

ABSTRACT
The phospholipid cardiolipin (CL) has been proposed to play a role in selective mitochondrial autophagy, or mitophagy. CL externalization to the outer mitochondrial membrane would act as a signal for the human Atg8 ortholog subfamily, MAP1LC3 (LC3). The latter would mediate both mitochondrial recognition and autophagosome formation, ultimately leading to removal of damaged mitochondria. We have applied quantitative biophysical techniques to the study of CL interaction with various Atg8 human orthologs, namely LC3B, GABARAPL2 and GABARAP. We have found that LC3B interacts preferentially with CL over other di-anionic lipids, that CL-LC3B binding occurs with positive cooperativity, and that the CL-LC3B interaction relies only partially on electrostatic forces. CL-induced increased membrane fluidity appears also as an important factor helping LC3B to bind CL. The LC3B C terminus remains exposed to the hydrophilic environment after protein binding to CL-enriched membranes. In intact U87MG human glioblastoma cells rotenone-induced autophagy leads to LC3B translocation to mitochondria and subsequent delivery of mitochondria to lysosomes. We have also observed that GABARAP, but not GABARAPL2, interacts with CL in vitro. However neither GABARAP nor GABARAPL2 were translocated to mitochondria in rotenone-treated U87MG cells. Thus the various human Atg8 orthologs might play specific roles in different autophagic processes.
KEYWORDS: CL, GABARAP, GABARAPL2, glioblastoma, LC3B, mitophagy
Introduction
Macroautophagy, ‘autophagy’ in the context of this work, is an intracellular degradation pathway conserved in all eukaryotes.1 It involves the formation of a double-membrane structure, the phagophore, and its subsequent maturation to form an autophagosome (AP) that fuses with lysosomes where the autophagosomal components are degraded.2 This process does not only provide nutrients under amino-acid limiting conditions (nonselective autophagy) but works as well as a quality control system by selectively removing misfolded aggregate-prone proteins and damaged or superfluous organelles (selective autophagy).3 Thus far several cargo-specific autophagy processes have been reported, including the specific removal of mitochondria known as mitophagy.4,5 Beyond quality control, mitophagy is required for steady-state turnover of mitochondria,6 for the adjustment of mitochondrion numbers to changing metabolic requirements,7 and during specialized developmental stages in mammalian cells.8 This cellular process is thought to delay aging and its dysregulation has been linked to multiple human pathological states including neurodegeneration,9 myopathies10 and cancer.11
Several molecular components involved in mitophagy have been identified both in yeast12 and higher eukaryotes,3,4 including members of the Atg8 protein family (mammalian LC3 and GABARAP subfamilies). Specific mitophagy receptors, by binding on one side to mitochondria and on the other end to those autophagosome-specific proteins, physically link mitochondria to the growing phagophores. However one outstanding question is how damaged or superfluous mitochondria are primarily recognized by that autophagic machinery. Several non-mutually exclusive mechanisms have been reported including changes in the nutrient conditions of yeast media,13 damage- or drug-induced loss of mitochondrial membrane potential,14 decreased ATP production15 or alteration of mitochondrial Ca2+ homeostasis.16 In addition, cardiolipin (CL) externalization to the outer mitochondrial membrane (OMM) has recently been shown to function as a signal for LC3 that mediates both mitochondrial recognition and AP formation, ultimately leading to targeted removal of damaged mitochondria.17
On the basis of these observations, we have attempted to gain further insight into the molecular basis of the interaction of LC3B with CL. Here we show that LC3B interacts preferentially with CL over other di-anionic lipids such as phosphatidylinositol-4-phosphate (PtdIns4P) and that the LC3B-CL interaction relies on both electrostatic forces and CL-specific changes in membrane properties. We also provide evidence indicating that the LC3B C terminus remains exposed to the hydrophilic environment after protein binding to CL-enriched membranes. The relevance of the LC3-CL interaction was further evaluated in intact U87MG human glioblastoma cells, where rotenone, but not other autophagy-inducing signals induces LC3B translocation to mitochondria and its subsequent delivery to lysosomes. Finally, we extended our analysis to the LC3 paralogs GABARAPL2 and GABARAP. We found that GABARAP, but not GABARAPL2, interacts with CL in vitro. However, unlike the situation found with LC3B, neither GABARAPL2 nor GABARAP were translocated to mitochondria upon rotenone treatment in U87MG cells. Altogether, our study indicates that (i) LC3B-cardiolipin interaction is modulated by particular membrane properties that could allow the specific recognition of impaired mitochondria and (ii) the various human orthologs and paralogs of yeast Atg8 may play specific roles in different autophagic processes.
Results
Human orthologs and paralogs of yeastAtg8
Atg8 family proteins are essential factors in the execution phase of autophagy. Whereas the yeast Saccharomyces cerevisiae contains a single ATG8 gene, in humans the ATG8 family contains 4 MAP1LC3 (LC3) subfamily genes (MAP1LC3A, MAP1LC3B, MAP1LC3B2 and MAP1LC3C) and 3 GABARAP subfamily paralog genes (GABARAP, GABARAPL1 and GABARAPL2); another human paralog of this subfamily, GABARAPL3, is currently considered to be a pseudogene, and no data at present link it with autophagy.
Originally implicated in intracellular trafficking processes all Atg8 homologs are found within autophagosomes through binding to PE via a conserved C-terminal glycine (see Fig. 1A, black arrow). Each of the 3 families is very well conserved across its entire length and all are very similar to each other in most of their positions (Fig. 1A). However, the amino-terminal region differs between the various mammalian Atg8 proteins—the N terminus of LC3 subfamily members is strongly basic, whereas in the GABARAP subfamilies this region is acidic. It has been proposed that differences between the various human Atg8 proteins in this N-terminal region might reflect their distinct functions. Moreover, in spite of being ubiquitously expressed, some subfamily members are expressed at increased levels in certain tissues.
Figure 1.
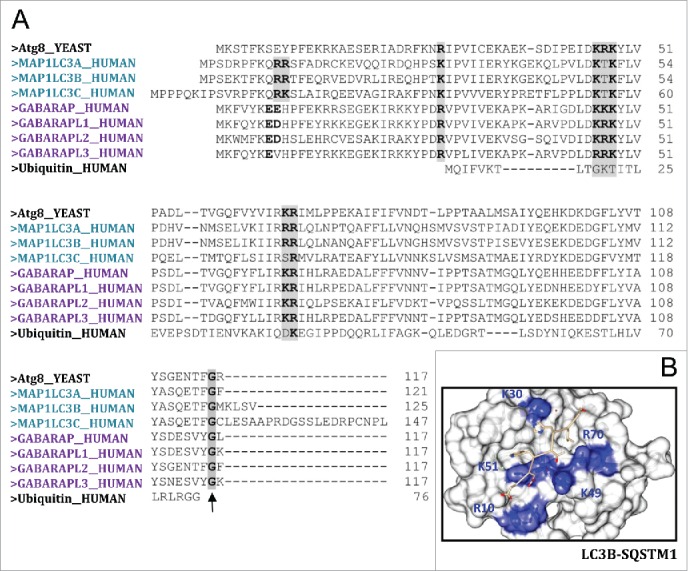
Human Atg8 family members. (A) Sequence alignment (Clustal Omega) of yeast Atg8, Atg8 human orthologs and ubiquitin. Highlighted in gray are the highly conserved basic amino acids, which have the potential to bind negatively charged amino acids located at the C- and N-terminal regions of the conserved LC3-interacting region (LIR; consensus: W/Y/FxxL/I/V) present in the different cargo receptors during selective autophagy. (B) View of LC3B bound to the SQSTM1 LIR-motif (PDB: 2ZJD). Highlighted in blue are the positively charged residues in LC3B that stabilize binding to SQSTM1 LIR motif. Picture was created by UCSF Chimera.
All Atg8 orthologs share a strong structural similarity containing 2 amino-terminal α-helices (N-terminal arm) in addition to their C-terminal ubiquitin core (ubiquitin-like domain). The ubiquitin core contains a hydrophobic feature that is conserved among family members and is suggested to have a role in protein-protein interactions (Fig. 1B), namely binding to the conjugation machinery proteins.
Interaction of LC3B with CL-containing membranes
Cardiolipin (CL), the signature lipid of mitochondria, is a unique glycerol-based phospholipid comprising 2 phosphate moieties and 4 acyl groups of which linoleic acid is predominant.18 CL is essential not only for normal mitochondrial function19 but it also plays a prominent role in disease-related processes such as apoptosis20 or mitophagy.17 In fact, although CL is usually considered to be a specific component of the inner mitochondrial membrane, the intramitochondrial location of CL is highly dynamic and CL externalization to the outer mitochondrial membrane has recently been shown to act as a signal for LC3, resulting in LC3 translocation to the OMM and targeted removal of damaged mitochondria via autophagy.17 Based on these observations, we decided to define in more detail the molecular mechanisms underlying LC3-CL interaction.
First we performed a protein-lipid overlay (PLO) assay in which recombinant LC3B was incubated with a nitrocellulose membrane containing some of the most common glycerolipids and sphingolipids present in mammalian cells (Fig. 2A). Among the lipid species examined, LC3B bound most strongly CL and cholesterol (Chol) and less strongly the anionic lipids PS and PA. Interestingly, LC3B binding was not detectable for PtdIns4P, which has the same net negative charge as CL, nor with the polyanionic lipids phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5]P2) or phosphatidylinositol-3,4,5-trisphophate (PtdIns[3,4,5]P3), with even higher net negative charges. Dose-response experiments indicated that LC3B bound CL with 5-fold higher apparent affinity than PS and PG, and 2-fold higher affinity than PA (Fig. 2B, D). These results indicate that LC3B recognizes different anionic phospholipid species and displays a preference for CL. The dose-response data for LC3B-CL binding (Fig. 2C) can be fitted to a sigmoidal curve, as detailed in the figure legend, showing a high positive cooperativity for the binding, apparent Hill coefficient n = 5.3. This might mean that binding of LC3B to one of the phosphate groups of CL would make easier the binding of the second phosphoryl moiety.
