measurement has been used in numerous recent studies to distinguish between active sites that are well organized for H-tunneling at their tunneling-ready-state (TRS, the QM-delocalized transition state), and those that have poorly organized TRS [12, 30-37]. This method is a very sensitive probe of changes in the organization and dynamics of the transition state of the chemical step under study.
Both WT ecTSase and bsTSase enzymes show temperature independent KIEs (see fig.4), but bsTSase has smaller KIEs (AH/AT = 6.8 ± 2.8 and 4.2 ± 1.4). In contrast, the KIEs of H147V are temperature dependent (ΔEa= 3.7 ±0.4 kcal/mol) with an inverse pre-exponential factor (AH/AT = 0.015 ± 0.009).
Formation of thiol-trapped intermediate for H147V ecTSase
Previous studies of ecTSase and its mutants [25, 38] have used different thiols, including DTT and ß-ME, to prevent oxidative damage to the enzyme. Our experiments on H147V ecTSase with ß-ME indicated the formation of 5-(hydroxyethyl)thiomethyl-dUMP (HETM-dUMP) by the nucleophilic attack of ß-ME at C7 of the exocyclic methylene intermediate (complex D in Scheme 1). This trapped intermediate has been previously observed in the Y209W and W80M mutants of ecTSase [22, 7/20/201638-40]. In the case of Y209W, a series of thiol agents of various sizes diffused into the active site, and the percentage of thiol byproduct reflected the accessibility of the active site to different thiols competing with the hydride transfer after the formation of intermediate D (Scheme 1). ß-ME leads to 73% byproduct (HETM-dUMP) after 150 min incubation with Y209W, while under the same conditions H147V generated only ~10% byproduct. In H147V, larger thiols did not trap the intermediate at all.
Effect of H147V ecTSase mutation on steady state kinetics
Steady-state kinetic experiments were carried out over a temperature range of 5-35 °C. We employed TCEP, a non-thiol reducing agent, to avoid any HETM-dUMP formation and to ensure more precise kinetic measurements. As with WT ecTSase [25], the rates of H147V with varying dUMP concentrations were fitted to the Michaelis-Menten equation, whereas the rates with varying CH2H4folate concentrations show substrate inhibition and were fitted accordingly (see fig. 3 and table 1). In accordance with ref. [41], there was 5-fold decrease in kcat and approximately 2-fold increase in both and . As can be seen in table 2, the free energy of activation increased for the slower H147V ecTSase mutation, mostly due to increased enthalpy of activation. This finding may reflect the alteration of the electronic potential of the active site, as the cationic histidine has been changed to the non-charged valine.
Comparison of the kinetic complexity in bsTSase, ecTSase, and its H147V mutant
Previous studies on the WT ecTSase at 20 °C reported observed H/T and D/T KIEs on V/K of 6.91 ± 0.05 and 1.78 ± 0.02, respectively, which were within error of the intrinsic KIEs, suggesting that the hydride transfer is rate limiting at that temperature [25]. In contrast to those of the WT ecTSase, the observed KIEs (triangles in fig. 4) of both H147V ecTSase and WT bsTSase were smaller than the intrinsic values at all temperatures. This observation indicates a kinetic complexity that masks intrinsic KIEs, as shown in equation 7 [29]:
| (7) |
where Cf and Cr are forward and reverse commitments to catalysis respectively, and EIE is the equilibrium isotope effect. Since the hydride transfer is an irreversible kinetic step, Cr is zero, which simplifies equation 7 to an equation with only one unknown Cf. The expression Cf is the ratio of the hydride transfer rate to the net rate of the preceding isotopically insensitive steps in the reverse direction.
Fig. 5 presents the forward commitment (Cf) values of the WT and H147V ecTSases and the WT bsTSase vs. the reciprocal of the absolute temperature (1/T). Intriguingly, both the WT TSases show similar temperature dependence of Cf, but with much higher magnitude in bsTSase. The H147V mutant, on the other hand, has a linear Arrhenius plot for Cf, which indicates a single step is responsible for the Cf. While at this time it is not clear which step that is, it is obvious that its behavior is substantially different from that of the two WT enzymes.
Fig. 5.