Figure 2.
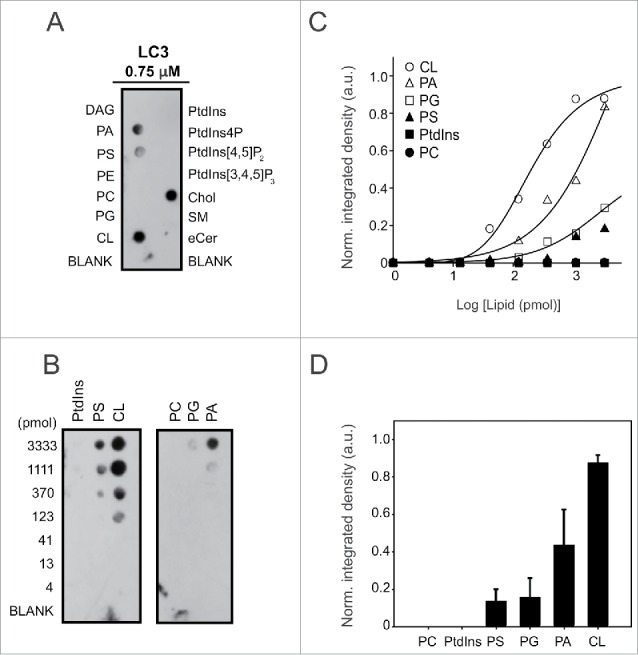
LC3B binds CL directly and preferentially over other anionic lipids. LC3B immunoreactivity is shown from a representative protein:lipid dot-blot experiment with some of the most common glycerolipids and sphingolipids present in mammalian cells (1 nmol of each lipid) (A) and with increasing amounts of some of those lipids (B). (C) Integrated LC3B immunoreactivity was measured at each spot and normalized for each lipid, and the data were fitted with a sigmoidal equation of the type f = (y0+a*xb)/(cb+xb), from which an apparent Hill coefficient (b) of 5.3 ± 1.2 for CL binding could be estimated (R2 = 0.991). (D) Quantitative assessment of bound LC3B (0.75 µM) with 1.11 nmol of each lipid. LC3B immunoreactivity was measured at each spot by immunoblotting using anti-LC3 antibody and densitometry. Data shown as mean ± SEM (n = 3). a.u., arbitrary units. Norm., normalized.
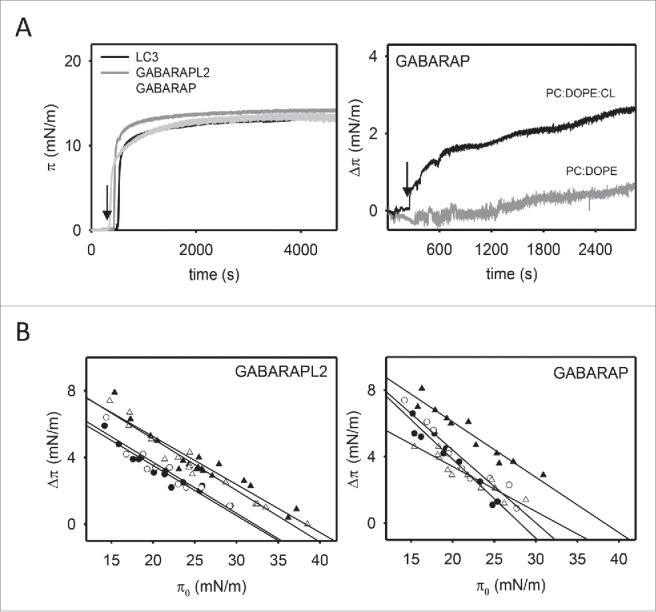
Next, we analyzed the interaction of LC3B with lipid monolayers using a Langmuir balance. Injection of LC3B into the aqueous phase led to a rapid, dose-dependent increase of lateral pressure (π) at the air-water interface (Fig. 3A, left-hand panel). An increase in π is an indication that the protein is adsorbing onto the air-water interface, i.e. that it has surface-active properties. The increase in surface pressure appears to reach a plateau value of ∼15 mN/m at 1.5 µM LC3B.
Figure 3.
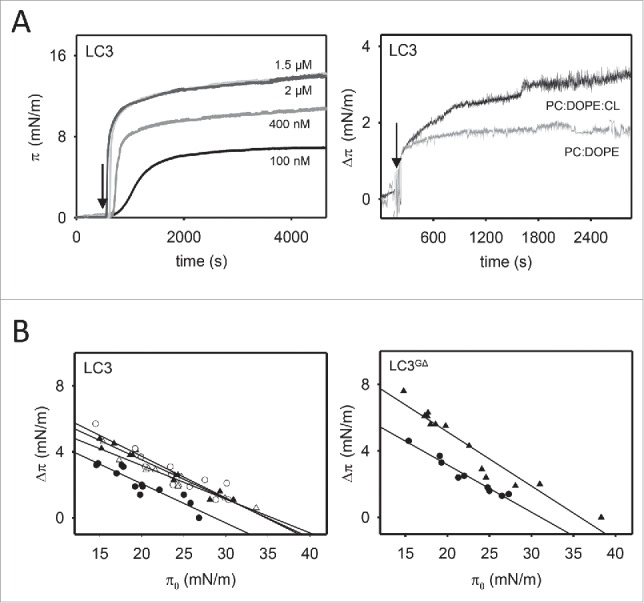
LC3B and LC3BGΔ insertion into lipid monolayers. (A) Representative time courses of LC3B adsorption at the air-water interface (left-hand panel) and representative time courses of increase in lateral pressure after LC3B (1.5 μM) insertion into PC:DOPE (80:20 mol ratio) and PC:DOPE:CL (55:20:25 mol ratio) monolayers (right-hand panel). (B) Maximum increase in lateral pressure after LC3B and LC3BGΔ insertion into lipid monolayers. Lipids were: [•] PC:DOPE (80:20 mol ratio), [▴] PC:DOPE:CL, [○] PC:DOPE:PS and [Δ] PC:DOPE:PtdIns4P (55:20:25 mol ratio). Data reported as a function of initial lateral pressure π0. Very similar results were obtained at 37°C, data not shown.
Lipid monolayers composed of either phosphatidylcholine (PC): 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (80:20 mol ratio), PC:DOPE:CL (55:20:25 mol ratio), PC:DOPE:PS (55:20:25 mol ratio), or PC:DOPE:PtdIns4P (55:20:25 mol ratio) were prepared with constant surface area at an initial pressure of 19 mN/m, i.e., above the surface pressure measured for the pure protein (∼15 mN/m). Recombinant LC3B was added to the subphase, and the resulting increase in monolayer surface pressure was monitored in real-time (Fig. 3A, right-hand panel). As shown in Fig. 3B, the increase in surface pressure observed with PC:DOPE:CL monolayers under near-equilibrium conditions was higher than the value observed with electrically-neutral PC:DOPE monolayers, indicating that LC3B inserted more favorably into negatively charged monolayers.
For a quantitative measure of the LC3B ability to penetrate into lipid monolayers, critical surface pressure values were determined. In these experiments, the increase in surface pressure (Δπ) upon LC3B addition was measured as a function of the initial surface pressure (π0). The data were fitted to a straight line, whose x-intercept corresponded to the monolayer critical surface pressure (πc). The results in Fig. 3B show that LC3B insertion was highest when anionic lipids were present in the monolayer. The LC3BGΔ mutant was also examined, lacking the C-terminal Gly of LC3B, to test whether differences observed in LC3B also appeared in its inactive mutant form. LC3BGΔ was found to be surface-active and it also exhibited a higher insertion into PC:DOPE:CL monolayers.
The critical surface pressure is a measure of the penetration capacity of a protein entering a monolayer: if this pressure exceeds 30 to 32 mN/m, then the protein is considered to be capable of membrane insertion (see Table 1). This is based on the estimation of ∼30 mN/m as the average physiological surface pressure in cell membranes.21
Table 1.
Critical pressures (πc) for LC3B and LC3BGΔ insertion into lipid monolayers.
| Protein | PC:DOPE (80:20) | PC:DOPE:CL (55:20:25) | PC:DOPE:PtdIns4P (55:20:25) | PC:DOPE:PS (55:20:25) |
|---|---|---|---|---|
| LC3B | 28.7 mN/m (±0 .40) | 35.0 mN/m (±0 .30) | 35.6 mN/m (±0 .29) | 34.7 mN/m (±0 .53) |
| LC3BGΔ | 31.0 mN/m (±0 .23) | 35.8 mN/m (±0 .64) |
Data calculated from the straight lines in Fig. 3B. The tendency line-associated standard error is given for each πc.
Thus, our data indicate that LC3B inserts at least part of its mass into the monolayer and that negatively charged groups favor the insertion. This is compatible with the idea of CL stabilizing initial electrostatic interactions leading to a partial insertion of LC3B into the bilayer.17
To test whether LC3B-CL interaction would occur in a bilayer membrane vesicle in which lipids maintain a certain bilayer curvature and could form domains, we performed liposomal float-up assays. In these experiments, pure LC3B was incubated with liposomes of a defined curvature and lipid composition and protein association with liposomes was assessed by the protein ability to float with the vesicles after equilibrium sucrose gradient centrifugation. Equivalent amounts of the top (T) and bottom (B) fractions of the gradient, corresponding respectively to proteoliposomes and to unbound protein, were then subjected to SDS-PAGE and western blot analysis. The immunoblot response of LC3B was quantified at different protein concentrations by densitometry and the data were linearly fitted by the least-squares method (R2 = 0.972; data not shown). The LC3B maximal binding response was observed with CL-containing large unilamellar vesicles (LUVs), while it did not bind PC:DOPE vesicles. Interestingly, LC3B did not bind either PtdIns4P-containing liposomes, although those vesicles had the same net negative charge as the CL-containing ones (Fig. 4A). Furthermore, considering that LC3B exhibited strong binding affinity for Chol in PLO assays (Fig. 2A), we also analyzed the binding of LC3B to model membranes containing both CL and Chol. However cholesterol inclusion in CL-enriched membranes did not lead to a higher binding response of the protein (Fig. 4A). Moreover, no LC3 binding was observed with LUVs or small unilamellar vesicles (SUVs) composed of PC:DOPE:Chol (50:20:30) (data not shown). Thus the presence of Chol in those lipid compositions did not significantly affect LC3 binding to liposomes. We also analyzed the protein dose-dependence of the interaction by incubating liposomes with LC3B at 3 different protein concentrations. As shown in Fig. 4B, little or no increase in LC3B binding to CL-containing vesicles was detected above 10 μM protein. Finally, Fig. 4C shows the dose-dependent binding of LC3B to CL-containing membranes; half-maximal binding occurred at 1.65 ± 0.2 mM total lipid. This suggests that the observed association of LC3B with PC:DOPE:CL vesicles is specific. Indeed, no protein binding was observed with PC:DOPE vesicles even at the highest lipid concentration. These results suggest that LC3B is not an integral membrane protein but that it binds membranes through an interaction with acidic phospholipids, in agreement with the hypothesis proposed above that LC3B inserts only part of its mass into the membrane and that negatively-charged groups favor its insertion.
Figure 4.

LC3B interacts preferentially with CL-containing liposomes. The liposome-bound protein fraction was analyzed after flotation in a sucrose density gradient by SDS-PAGE/immunoblot analysis and quantified by densitometric integration of the dots. (A) 10 µM LC3B was incubated with 3 mM LUVs composed of either PC:DOPE (80:20 mol ratio), PC:DOPE:CL (50:20:30 mol ratio), PC:DOPE:Chol:CL (30:14:33:23 mol ratio) or PC:DOPE:PtdIns4P (50:20:30 mol ratio). Molecular mass is shown in kDa on the left-hand side. Data shown as mean ± SEM (n = 3); **P = 0.001 to 0.01, ***P< 0.001. (B) LC3B dose-dependence analysis using 3, 10 and 20 µM protein and 3 mM liposomes. Data shown as mean ± SD (n = 3); **P = 0.001 to 0.01, ***P < 0.001. (C) Protein:lipid ratio effect on LC3B binding to PC:DOPE or PC:DOPE:CL vesicles. The continuous line represents the best fit of the data, assuming an EC50 (half maximal effective concentration) of 1.65 ± 0.2 mM for binding to PC:DOPE:CL vesicles. Data shown as mean ± SEM from at least 3 independent experiments. (D) 10 µM LC3B was incubated with 3 mM LUVs composed of PC:DOPE (80:20 mol ratio), PC:DOPE:CL, PC:DOPE:Cer16, PC:DOPE:Cer18 or PC:DOPE:Cer24:1 (50:20:30 mol ratio). Data shown as mean ± SEM from at least 3 independent experiments. (E) 10 µM LC3B was incubated with 3 mM LUV or SUV to analyze vesicle size effect on the interaction. Data shown as mean ± SD (n = 3); *P = 0.01 to 0.05. (F) Comparison between heart bovine (CL) and E. coli (CL*) cardiolipins in the interaction. 10 µM LC3B was incubated with 3 mM LUV of either composition. Data shown as mean ± SD (n = 3); **P = 0.001 to 0.01, ***P < 0.001. (G) Effect of pH on LC3B (10 μM) binding to PC:DOPE:CL large vesicles (3 mM). Data shown as mean ± SEM (n = 3); *P = 0.01 to 0.05, **P = 0.001 to 0.01.
n-octadecanoyl-d-erythro-sphingosine/C18-ceramide (Cer18) has been recently reported as being a receptor for anchoring lipidated and AP-associated LC3 (LC3-II) to mitochondrial membranes, through direct interaction of Cer18 with LC3-II, but not with the nonlipidated LC3-I.22 Based on these data, we decided to examine the binding of the soluble form of LC3B to ceramides with different fatty acid chain lengths using the flotation assay. LC3B-I binding was not detectable neither with Cer18 nor with n-palmitoyl-D-erythro-sphingosine (Cer16) or n-nervonoyl-D-erythro-sphingosine (Cer24:1) (Fig. 4D). Thus while the lipidation of LC3 appears to be important for Cer18 interaction, it is not required for LC3B binding to CL. This may suggest that ceramide interaction involves the central hydrophobic domain of LC3 while CL would bind the N-terminal domain, as predicted by docking analysis.17,22
Membrane curvature is increasingly being reported to play an important role during autophagy.23,24 Mitophagy is preceded by mitochondrial fission, which divides elongated mitochondria into smaller pieces of manageable size for encapsulation, and lipidation of LC3 has been recently proposed to occur preferentially on small, highly curved membranes in vivo and in vitro.25 To address the effect of vesicle radius on LC3 membrane binding, we examined the interaction of LC3B with PC:DOPE and PC:DOPE:CL vesicles of 2 different sizes: SUV (∼60 nm) and LUV (∼100 nm). As shown in Fig. 4E, we observed almost no binding of LC3B to PC:DOPE vesicles independently of their size. However LC3B bound CL-enriched SUVs better than LUVs. This result supports the notion that interaction of LC3B with CL is promoted in the context of a curved membrane.
Apart from containing net negative charges CL is also characterized by a high degree of unsaturation of its acyl chains. This confers a high degree of flexibility to the membrane and the latter may in turn promote LC3 association. To evaluate this particular issue we analyzed LC3B binding to liposomes containing CL from E.coli which presents relatively short and fully-saturated or mono-unsaturated acyl chains. As shown in Fig. 4F, LC3B bound significantly less the more rigid vesicles containing bacterial CL. This is in agreement with a decrease in LC3B (but not GABARAP) membrane association with a higher saturation degree of CL fatty acyl chains (Fig. S1).
Moreover we have investigated the possible effect of changes in environmental pH on LC3B binding to model CL-containing membranes. CL is supposed to have only one negative charge below pH 7.5.26 In that case the electrostatic repulsion between CL phosphate groups might be reduced which together lowers the effective size of the headgroup and the smaller size of the polar headgroup should enhance the propensity of CL to form inverted nonlamellar lipid phases.27 Regarding the pH effect on LC3B, the protein has a theoretical pI of 8.89 thus a net positive charge under our experimental conditions, with the charge becoming more positive as pH is lowered. As predicted, in vitro interaction of LC3B with CL-containing bilayers appeared to be highest at lower pH (Fig. 4G).
Additionally to the float-up assays, we used 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-conjugated monocysteine LC3B mutants (LC3BQ15C and LC3BS101C) for studying LC3B interaction with CL-containing membranes. Since the protein was labeled in each case with a unique NBD moiety, this approach allowed us to characterize the association to the membrane of specific sites of the protein. We first examined the spectral properties of the NBD-labeled LC3BQ15C mutant in the absence (black line) and presence (green line) of PC:DOPE, PC:DOPE:CL and PC:DOPE:PtdIns liposomes (Fig. 5A). In the absence of liposomes, LC3BQ15C-NBD displayed NBD λem(max) values around 550 nm indicating a predominantly solvent-exposed NBD environment. No changes in the NBD spectrum were observed upon incubation with SUVs composed of PC:DOPE or LUVs composed of PC:DOPE:PtdIns, in agreement with the low affinity of the protein observed for liposomes that did not contain CL (Fig. 4A). However incubation with CL-containing liposomes produced a substantial increase in LC3BQ15C-NBD fluorescence intensity and blue-shifts in λem(max) values, the change observed in the presence of CL-enriched SUVs being larger than with LUVs (Fig. 5A). As shown in Fig. 5B, virtually no change in the emission spectrum of LC3BS101C-NBD was observed upon addition of large or small vesicles containing CL (red line), unlike the situation found with the LC3BQ15C mutant. Based on these data, we propose a structural model in which residue C101, which is located within the C terminus of the protein, remains in a hydrophilic environment before and after LC3B binding to CL-containing liposomes, while residue C15, which is localized within the N terminus, moves from a hydrophilic to a hydrophobic environment upon LC3B membrane binding (Fig. 5C).
Figure 5.
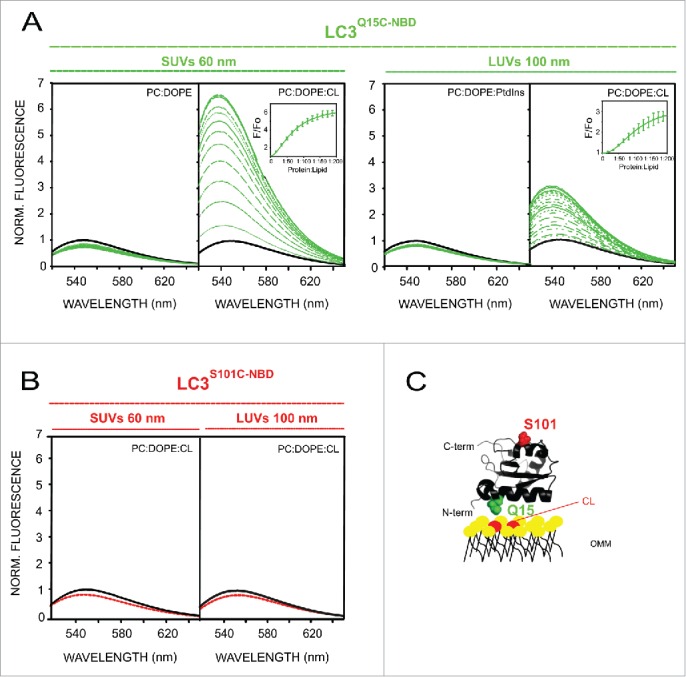
LC3B C terminus remains exposed to the hydrophilic environment after protein binding to CL-enriched membranes. Representative NBD fluorescence emission spectra of (A) LC3BQ15C-NBD (1 µM) in the absence or presence of increasing amounts of liposomes containing either PC:DOPE (80:20 mol ratio), PC:DOPE:CL or PC:DOPE: PtdIns (50:20:30 mol ratio); and (B) LC3BS101C-NBD (1 μM) in the absence or presence of PC:DOPE:CL liposomes. In each case, fluorescence was normalized to the peak intensity of the protein spectrum in the absence of liposomes. (C) Structural model generated with PyMol depicting the 2 LC3B residues that were individually mutated to cysteine obtaining single-cysteine LC3B mutants. The environmentally sensitive fluorophore NBD was used to label each of these single cysteine residues. PDB: 1UGM. OMM, outer mitochondrial membrane. Norm., normalized.