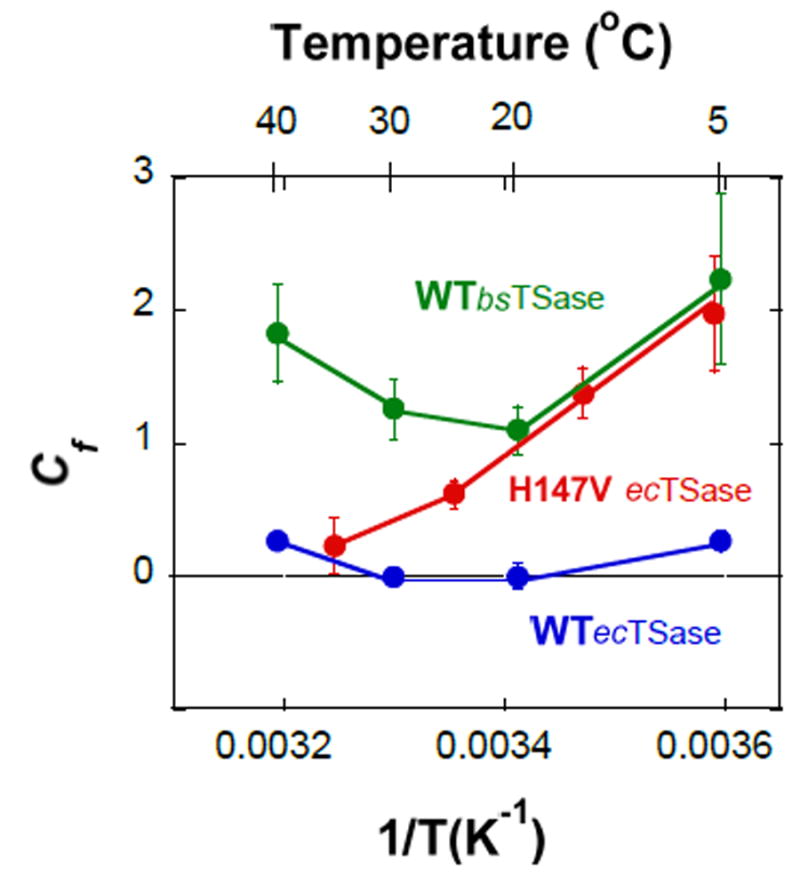
Temperature dependence of the forward commitments to catalysis (Cf) on V/K for the WT (blue) and H147V (Red) ecTSases, and WT bsTSase (Green).
DISCUSSION
The current work compares the hydride transfer steps in the evolutionarily divergent ecTSase, bsTSase, and the H147V mutant of ecTSase, which “bridges” between these isozymes. This study examines the detailed mechanism of the C-H→C hydride transfer, and compares the nature of the same chemical transformation in two divergent isozymes that, despite lack of genetic or active site similarities, catalyze the same reaction [15].
Steady state kinetic parameters
Table 1 lists the steady state kinetic parameters for H147V mutant and the two isozymes. The fact that the mutation increased the KM for both CH2H4folate and dUMP was unexpected, as this His is 3.7 Å from the C4 carbonyl of dUMP but more than 8 Å from the nearest atom of the folate. While H147 does not bind directly to CH2H4folate, the mutation of this residue indirectly affects the , presumably by changing the order of neighboring residues in the active site or by changing the conformation of dUMP that collectively forms the binding site for the cofactor.
The KM values for WT ecTSase and bsTSase are comparable to each other, despite the fact that the active site residues are dramatically different (fig. 6), but this may correlate with the similar backbone overlap of the two crystal structures (fig. 2 B). The similarities in kinetic parameters of these genetically unrelated WT TSases might also reflect the evolutionary pressure that leads enzymes to divergently evolve to have similar biological activity.
Fig. 6.