To sum up, results obtained using 4 independent lipid-interaction assays (lipid dot-blot, lipid monolayer surface pressure measurements, liposome flotation and NBD fluorescence spectroscopic analysis) concur in suggesting that (i) LC3B interacts preferentially with CL over other anionic phospholipids, (ii) LC3B-CL interaction relies on both electrostatic and other CL-mediated changes in membrane properties, e.g. bilayer fluidity and (iii) LC3B C terminus remains exposed to the hydrophilic environment after protein binding to CL-enriched membranes.
CL translocation to the OMM in U87MG glioblastoma cells leads to LC3B localization in mitochondria
The conversion of the soluble form of LC3 (LC3-I) to the lipidated and autophagosome-associated form (LC3-II) is considered one of the hallmarks of autophagy. In intact cells, formation of LC3-II is associated with the appearance of LC3 puncta readily detected by conventional fluorescence microscopy. The mitochondrial complex I inhibitor rotenone and other promitophagy stimuli have been shown to increase the number of LC3 puncta and the level of colocalization with mitochondria. Interestingly, rotenone treatment also induced externalization of CL to the mitochondrial surface.17
U87MG cells were selected as a cellular model to investigate the activation of autophagy and mitophagy by different stimuli. In addition it is known that autophagy plays an important role in cancer cells and therefore the mechanism of regulation of mitochondria degradation studied in this cell line could also be relevant in the control of cancer cell survival. Cells were treated with rotenone for 4 h and colocalization of mitochondria with endogenous LC3B puncta was examined. In control experiments, nutrient starvation or Δ9-tetrahydrocannabinol (THC) were used as nonselective autophagy inducers. In agreement with Kagan and coworkers, rotenone treatment elicited robust colocalization of mitochondria with LC3B puncta, indicative of LC3B translocation to the organelle (Fig. 6A). In parallel experiments, both THC and nutrient starvation caused potent LC3B puncta formation consistent with reports indicating that THC and nutrient deprivation induce autophagy-mediated death and protective autophagy, respectively.28,29 Nevertheless, LC3B puncta formed under these treatment conditions did not exhibit mitochondrial colocalization unlike the situation observed with rotenone. Rotenone-induced mitophagy was further confirmed by colocalization of both mitochondria (Fig. 6B) and LC3B (Fig. 6C) with lysosomes. Finally, consistent with THC and nutrient starvation not inducing mitophagy, LC3B puncta (Fig. 6C) but not mitochondria (Fig. 6B) exhibited lysosomal colocalization. Altogether, these results indicate that rotenone, but not other well-characterized autophagy inducers (THC, nutrient starvation) elicited mitophagy in U87MG human glioblastoma cells.
Figure 6.
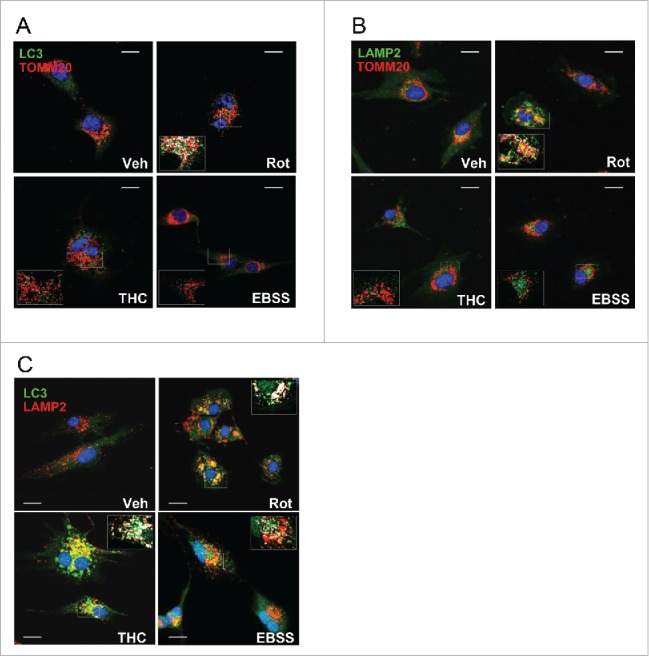
Rotenone, but not THC or nutrient starvation, elicits mitophagy in U87MG human glioblastoma cells. (A) Analysis of endogenous LC3B puncta colocalization with TOMM20-stained mitochondria in response to rotenone (Rot) (1 μM; 4 h) or THC (4 μM; 6 h) treatments, or to nutrient deprivation (EBSS) conditions in U87MG cells. (B) Rotenone, but not THC or EBSS, increased delivery of TOMM20-stained mitochondria to LAMP2-stained lysosomes in U87MG cells. (C) LC3B puncta colocalization with lysosomes (LAMP2) in response to rotenone (Rot) (1 μM; 4 h) or THC (4 μM; 6 h) treatments, or to nutrient deprivation (EBSS) conditions in U87MG cells. Bar: 20 µm. Veh, vehicle.
GABARAPL2 and GABARAP interaction with CL-containing membranes
To determine whether mechanisms underlying LC3-CL interaction are protein-specific or whether they are the same for all human orthologs, we analyzed GABARAPL2 and GABARAP interaction with CL-enriched membranes.
First we observed that GABARAPL2 and GABARAP injection into the aqueous phase led to an increase in π at the air-water interface similar to that of LC3B (Fig. 7A, left-hand panel), also reaching a plateau value of ∼15 mN/m at 1.5 µM protein concentration. Moreover, monolayer insertion of GABARAPL2 and GABARAP was also facilitated by the presence of CL or PtdIns4P (Fig. 7A (right-hand panel) and B). PS however did not distinctly modify the insertion of these 2 orthologs, resulting in a change in lateral pressure equivalent to that observed with PC:DOPE (see Table 2).
Figure 7.
GABARAPL2 and GABARAP insertion into lipid monolayers. (A) Representative time courses of adsorption of LC3B, GABARAPL2 and GABARAP (1.5 µM) at the air-water interface (left-hand panel) and representative time courses of increase in lateral pressure after GABARAP (1.5 μM) insertion into PC:DOPE (80:20 mol ratio) or PC:DOPE:CL (55:20:25 mol ratio) monolayers (right-hand panel). (B) Maximum increase in lateral pressure after GABARAPL2 and GABARAP insertion into lipid monolayers. Lipids were: [•] PC:DOPE (80:20 mol ratio), [▴] PC:DOPE:CL, [○] PC:DOPE:PS and [Δ] PC:DOPE:PtdIns4P (55:20:25 mol ratio). Data reported as a function of initial lateral pressure π0. Very similar results were obtained at 37°C, data not shown.
Table 2.
Critical pressures (πc) for GABARAPL2 and GABARAP insertion into lipid monolayers.
| Protein | PC:DOPE (80:20) | PC:DOPE:CL (55:20:25) | PC:DOPE:PtdIns4P (55:20:25) | PC:DOPE:PS (55:20:25) |
|---|---|---|---|---|
| GABARAPL2 | 31.8 mN/m (±0 .39) | 38.2 mN/m (±0 .57) | 36.6 mN/m (±0 .48) | 32.1 mN/m (±0 .49) |
| GABARAP | 28.1 mN/m (±0 .43) | 38.3 mN/m (±0 .48) | 32.5 mN/m (±0 .42) | 30.0 mN/m (±0 .55) |
Data calculated from the straight lines in Fig. 7B. The tendency line-associated standard error is given for each πc.
Furthermore, estimating the electrostatic surface potential of Atg8 orthologs in solution revealed that the predicted CL-interacting region for LC3B was markedly less basic in the case of GABARAPL2 and GABARAP (Fig. 8A).To test whether these structural features affected the lipid-binding profile of each LC3 ortholog, we assessed the binding of GABARAPL2 and GABARAP to PC:DOPE and PC:DOPE:CL vesicles by liposomal float-up assays. As depicted in Fig. 8B, GABARAP displayed a significantly lesser CL-binding response compared with that of LC3B and no differences were observed with vesicles containing mono-unsaturated or fully saturated CL (Fig. S1). Additionally, even less GABARAPL2 was observed to bind to CL-containing liposomes. We also compared the effect of liposomes containing CL on GABARAPL2 and GABARAP labeled at equivalent positions with NBD. To this aim, GABARAPS16C and GABARAPS88C mutants and GABARAPL2, on its endogenous C15 cysteine, were specifically labeled with NBD. For each monocysteine mutant, a structural model is shown in Fig. 8C indicating the location of the Cys-substituted amino acid which was labeled with NBD. A representative NBD spectrum for each mutant in the absence (black line) or presence (green line) of PC:DOPE and PC:DOPE:CL is also shown. In solution, the λem (max) values of NBD attached to monocysteine GABARAPL2 or GABARAP mutants were similar to those of LC3B mutants. The magnitude of NBD spectral changes induced by the presence of CL-enriched liposomes was somewhat smaller for GABARAPS16C than for LC3BQ15C (Fig. 8C) and even smaller for GABARAPL2C15, in correlation with the electrostatic surface potential of each protein and the flotation experiments shown above (Fig. 8A, B). Moreover, no increase in the emission spectrum of GABARAPS88C-NBD was observed upon its incubation with CL-containing large vesicles (red line) (Fig. 8C). On the basis of the previously observed NBD spectral changes upon moving from solution to CL-containing liposomes, both LC3B and GABARAP would interact with CL-enriched membranes while exposing their C terminus to the hydrophilic environment (Figs. 5C and 8D). Taken together, these results strongly suggest that differences shown in the electrostatic surface potential of LC3 family proteins may lead to lipid-binding specificities among them.
Figure 8.

Different CL-interacting properties among human Atg8 orthologs. (A) Electrostatic potential surface of each LC3 ortholog in solution, calculated using the Poisson-Boltzmann equation and displayed with PyMOL. PDB: 1UGM (LC3), 1EO6 (GABARAPL2), 1GNU (GABARAP). (B) Each ortholog (10 µM) was incubated with 3 mM LUVs composed of PC:DOPE (80:20 mol ratio) or PC:DOPE:CL (50:20:30 mol ratio) followed by flotation of the liposomes by gradient centrifugation. Bound protein fraction was quantified by SDS-PAGE/immunoblot analysis using anti-LC3, GABARAPL2 and GABARAP antibodies. Molecular masses are shown in kDa on the left-hand side. Data shown as mean ± SD (n ≥ 3); ***P< 0.001. (C) Representative NBD fluorescence emission spectra of GABARAPL2C15-NBD and GABARAPS16C-NBD (1 µM) in the absence or presence of increasing amounts of liposomes containing PC:DOPE (80:20 mol ratio) or PC:DOPE:CL (50:20:30 mol ratio); and GABARAPS88C-NBD (1 μM) in the absence or presence of PC:DOPE:CL liposomes. In each case, fluorescence was normalized to the peak intensity of the protein spectrum in the absence of liposomes. (D) A structural model generated with PyMol is also shown depicting the 2 GABARAP residues that were individually mutated to cysteine obtaining single-cysteine GABARAP mutants and the endogenous cysteine of GABARAPL2. The environmentally sensitive fluorophore NBD was used to label each of these single cysteine residues. PDB: 1EO6 (GABARAPL2), 1GNU (GABARAP). CL is colored in red. Norm, normalized.
In their study, Kagan and colleagues17 have determined that cardiolipin recognition for mitophagy depended on N-terminal amino acids (R10 and R11) of LC3B. To further validate our liposome-binding assay, we mutated those residues to alanine in LC3B and evaluated the LC3BR10,11A capacity to bind CL-enriched liposomes (Fig. S2A). Substitution of those residues did not produce structural changes in the protein as tested by secondary structure examination of both wild-type (WT) and mutant LC3B forms using far-UV circular dichroism spectroscopy (data not shown). Mutating those 2 residues made the anti-LC3 antibody unable to recognize LC3B in western blots (data not shown). To circumvent this we decided to label both LC3B and LC3BR10,11A with Alexa Fluor 488. The Alexa Fluor 488 fluorescence of LC3B-A488 was quantified at different protein concentrations by densitometry and the data were linearly fitted by the least-squares method (R2 = 0.997; data not shown). Consistent with a critical role of these 2 basic amino acids in the interaction of LC3 with CL, the RR to AA mutation clearly decreased LC3BR10,11A affinity for CL (Fig. S2B and C). In order to test whether these N-terminal residues are also involved in GABARAP interaction with CL-containing membranes, R14 and R15 GABARAP amino acids equivalent to the ones in LC3B, and K46, K47, K48 amino acids, that are also within the proposed interaction patch (see Fig. 8A), were mutated to alanines (Fig. S2D). Note that K46, K47 and K48 are highly conserved basic amino acids (see Fig. 1A) that have been shown to interact with amino acids present in the conserved LC3-interacting region (LIR) of the different cargo receptors during selective autophagy. Notably, all GABARAP mutants were still capable of recognizing CL, some of them with an even higher affinity than wild-type GABARAP (Fig. S2E). Taking into account that substitution of the positively charged residues R10 and R11 had a negative effect on LC3B but not on GABARAP binding to CL, the contribution of electrical charge to each protein-CL interaction was further analyzed. To this aim, the interaction of LC3B and GABARAP with CL-enriched liposomes was examined in the presence of 300 mM NaCl (Fig. S2F). We found that increasing the ionic strength of the medium both LC3B and GABARAP binding to CL-containing liposomes decreased, suggesting that in both cases the interaction depends at least in part on an electrostatic component. Given that R14, R15, K46, K47 and K48 residues are not essential for GABARAP binding to CL, other N-terminal residues may be involved in the protein interaction with CL.
Next, in order to analyze the functional implications of the differences observed in vitro among human orthologs, we examined whether rotenone treatment induced GABARAPL2 and GABARAP translocation to mitochondria in glioblastoma cells. To this aim, U87MG cells were transiently transfected with plasmids expressing human GABARAPL2- or GABARAP-GFP and subjected to either nutrient starvation, THC or rotenone treatments. As shown in Fig. 9A and B, all treatments elicited an increase in GABARAPL2- and GABARAP-GFP puncta within cells. However GABARAP- or GABARAPL2-GFP puncta did not exhibit mitochondrial colocalization after rotenone treatment. Thus, LC3B translocation to mitochondria in response to externalized CL might play a specific role in this cell line, different from that of GABARAPL2 and GABARAP.
Figure 9.
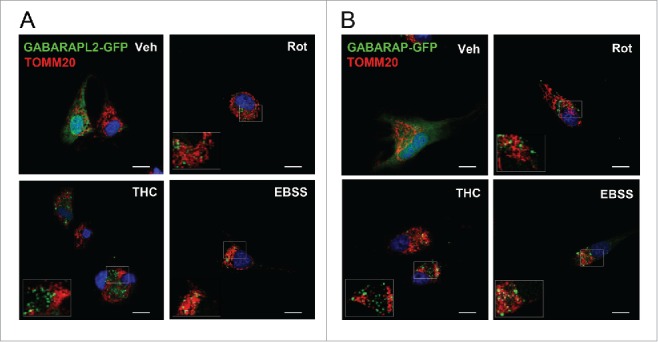
GABARAPL2 and GABARAP do not exhibit mitochondrial colocalization after rotenone treatment in U87MG cells. (A-B) Analysis of GABARAPL2-GFP or GABARAP-GFP puncta colocalization with TOMM20-stained mitochondria in response to rotenone (1 μM; 4 h) or THC (4 μM; 6 h) treatments, or to nutrient deprivation conditions in U87MG cells. Bar: 20 µm. Veh, vehicle.
Discussion
The expansion of the Atg8 family proteins in higher eukaryotes has been proposed to facilitate differential cargo handling during selective autophagy.30 The components of the ULK1 (unc-51-like autophagy activating kinase 1)-ULK2 complex, for instance, bind preferentially GABARAP-like over LC3-like proteins31 whereas the receptor protein FYCO1 (FYVE and coiled-coil domain-containing 1) displays a strong preference for LC3.32 Structural studies have highlighted the importance of specific features of individual members in determining these preferential bindings. Importantly while most ubiquitin-directed autophagy receptors (SQSTM1, NBR1 [NBR1, autophagy cargo receptor] and OPTN [optineurin]) interact with both GABARAP and LC3 subfamily proteins (Fig. 1),33-35 specific binding of CALCOCO2 to LC3C (and not the other LC3 isoforms) is crucial for innate immunity during xenophagy.36 Again structural analysis revealed that the selectivity of CALCOCO2 for LC3C is conferred by a noncanonical LIR in CALCOCO2, in which the lack of an aromatic residue is balanced by specific interactions within the N-terminal region of LC3C. In addition to its role in microbial clearance, LC3C has been related to a unique anticarcinogenic activity37 whereas LC3B promotes tumor growth. While it is very likely that this opposite effect is related to differential recruitment of critical autophagy targets, the identity of these cargos and the structural basis for these specific interactions are still elusive.
Selective elimination of mitochondria by mitophagy regulates not only the steady-state mitochondrial number required for changing metabolic demands, but also maintains cell quality control by removing damaged organelles. Although mitochondria can be engulfed nonselectively along with other cytosolic contents during bulk autophagy, different lines of evidence indicate that mitophagy can be mechanistically differentiated from other forms of autophagy, and mitophagy-specific mechanisms are now beginning to emerge.
Yeast genetic screening identified Atg11 as an important mitophagy regulator. By binding to Atg32 at the OMM and to Atg8 at the growing phagophore, Atg11 recruits the canonical autophagic machinery to the mitochondria and primes mitochondria for degradation.38 In mammals, the ubiquitin-binding receptor SQSTM1 (sequestosome 1) is thought to act in the same way as Atg1139 and during erythrocyte differentiation mitochondrial resident BNIP3L (BCL2 interacting protein 3 like) functions as a mitophagy receptor at least in part by binding to LC3 orthologs.40
Alternatively, upon mitochondrial membrane depolarization, PINK1 accumulates at the OMM and recruits the E3 ubiquitin-protein ligase PARK2 from the cytosol specifically to the damaged mitochondrion. PARK2 then induces mitochondrial degradation through both ubiquitin-dependent and -independent pathways. The genes encoding PINK and PARK2 were found to be mutated in certain forms of autosomal recessive Parkinson disease (PD)41,42 and loss of either protein in Drosophila results in mitochondrial dysfunction.43 Nevertheless, pink1- or park2-knockout mice appear largely normal and have only subtle phenotypes, suggesting that in the absence of PINK1or PARK2 other factors can sustain effective mitophagy. In this line, it has been recently shown that exposed CL on the surface of the OMM combines with LC3 and gives rise to the recognition of injured mitochondria.17
The process was not accompanied by membrane depolarization and was independent of PINK1 or PARK2. Interestingly, CL has also been reported to interact with other mitophagy-related proteins including DNM1L (dynamin 1 like),44 human IRGM (immunity-related GTPaseM)45 and BECN1 (Beclin 1),46 making CL an essential lipid in this process for both cargo reception and protein function regulation.