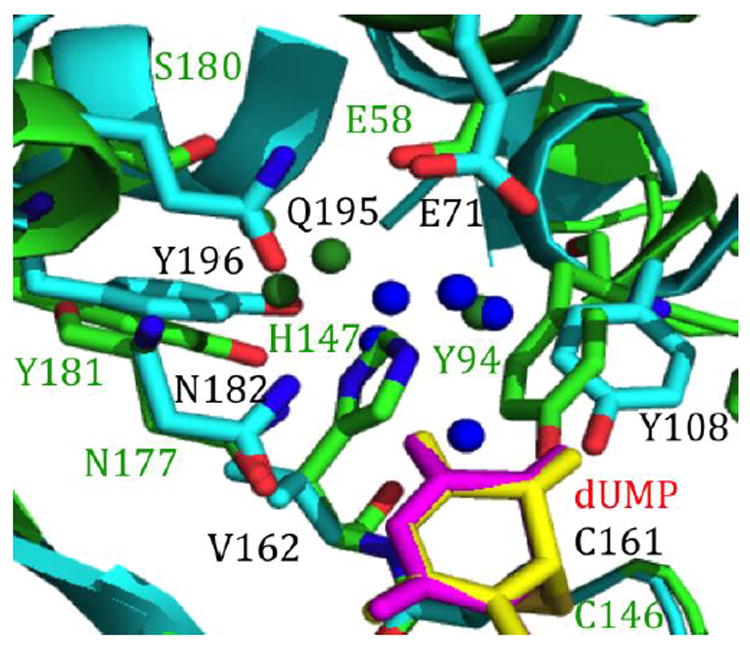
Crystal structures showing the active sites of the WT ecTSase (green; PDB ID 1TSN) and WT bsTSase (cyan; PDB ID 1B02) with FdUMP (magenta and yellow). Most of the amino acids of WT ecTSase (labeled in green) and WT bsTSase (labeled in black) align well. The water molecules of WT ecTSase (green) and WT bsTSase (blue) show a significantly different solvent structure in the two enzymes.
What accounts for the 15-fold decrease in the H147V activity compared to that of ecTSase? The finding that ß-ME trapped ~10% of HETM-dUMP suggested impaired closing of the active site cavity. Thus, the H147-controlled electrostatics and H-bonding could be essential in maintaining the proper enzyme conformation for the hydride transfer [41]. In addition, the inflated commitment of H147V for the V/K of hydride transfer (compared to WT ecTSase), discussed below, suggests that the mutation affects one or more steps preceding the hydride transfer (steps 1-4).
The C-H →C hydride transfer step
The comparison of the same chemical transformation catalyzed by two enzymes that evolved from different origins and share little genetic and structural similarities is of general interest in biology. Here we employed the temperature dependence of the KIEs as a tool to investigate the effect of two variants of WT TSase and a “bridging” mutant (H147V ecTSase) on the nature of the hydride transfer step. Both ecTSase and bsTSase exhibit temperature-independent KIEs on the hydride transfer step (fig.4). According to phenomenological models known as Activated Tunneling Models [42], the lack of temperature dependence of the KIEs for the WT TSases indicates a narrow and well defined distribution of donor-acceptor distances (DADs) at the reaction’s tunneling ready state (TRS) [35, 37], or in other words, a well defined TS (TRS is just the quantum mechanically delocalized TS). Temperature independent KIEs are typical in WT enzymes where the DAD is optimized for tunneling at the TRS. Comparison of the two isozymes indicates variance in multiple active site residues, leading to a very different network of H-bonds involving the bound substrates. It is remarkable that such different active site arrangements lead to the same narrowly distributed DADs at the TRS of these two enzymes (as indicated by the lack of temperature effect on intrinsic KIEs).
The findings with the H147V ecTSase meant to examine the effect of a single mutation of mechanistically important residue that brings the ecTSase closer to the bsTSase. It is apparent from fig. 4 and table 1 that in contrast to the two WT enzymes, the mutant is slow and has temperature-dependent intrinsic KIEs. To quantify the effect of the H147V on the DAD we fitted the data to the Marcus-like model, as recently described [43]. The two WT TSases can be fitted to a single, narrowly distributed population of DADs with average of 3.06 and 3.05 Å for ecTSase and bsTSase, respectively. In contrast, the temperature dependence of KIEs for H147V is too steep to be fitted to one population model, and has to be fitted to a two-population model. A vast majority of the TRS population has a DAD of 3.36 Å, which is much longer than that of WT ecTSase. The longer average DAD and its much broader distribution for the H147V mutant indicate it has a poorly reorganized TRS (i.e., a broad ensemble of poorly organized TSs) compared with both WT TSases.
The KIE data suggest that the H147V mutation directly affects the hydride transfer step, possibly by deteriorating the protein motions and the H-bond network at the active site needed for the chemical conversion. Since the nearest dUMP moiety to H147 is carbonyl 4 (H-bonded through one water molecule), the findings could be interpreted as supporting a mechanism where negative charge is accumulating at that carbonyl as the hydride is transferred. Such negative charge has to be partial and cannot account for a fully formed enol intermediate at the C4 position of dUMP (step 5 in Scheme 1), because in such case, no 2° KIE on C6 of dUMP would be observed as reported recently for WT ecTSase [12]. Additionally, it is likely that the H147 is involved in the network of H-bonds that span the active site across several residues and water molecules (see fig. 6), including C146, Y94, and other residues and water molecules. The mutation thus may enhance the dissociation of C146 from C6 in concert with the hydride transfer (step 5’).