LC3B-CL interaction in vitro
The aim of this study was to characterize the association of the autophagy protein LC3 with CL-containing membranes. In this work, we have described novel molecular mechanisms underlying this protein-lipid interaction. Using several compositionally defined in vitro reconstituted systems we show that LC3B avidly interacts with CL, both when CL was spread onto nitrocellulose membranes in the context of a PLO assay and in CL-enriched model membranes (Figs. 2 to 5). LC3B binds preferentially CL over other anionic lipids such as PtdIns4P, even though the latter also carries 2 negative charges and has been related to certain types of selective autophagy. Furthermore, we show that association of LC3B with CL predominantly relies on electrostatic interactions (Fig. 8). These results are consistent with the notion that the interaction is mediated by basic patches at LC3 surface that stabilize the phosphate moieties of CL.17 Our results with the LC3BR10,11A mutant (Fig. S2) strongly support the idea that some of the residues within those patches are essential for LC3-CL interaction, modulating the binding capacity of the protein. Moreover, molecular docking analyses of LC3B upon binding to CL-containing membranes implicate those N-terminal Arg10 and Arg11 in the initial interactions of LC3 with CL.17 In the in silico model the C terminus of LC3, which becomes crosslinked to autophagosome membranes through a conserved glycine residue (see Fig. 1A, black arrow) is predicted to remain exposed to the solvent. Here, using site-specific NBD labeling coupled to NBD fluorescence spectroscopic analysis, we provide experimental data supporting this model in which the N- and C-terminal regions would bind mitochondria and autophagosomal membranes respectively (Fig. 5). This membrane-interacting surface of LC3B located at the N-terminal domain would approach the hydrophobic region of the bilayer and directly interact with CL in the lipid bilayer. This N-terminal interaction with CL may induce a conformational rearrangement in LC3 that would lead to an enhancement of LC3 autophagic activity via its C terminus binding the autophagosomal membrane. Moreover, our previous study has found that fusion induced by LC3 human orthologs is facilitated by the presence of CL in model membranes.47 Therefore, CL-mediated modulation of LC3B function would occur via changes in both membrane and protein properties. However, whether or not the CL effects on LC3B will affect the role of the LC3 conjugation system in autophagy remains to be clarified.
CL isolated from heart cells displays not only a unique tandem head group arrangement but also a high degree of unsaturation in the acyl chains, linoleic (C18:2) being the most abundant fatty acid. This structural feature contributes to a high degree of flexibility within the membrane that may facilitate LC3 insertion and association. In support of this idea, LC3B binding to CL was significantly lower with vesicles enriched in bacterial CL that contains more saturated acyl chains leading to a more rigid membrane (Fig. 4F). Thus bilayer rigidity would be another regulative mechanism in LC3B association with CL-enriched membranes. Indeed, lipid oxidation increases markedly mitochondrial membrane rigidity observed in aging animals.48 Hence free radical reaction-induced decrease in membrane fluidity would affect LC3 recognition of damaged mitochondria and this would be an aging-related process.
LC3B preferential binding to smaller and more curved CL-containing liposomes may suggest that vesicle curvature is another factor increasing LC3 affinity for CL. This could be a mechanism of recognition of previously processed mitochondria, which would have been converted into pieces of manageable size for their engulfment by autophagosomes. Studies in different mammalian cells have shown that after DNM1L-mediated fission, depolarized mitochondrial products are much less likely to fuse and are eventually autophagocytosed.49 Moreover, inhibition of DNM1L prevents both mitochondrial fragmentation and mitophagy, indicating that fission is related to efficiency of mitophagy. Therefore, mitochondrial shape and size appear to be critical for LC3-CL interaction and targeted mitochondrial degradation. Note in this respect that the negative intrinsic curvature of the lipid CL is conceptually different from the geometrical curvature of a vesicle, in fact lipids with an intrinsic negative curvature can exist in vesicles, e.g., SUVs, with a high geometrical curvature (small radius).50
Additionally, LC3B interaction with CL-containing membranes would be facilitated in an acidic environment (Fig. 4G), which is in agreement with LC3 interacting with CL in the OMM of damaged mitochondria. Indeed inhibition of the mitochondrial ATP synthase could induce a higher proton concentration in the intermembrane space that would finally cause a local cytosolic pH decrease. When the mitochondrial respiratory chain is blocked lactic acid fermentation could also be activated as an alternative for the cell to obtain energy, and lactic produced in that process would tend to acidify the intracellular environment, thus to facilitate LC3-CL interaction. However the relationship between pH, CL net charge and LC3-CL binding may not be straightforward. Changing the environmental pH would change the protonation state of CL, but also the protein net charge, and CL packaging in the membrane.51,52 Since at lower pH CL would have a smaller negative charge, less binding of LC3B to CL would be expected, but at the same time LC3 charge would become more positive, and this would facilitate LC3 electrostatic interaction with CL. Moreover we must take into account that this interaction relies only partially on electrostatic forces, thus CL negative charge is just one of the several membrane properties that facilitate the interaction. Instituting an artificial pH gradient in CL-containing giant unilamelar vesicles (pH 4 to 5 outside) generates profound invaginations, potentially enriched in CL, that would mimic mitochondrial cristae.53 If these are indeed CL-enriched microdomains, they would facilitate the local binding of LC3.
LC3B translocation to mitochondria
In contrast to the protonophores carbonyl cyanide p-trifluoromethoxyphenyl hydrazone (FCCP) or carbonyl cyanide m-chlorophenyl hydrazine (CCCP), that have been found to cause a severe loss of mitochondrial membrane potential in neuronal cells, sublethal doses of rotenone and 6-hydroxydopamine (6-OHDA) have no effect on membrane potential although they induce LC3, but not PINK1, translocation to mitochondria.17 Consistent with this, results presented in this work show that LC3B translocates to mitochondria upon mild rotenone treatment in U87MG glioblastoma cells, resulting in targeted removal of damaged mitochondria through the action of lysosomes (Fig. 6). Nevertheless, nutrient deprivation did not elicit translocation of LC3B to mitochondria or targeting of mitochondria to lysosomes. These results are consistent with reports indicating that in higher eukaryotes mitochondria are in fact spared from autophagy during starvation. In yeast however degradation of mitochondria occurs upon both nutrient depletion and/or mitochondrial damage54 and there is no experimental evidence that mitophagy selects damaged, but not intact, mitochondria for degradation. Moreover, yeast proteins mediating this process have no mammalian orthologues. Therefore the molecular mechanisms involved in selective mitophagy appear to be fairly different in yeast and mammals.
THC induced potent bulk autophagy, however, no mitochondria were targeted to degradation in U87MG cells (Fig. 6B). THC increases sphingolipid synthesis in the ER, eventually contributing to autophagy-mediated cancer cell death.27 In parallel, C18-ceramide anchors LC3-II-autolysosomes to mitochondrial membranes, inducing lethal mitophagy.22 Furthermore, mitochondrial localization of endogenous ceramides and previous lipidation of LC3 have been suggested to be essential for LC3-ceramide association during mitophagy,22 the latter being in agreement with our experimental data. Moreover during THC-induced autophagy-mediated cell death, ceramide accumulation has been reported to stimulate an ER stress-related signaling route. However, during bulk THC-promoted autophagy, direct LC3 interaction with ceramide has not yet been described.
The experimental method above could also be used to measure the mitophagy flux via LC3B-CL interaction but in this case with or without the presence of a compound such as cloroquine to inhibit mitochondrial degradation in the lysosome. In this way we could measure an increment in the level of mitochondria colocalization with LC3 together with a decrease in its colocalization with lysosomes.
To sum up, CL externalization promotes LC3B translocation to mitochondria and the subsequent induction of mitophagy in U87MG glioblastoma cells, while other proautophagy stimuli lead to nonselective autophagy. The specific factors, distinct from bulk autophagy, required for individual mitochondria to be distinguished within the mitochondrial network are still to be determined.
GABARAPL2 and GABARAP in vitro and in vivo
Since most studies have been focused on LC3, the role of other human Atg8 orthologs and paralogs in mitophagy is less well characterized. LC3 family proteins have different specificities guiding the selective recruitment of different LIR-containing proteins.55 CL-binding assays and electrostatic surface potentials shown in this study revealed that human Atg8 orthologs also differ in their lipid-binding affinities (Figs. 7 to 9). In contrast to LC3B and GABARAP, GABARAPL2 hardly binds CL-containing membranes (Fig. 8). Furthermore, unlike LC3B, GABARAPL2 and GABARAP do not translocate to mitochondria upon mitophagy induction in U87MG cells (Fig. 9), suggesting different regulatory mechanisms between orthologs. It is noteworthy that even though GABARAP is able to interact with CL in vitro, though with less affinity than LC3B, it does not show mitochondrial localization after rotenone treatment. Thus the possibility remains that its capacity to interact with CL simply relies on its structural homology with LC3B, in such a way that translocation of GABARAP to mitochondria would occur via a different mechanism. LC3B, GABARAPL2 and GABARAP may play different roles during selective and nonselective autophagy, and there might be different mechanisms involved in the regulation of mitophagy pathways depending on the cellular and physiological context.
Concluding remarks
The quantitative data on LC3B-CL interaction presented in the current study should help to clarify the molecular mechanisms by which this phospholipid acts as a receptor for mitochondrial targeting of the human autophagic machinery. LC3B targeting to the OMM upon CL externalization is clearly modulated by factors including ionic strength and bilayer fluidity. Moreover, LC3B may recognize more efficiently smaller mitochondrial fragments for their encapsulation, consistent with the targeted removal of mitochondria being closely linked to mitochondrial fission. The lipid-protein electrostatic interaction would be a mitophagy-specific mechanism that may not occur in protective nonselective autophagy or THC-induced autophagy. Our data also imply that the individual human Atg8 orthologs respond differently to the presence of CL in model membranes, in accordance with the differences observed for their electrostatic surface potential. Furthermore, GABARAPL2 and GABARAP do not respond to CL externalization during mitophagy in U87MG cells, indicating different cargo-targeting mechanisms among human Atg8 orthologs that could also be cell type-dependent. However, the possibility of this process being cell, tissue or organ specific and the possible crosstalk between the different selective types of autophagy need to be further investigated for a better understanding of mechanisms triggering mitophagy and therefore of neurodegenerative diseases such as Parkinson or Alzheimer diseases.
Materials and methods
Materials
Phosphatidylcholine from egg yolk (PC) (Lipid Products, Grade 1), cholesterol (Chol) (Avanti Polar Lipids, 700000), heart bovine cardiolipin (CL; 90% tetralinoleoylcardiolipin, 5% tetraoleoylcardiolipin, 5% unknown; Avanti Polar Lipids, 840012), tetraoleoylcardiolipin (CL 18:1; Avanti Polar Lipids, 710335), tetramyristoylcardiolipin (CL 14:0; Avanti Polar Lipids, 750332), E.coli cardiolipin (CL*; Avanti Polar Lipids, 841199), phosphatidylinositol-4-phosphate (PtdIns4P; Avanti Polar Lipids, 840045), phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5]P2; Avanti Polar Lipids, 840046), phosphatidylinositol-3,4,5-trisphophate (PtdIns[3,4,5]P3; Avanti Polar Lipids, 850156), brain phosphatidylserine (PS; Avanti Polar Lipids, 840032), liver phosphatidylinositol (PtdIns; Avanti Polar Lipids, 840042), egg phosphatidylglycerol (PG; Avanti Polar Lipids, 841138), egg phosphatidic acid (PA; Avanti Polar Lipids, 840101), 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (Rho-PE; Avanti Polar Lipids, 810150), 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE; Avanti Polar Lipids, 850725), egg ceramide (eCer; Avanti Polar Lipids, 860051), N-palmitoyl-D-erythro-sphingosine (Cer16; Avanti Polar Lipids, 860516), N-octadecanoyl-D-erythro-sphingosine (Cer18; Avanti Polar Lipids, 860518), N-nervonoyl-D-erythro-sphingosine (Cer24:1; Avanti Polar Lipids, 860525). Anti-LC3 monoclonal antibody (Medical & Biological laboratories Co. Ltd., M186-3), anti-GABARAP monoclonal antibody (Medical & Biological laboratories Co. Ltd., M135-3) and anti-GABARAPL2 polyclonal antibody (Medical & Biological laboratories Co. Ltd., PM038). Anti-LAMP2 (lysosomal-associated membrane protein 2) antibody (1:100; BD Biosciences, 555803), anti-LC3 antibody (1:1000; Sigma Aldrich, L8918) and anti-TOMM20 antibody used in immunofluorescence (Santa Cruz Biotechnology, sc-17764).
DNA constructs and site-directed mutagenesis
Plasmids for expression of several of the various orthologs of Atg8 (human LC3B, human GABARAPL2 and human GABARAP) and human LC3B lacking the C-terminal Gly (LC3BGΔ) were kindly provided by Dr. Isei Tanida (National Institute of Infectious Diseases, Tokyo, Japan). Note that each Atg8 ortholog is a truncated form ending in the reactive C-terminal Gly such that no ATG4-mediated preprocessing is necessary. These plasmids were used as templates for site-directed mutagenesis using QuikChange site-directed mutagenesis kit (Stratagene, 200514) following the manufacturer's instructions. All primers designed to introduce site-directed mutations were synthesized and purified by Sigma-Aldrich. The polymerase chain reaction-amplification products were evaluated by agarose gel electrophoresis and the parental methylated and hemimethylated DNA was digested by DpnI endonuclease (New England Biolabs, R0176). After inactivation of DpnI (80°C for 20 min), the digested polymerase chain reaction product was transformed into DH5-α E. coli chemo-competent cells and inoculated on Luria-Bertani (LB; Sigma Aldrich, L3022) plate containing 100 mg/ml ampicillin (Sigma Aldrich, A0166). A total of 5 colonies were selected and their plasmids were isolated by mini-prep (GeneJET Plasmid Miniprep Kit, Thermo Scientific, K0502) and used for DNA sequencing (Secugen S.L., Madrid, Spain).
The double mutant for arginine (Arg) to alanine (Ala) at positions 10 and 11 of LC3B (LC3BR10,11A) was encoded using sense primer (5′-GAAGGTGGCGGCCTGCTTGAA-3′) and antisense primer (5′-TTCAAGCAGGCCGCCACCTTC-3′) respectively. The double mutant for arginine (Arg) to alanine (Ala) at positions 14 and 15 of GABARAP (GABARAPR14,15A) was encoded using sense primer (5′-CTTCTCGCCCTCAGAGGCGGCCTTCTCGAACGGATGC-3′) and antisense primer (5′-GCATCCGTTCGAGAAGGCCGCCTCTGAGGGCGAGAAG-3′) respectively.
The single mutant for lysine (Lys) to alanine (Ala) at position 47 of GABARAP (GABARAPK47A) was encoded using sense primer (5′- AGAAGGCACCAGGTATTTCGCTTTGTCCAGGTC-3′) and antisense primer (5′-GACCTGGACAAAGCGAAATACCTGGTGCCTTCT-3′) respectively. The double mutant for lysine (Lys) to alanine (Ala) at positions 46 and 47 of GABARAP (GABARAPK46,47A) was encoded using sense primer (5′-ATAGGAGACCTGGACGCAGCGAAATACCTGGT-3′) and antisense primer (5′-ACCAGGTATTTCGCTGCGTCCAGGTCTCCTAT-3′) respectively. The triple mutant for lysine (Lys) to alanine (Ala) at positions 46, 47 and 48 of GABARAP (GABARAPK46,47,48A) was encoded using sense primer (5′-GCACCAGGTATGCCGCTGCCTCCAG-3′) and antisense primer (5′-CTGGAGGCAGCGGCATACCTGGTGC-3′) respectively.
Single cysteine (Cys) point mutants of LC3B (LC3BQ15C and LC3BS101C) were encoded with sense primers [5′-CGGACATCTTCTACTCTGCATTCGAAGGTGCGGCGCTGCT-3′ and 5′- AGCAGCGCCGCACCTTCGAATGCAGAGTAGAAGATGTCCG-3′] and antisense primers [5′-CCATCTTCATCTTTCTCACACTCATACACCTCTGAGATT-3′ and 5′-AATCTCAGAGGTGTATGAGTGTGAGAAAGATGAAGATGG-3′] respectively. Single Cys point mutants of GABARAP (GABARAPS16C and GABARAPS88C) were encoded with sense primers [5′-CTCGCCCTCACAGCG GCGCTTCTC-3′ and 5′-CCCATTGTGGCACAGGTGGGTGGAAT-3′] and antisense primers [5′-GAGAAGCGCCGCTGTGAGGGCGAG-3′ and 5′- ATTCCACCCACCTGTGCCACAATGGG-3′] respectively.
Recombinant protein expression and purification
All proteins were purified from soluble fractions of bacterial extracts obtained in the absence of detergents, and were >90% pure as evaluated by Coomassie Blue-stained SDS-PAGE. E.coli BL21(λDE3) cells were transformed with appropriate plasmids and cells were grown to OD600 (optical density at 600 nm) of 0.8 and induced with 0.5 mM IPTG (isopropyl-β-D-1-thiogalactopiranoside; Sigma Aldrich, I6758) for 4 h at 20°C in the case of GABARAP, GABARAPR14,15A, GABARAPK47A, GABARAPK46,47A, GABARAPK46,47,48A, GABARAPS16C, GABARAPS88C and 3 h at 37°C for GABARAPL2 and LC3B, LC3BGΔ, LC3BR10,11A, LC3BQ15C, LC3BS101C. Following centrifugation at 4,500 × g for 15 min, the pellet was resuspended and sonicated in breaking buffer (10 mM phosphate-buffered saline [PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4], 20 mM Tris-HCl, pH 7, 150 mM NaCl, supplemented with freshly prepared 1 mg/ml lysozyme [Sigma-Aldrich, L6876], 1 mM DTT [Sigma-Aldrich, D0632], bacterial protease inhibitors [Roche, 11697498001] and 2.5 μg/ml DNAase [Sigma-Aldrich, DN25]). After removal of cellular debris by centrifugation at 30,000 x g for 30 min at 4°C, the sample supernatant fraction was incubated with 1 ml glutathione Sepharose 4B (GE Healthcare, 17-0756-01) for 3 h at 4°C to bind glutathione S-transferase tagged proteins. Then PreScission Protease (GE Healthcare, 27-0843-01) was added at 100 units/ml in a 2-bed volume of PreScission Buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA) freshly prepared with 1 mM DTT and cleavage was performed for 4 h at 4°C. Cleaved protein was eluted and concentrated. Proteins were stored in 20% glycerol at −80°C.