The fact that the intrinsic KIEs of WT bsTSase has temperature-independent KIEs similar to, and even smaller than the WT ecTSase, indicates that the presence of Val rather than His in bsTSase’s position 162 is compensated for other residues in the active site of this highly evolved enzyme, leading to well oriented DADs with the narrow distribution found in most WT enzymes. This kind of conservation of structural dynamics that leads to temperature-independent KIEs in evolution has been recently observed for the enzyme dihydrofolate reductase (DHFR) from bacteria to human [44]. In the DHFR study, it was found the mutations and insertions that are introduced prematurely (i.e., not in the same order they were introduced by evolution) lead to temperature-dependent intrinsic KIEs. This observation indicated disturbance of the well-organized TRS of the evolved WT enzymes by those mutations, but could be corrected by introducing other mutations that better mimic a naturally evolved enzyme. Here we report a similar effect in a case of divergent evolution, as it is apparent that the ecTSase and TSase-A (similar to TSase from phage Φ3T) branched early in evolution [15]. As shown in fig. 6, bsTSase has a dramatically different H-bond network and this change results not only from His to Val, but also Ser to Gln in bsTSase. Most importantly, those changes lead to a very different network of active site residues and water molecules that could affect both enol formation at carbonyl 4 of dUMP (affecting step 5, Scheme 1) and activation of the thiol at Cys146, making it a better leaving group (steps 5’ and 6, Scheme 1).
The temperature-dependent KIE on the hydride transfer of H147V ecTSase indicates that a single mutation of a highly conserved and mechanistically important residue is not sufficient to mimic the active site of bsTSase, and the whole active site (if not the whole protein) must evolve to reach the temperature-independent KIEs typical of highly evolved enzymes.
Events that precede the C-H→C hydride transfer
Other kinetic steps but the C-H→C hydride transfer seem to also be affected by the mutation. The inflated Cf of the observed KIE on the second order rate constant (kcat/KM or V/K) (fig. 5) suggests that a backward step preceding the hydride transfer is slower for both WT bsTSase and the H147V mutant than it is for WT ecTSase.
Moreover, the effect of the mutation on the last step before the hydride transfer became evident by another experimental observation. The thiol-trapping experiments for H147V ecTSase suggest that the disturbances to the kinetics and the active site dynamics and structure, after the formation of the exocyclic methylene intermediate, in H147V are not as severe as in Y209W [22]. The thiol-trapping experiments, together with kinetic data with Y209W, suggest that the mutation appears to perturb kinetic steps at a slow timescale (millisecond-second) and alter the preorganization of the system before the hydride transfer. In ecTSase, H147 is hydrogen-bonded through a water molecule to the O4 of dUMP. This water molecule also has H-bonds to E58, which interacts with W80 [2, 45], the amino acid that serves as a key residue isolating the active site cavity from solvent [3]. Consequently, the mutational effect of H147V may impair the closing of the active site cavity after the ternary complex formation and thus may account for the thiol-trapped intermediate. Previous studies of the W80G-dUMP-CB3717 crystal structure showed that the active site seals improperly, which creates an enlarged active site cavity [39]. In H147V no thiol larger than β-ME could trap the intermediate, suggesting that the volume of the cavity is not large enough to accommodate larger thiols such as DTT. This finding indicates altered kinetics and structural dynamics imposed by the mutation. The ability of thiols to trap the exocyclic intermediate demonstrates that they compete effectively with the hydride transfer from (6S)-H4folate, given the opportunity to attack that intermediate (D in Scheme 1). This would require both kinetic accumulation of the intermediate (i.e., delay in hydride attack, slower step 5, and altered kinetics) and better accessibility of thiols to the active site (suggesting altered structural fluctuation of the enzyme at that intermediate complex, resulting in an altered distribution of conformations at that intermediate state, which many would refer to as altered enzyme dynamics), enabling penetration of thiols into the active site.
By contrast, when reacting with either WT ecTSase or WT bsTSase in the presence of thiols, no byproducts were formed, indicating an active site that is well-evolved to protect the intermediate from solvent and orient the H4folate in the correct position for hydride transfer. These observations are in accordance with the studies of the temperature dependence of intrinsic KIEs on the hydride transfer discussed above.
CONCLUSION
The above studies bear significantly on both practical and intellectual aspects of enzymology. Practically, given that TSase is a common target for chemotherapeutic drugs, a more sophisticated understanding of its mechanism might assist in rational design of new leads or improvements of such drugs. Intellectually, the reason for the ideal rearrangement of DADs in chemical steps of many enzymes is intriguing, as many of these steps are not rate liming on the overall catalytic turnover [44, 46]. It is interesting to note that an evolutionary pressure seems to maintain that unique DADs reorganization on steps that are not rate limiting.
Acknowledgments
This work was supported by NIH GM065368 and NSF CHE-1149023, and ZW was supported by the University of Iowa, Center of Biocatalysis and Bioprocessing.
ABBREVIATIONS
- TSase
Thymidylate synthase
- bsTSase
Bacillus subtilis TSase
- ecTSase
Escherichia coli TSase
- dUMP
2’-deoxyuridine-5’-monophosphate
- dTMP
2’-deoxythymidine-5’-monophosphate
- CH2H4folate
(6R)-N5,N10-methylene-5,6,7,8-tetrahydrofolate
- H147V
ecTSase variant that has His substituted for Val at residue 147
- KIE
kinetic isotope effect
- DTT
dithiotheritol
- ß-ME
β-mercaptoethnol
- TCEP
tris(2-carboxyethyl)phosphine
- TS
transition state
- TRS
tunneling ready state
- DAD
donor-acceptor distance
- WT
wild-type
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors state no conflict of interest.
References
- 1.Carreras CW, Santi DV. Annu Rev Biochem. 1995;64:721. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 2.Stroud RM, Finer-Moore JS. Biochemistry. 2003;42:239. doi: 10.1021/bi020598i. [DOI] [PubMed] [Google Scholar]
- 3.Finer-Moore JS, Santi DV, Stroud RM. Biochemistry. 2002;42:248. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- 4.Huang W, Santi DV. J Biol Chem. 1994;269:31327. [PubMed] [Google Scholar]
- 5.Maley F, Pedersen-Lane J, Changchien L. Biochemistry. 1995;34:1469. doi: 10.1021/bi00005a001. [DOI] [PubMed] [Google Scholar]
- 6.Saxl RL, Changchien L-M, Hardy LW, Maley F. Biochemistry. 2001;40:5275. doi: 10.1021/bi002925x. [DOI] [PubMed] [Google Scholar]
- 7.Phan J, Steadman DJ, Koli S, Ding WC, Minor W, Dunlap RB, Berger SH, Lebioda L. J Biol Chem. 2001;276:14170. doi: 10.1074/jbc.M009493200. [DOI] [PubMed] [Google Scholar]
- 8.Kanaan N, Marti S, Moliner V, Kohen A. Biochemistry. 2007;46:3704. doi: 10.1021/bi061953y. [DOI] [PubMed] [Google Scholar]
- 9.Roston D, Islam Z, Kohen A. Molecules. 2013;18:5543. doi: 10.3390/molecules18055543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruice TW, Santi DV. In: Enzyme Mechanisms from Isotope Effects. Cook PF, editor. CRC Press; Boca Raton: 1991. p. 457. [Google Scholar]
- 11.Kanaan N, Marti S, Moliner V, Kohen A. J Phys Chem A. 2009;113:2176. doi: 10.1021/jp810548d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Islam Z, Strutzenberg TS, Gurevic I, Kohen A. J Am Chem Soc. 2014;136:9850. doi: 10.1021/ja504341g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanaan N, Ferrer S, Martí S, Garcia-Viloca M, Kohen A, Moliner V. J Am Chem Soc. 2011;133:6692. doi: 10.1021/ja1114369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox KM, Maley F, Garibian A, Changchien LM, Roey PV. Protein Science. 1999;8:538. doi: 10.1110/ps.8.3.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stout TJ, Schellenberger U, Santi DV, Stroud RM. Biochemistry. 1998;37:14736. doi: 10.1021/bi981270l. [DOI] [PubMed] [Google Scholar]
- 16.Tam NH, Borriss R. Mol Gen Genet. 1998;258:427. doi: 10.1007/pl00008625. [DOI] [PubMed] [Google Scholar]
- 17.Verhees CH, Kengen SWM, Tuininga JE, Schut GJ, Adams MWW, De Vos WM, Van Der Oost J. Biochem J. 2003;375:231. doi: 10.1042/BJ20021472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blakley RL. Nature. 1960;188:231. [Google Scholar]
- 19.Agrawal N, Kohen A. Analytical Biochemistry. 2003;322:179. doi: 10.1016/j.ab.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Changchien LM, Garibian A, Frasca V, Lobo A, Maley GF, Maley F. Protein Expression Purif. 2000;19:265. doi: 10.1006/prep.2000.1245. [DOI] [PubMed] [Google Scholar]
- 21.Agrawal N, Mihai C, Kohen A. Anal Biochem. 2004;328:44. doi: 10.1016/j.ab.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Abeysinghe T, Finer-Moore JS, Stroud RM, Kohen A. J Am Chem Soc. 2012;134:17722. doi: 10.1021/ja307859m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahba AJ, Friedkin M. J Biol Chem. 1961;236:PC11. [PubMed] [Google Scholar]
- 24.Wang Z, Sapienza PJ, Abeysinghe T, Luzum C, Lee AL, Finer-Moore JS, Stroud RM, Kohen A. J Am Chem Soc. 2013;135:7583. doi: 10.1021/ja400761x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agrawal N, Hong B, Mihai C, Kohen A. Biochemistry. 2004;43:1998. doi: 10.1021/bi036124g. [DOI] [PubMed] [Google Scholar]
- 26.Hong B, Maley F, Kohen A. Biochemistry. 2007;46:14188. doi: 10.1021/bi701363s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Northrop DB. Biochemistry. 1975;14:2644. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 28.Northrop DB. Univ Park Press; 1977. p. 122. [Google Scholar]
- 29.Northrop DB, Cook PF, editors. CRC Press; Boca Raton, FL: 1991. p. 181. [Google Scholar]
- 30.Singh P, Sen A, Francis K, Kohen A. J Am Chem Soc. 2014;136:2575. doi: 10.1021/ja411998h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luk LYP, Javier Ruiz-Pernía J, Dawson WM, Roca M, Loveridge EJ, Glowacki DR, Harvey JN, Mulholland AJ, Tuñón I, Moliner V, Allemann RK. Proc Natl Acad Sci U S A. 2013;110:16344. doi: 10.1073/pnas.1312437110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roston D, Kohen A. J Am Chem Soc. 2013;135:13624. doi: 10.1021/ja405917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pudney CR, Johannissen LO, Sutcliffe MJ, Hay S, Scrutton NS. J Am Chem Soc. 2010;132:11329. doi: 10.1021/ja1048048. [DOI] [PubMed] [Google Scholar]
- 34.Meyer MP, Tomchick DR, Klinman JP. Proc Natl Acad Sci U S A. 2008;105:1146. doi: 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klinman JP, Kohen A. J Biol Chem. 2014;289:30205. doi: 10.1074/jbc.R114.565515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan, Gadda G. J Am Chem Soc. 2005;127:17954. doi: 10.1021/ja0560377. [DOI] [PubMed] [Google Scholar]
- 37.Cheatum C, Kohen A. In: Dynamics in Enzyme Catalysis. Klinman J, Hammes-Schiffer S, editors. Vol. 337. Springer; Berlin Heidelberg: 2013. p. 1. [Google Scholar]
- 38.Hong B, Haddad M, Maley F, Jensen JH, Kohen A. J Am Chem Soc. 2006;128:5636. doi: 10.1021/ja060196o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fritz TA, Liu L, Finer-Moore JS, Stroud RM. Biochemistry. 2002;41:7021. doi: 10.1021/bi012108c. [DOI] [PubMed] [Google Scholar]
- 40.Variath P, Liu Y, Lee TT, Stroud RM, Santi DV. Biochemistry. 2000;39:2429. doi: 10.1021/bi991802d. [DOI] [PubMed] [Google Scholar]
- 41.Dev IK, Yates BB, Atashi J, Dallas WS. J Biol Chem. 1989;264:19132. [PubMed] [Google Scholar]
- 42.Kohen A. Acc Chem Res. 2015;48:466. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roston D, Cheatum CM, Kohen A. Biochemistry. 2012;51:6860. doi: 10.1021/bi300613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Francis K, Stojkovi V, Kohen A. J Biol Chem. 2013;288:35961. doi: 10.1074/jbc.M113.507632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finer-Moore JS, Montfort WR, Stroud RM. Biochemistry. 1990;29:6977. doi: 10.1021/bi00482a005. [DOI] [PubMed] [Google Scholar]
- 46.Klinman JP, Kohen A. Annu Rev Biochem. 2013;82:471. doi: 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]