Alexa Fluor 488 protein labeling
Purified proteins were first concentrated to 4 to 5 mg/ml (protein concentration should be at least 2 mg/ml for optimal results) and dialyzed against 0.1 M sodium bicarbonate (pH 8.2), 150 mM NaCl buffer to remove any amine-containing substances that would interfere with the conjugation reaction. Then 10 µl of the reactive dye solution (10 mg/ml of the Alexa Fluor® 488 dye [Molecular Probes, A20000] dissolved in dimethylsulfoxide [Sigma Aldrich, 472301] was slowly added to the protein solution. The reaction was incubated for 2 h at 37°C with continuous stirring. A Sephadex G-25 chromatography column (GE Healthcare, 17-0851-01) was used to separate the conjugate from unreacted labeling reagent with PreScission buffer used as the eluent. The degree of labeling was determined measuring the absorbance of the protein-dye conjugate at 280 nm and at 488 nm for the dye. Labeled samples were subjected to SDS-PAGE and visualized using a VersaDoc MP 4000 Imaging System (Bio-Rad laboratories, Hercules, CA, USA).
IANBD cysteine-substituted protein labeling
In a typical labeling reaction, cysteine-substituted proteins were first concentrated to 2–3 mg/ml and incubated with 1 mM TCEP (Sigma-Aldrich, C4706) for 30 min at room temperature. Then proteins were dialyzed against the labeling buffer (10 mM HEPES [Sigma-Aldrich, H3375], pH 7.3, 100 mM KCl, 1 mM EDTA) to remove TCEP. IANBD (Molecular Probes, I9) dissolved in dimethylsulfoxide was then added in a small volume (15 to 25 µl) to the protein solution to give a final protein:probe molar ratio of 1:10 (NBD stock solution concentration was 30 mM). The mixture was incubated overnight at 4°C with gentle stirring. Next the mixture was passed again through a PD-10 column (GE Healthcare, 17-0851-01) equilibrated with labeling buffer to separate the conjugate from unreacted labeling reagent. The stoichiometry of the NBD-protein conjugation reaction was calculated as the molar ratio of conjugated NBD and labeled proteins. The molar concentrations of the conjugated NBD and protein were determined from the absorbance at 488 nm (for NBD) and 280 nm (for proteins) using the molar extinction coefficients (eM) of 26500, 5960, 18450 and 11920 M−1cm−1 for NBD, LC3B, GABARAPL2 and GABARAP respectively.
Liposome preparation
The appropriate lipids were mixed in organic solution (chloroform:methanol [2:1 v/v] and the solvent was evaporated to dryness under a N2 stream. Then the sample was kept under vacuum for 2 h to remove solvent traces and the lipids were swollen in PreScission buffer. Large unilamellar vesicles were prepared from swollen lipids, subjected to 10 freeze/thaw cycles, and then extruded (LIPEX Liposome Extrusion System [Transferra Nanosciences, Burnaby, Canada]) using 0.1 µm pore size Nuclepore filters (Whatman, 110605) as described by Mayer et al.56 Small unilamellar vesicles were obtained by sonicating multilamellar vesicles (MLVs) with a probe tip sonicator (MSE Soniprep 150, MSE, UK) for 10 min (10 sec on, 10 sec off) on ice. Vesicle size was checked by quasi-elastic light scattering using a Malvern Zeta-Sizer 4 spectrometer (Malvern Instruments, Worcestershire, UK). LUVs had an average diameter of 100 nm and SUVs average diameter was 60 nm. Lipid concentration was determined by phosphate analysis.
Monolayer surface pressure measurements
Surface pressure experiments were carried out in a multi-well Delta Pi-4 Langmuir balance (Kibron Inc., Helsinki, Finland) under constant stirring. Protein-induced changes in surface pressure at the air-water interface and protein-lipid monolayer interactions were studied at either 25°C or 37°C. Monolayers were formed by spreading a small amount of the lipid mixtures in chloroform:methanol (2:1 v/v) solution on top of assay buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.2 mM DTT) until the desired initial surface pressure was reached. Proteins were injected with a micropipette through a hole connected to the subphase.
Protein-lipid overlay assay
Stock solutions of different lipids were solubilized in 2:1:0.8 MeOH:CHCl3:H2O, spotted onto Hybond C nitrocellulose (GE Healthcare, RPN2020D) and allowed to dry. To detect LC3B by immunoblotting, the nitrocellulose was first blocked with 5% fat-free milk in PBS for 1 h and further incubated for 1 h with LC3B in PreScission buffer at 37°C. The nitrocellulose was washed 4 times, 10 min each, with PBS, and soaked in 5% fat-free milk in PBS with an anti-LC3 monoclonal antibody at 1:1000 dilution overnight at 4°C. The nitrocellulose was washed 4 times with PBS and soaked in 5% fat free milk in PBS with horseradish peroxidase-conjugated anti-mouse antibody (Santa Cruz Biotechnology, sc-2031) at a 1:5000 dilution for 1 h at room temperature. After washing with PBS 4 × 10 min at room temperature, the protein was detected by chemiluminescence. Protein bands were digitalized and integrated densities were measured using Quantity One software (Bio-Rad laboratories, Hercules, CA, USA).
Sucrose gradient centrifugation of liposomes
Purified proteins as indicated (10 µM) were incubated with 3 mM liposomes (containing 0.05 mol percent Rho-PE for detection) for 1 h at 37°C in 200 µl PreScission buffer. 125 μl protein/lipid mix was diluted to 300 µl in PreScission buffer containing 2.4 M sucrose (Sigma-Aldrich, S0389). Then the reaction mix was transferred to a centrifuge tube. The 1.4 M sucrose layer was overlaid with 400 µl PreScission buffer containing 0.8 M sucrose and 300 µl of PreScission buffer containing 0.5 M sucrose. Sucrose step gradients were centrifuged in a TLA-120.2 rotor (Beckman Coulter, 357656, Brea, CA, US) at 356,160 × g for 3 h at 4°C. Four 250-µl fractions were pipetted, starting from the bottom. The top fraction (T), containing liposomes as indicated by the rhodamine fluorescence and the bottom fraction (B) containing the unbound protein were analyzed by SDS-PAGE and western blotting.
Fluorescence spectroscopy
NBD fluorescence measurements were performed in a QuantaMaster spectrofluorometer (Photon Technology International, Birmingham, NJ, US) in a thermostatically controlled 1-cm path length cuvette with constant stirring at 37°C. The excitation wavelength was 465 nm, the band-pass was typically 5 nm, and emission spectra of NBD were measured between 520 nm and 650 nm at a rate of 1 nm/sec. A 515 nm cut-off filter was placed between the sample and the emission monochromator to avoid scattering interference. In all cases, background intensities from samples lacking protein were substracted from the intensities measured in protein containing samples.
Cell culture
U87MG (human glioma cell line) cells were obtained from the American Type Culture Collection (Rockville, MD, USA; ATCC®, HTB-14™) and cultured in DMEM (Dulbecco's modified Eagle's medium) (Lonza, BE-12-604F) containing 10% fetal bovine serum (Linus, 91S1800) and penicillin/streptomycin (5 µg/ml) (Lonza, BE17-603E). When required, cells were seeded at a density of 5000 to 10000 cells/cm2 and transferred to medium containing 0.5% fetal bovine serum, 18 h before performing the different treatments. The latter included 1 μM rotenone (Sigma-Aldrich, R8875) or 4 μM THC (THC Pharm GmbH, THC-1099) for 4 or 6 h, respectively. For nutrient deprivation experiments, cells were incubated in Earle balanced salt solution (EBSS) medium (Lonza, BE10-502F).
Confocal laser scanning microscopy
Transfections of expression vectors GABARAPL2-GFP and GABARAP-GFP were performed with Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer's instructions.
Standard protocols for immunofluorescence microscopy were used. Briefly, cell cultures grown on 12 mm-coverslips (Menzel Glässer, P231.2) were washed in PBS, fixed with 4% paraformaldehyde (Sigma Aldrich, P6148) (20 min at room temperature) and permeabilized with 0.5% Triton X-100 (Sigma Aldrich, X100) (5 min at room temperature). Cells were then incubated with the appropriate primary antibodies (in case of LC3 the antibody recognizes the isoform B of the protein) diluted in PBS-0.1% bovine serum albumin (Sigma Aldrich, A6003) for 2 h and washed 3 times with this same buffer. Incubation with appropriate Alexa Fluor 488 or Alexa Fluor 594-conjugated secondary antibodies (Invitrogen, A11008, A11005) was performed in the dark at room temperature for 90 min. Cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (Roche, 010236276001) for 10 min at room temperature. Finally, coverslips were mounted in Mowiolmounting medium (Calbiochem, 475704) and observed in a Leica TCS SP2 confocal microscope (Leica Microsystems CMS GmbH, Mannheim, Germany).
Statistical analyses
Statistical analyses were performed by ANOVA with a post hoc analysis by the Student-Neuman-Keuls test.
Supplementary Material
Abbreviations
- Atg
autophagy-related
- CALCOCO2
calcium-binding and coiled-coil domain 2
- Cer16
n-palmitoyl-d-erythro-sphingosine
- Cer18
n-octadecanoyl-d-erythro-sphingosine
- Cer24:1
n-nervonoyl-d-erythro-sphingosine
- Chol
cholesterol
- CL
heart bovine cardiolipin
- DNM1L
dynamin 1 like
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DTT
dl-dithiothreitol
- EBSS
Earle's balanced salt solution
- GABARAP
GABA type A receptor-associated protein
- GABARAPL2
GABA type A receptor-associated protein like 2
- GFP
green fluorescent protein
- LIR
LC3-interacting region
- LUV
large unilamellar vesicle
- MAP1LC3A/LC3A
microtubule-associated protein 1 light chain 3 α
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- MAP1LC3C/LC3C
microtubule-associated protein 1 light chain 3 γ
- NBD
7-nitrobenz-2-oxa-1,3-diazol-4-yl
- OMM
outer mitochondrial membrane
- PBS
phosphate buffered saline
- PC
phosphatidylcholine
- PtdIns4P
phosphatidylinositol 4-phosphate
- PINK1
PTEN induced putative kinase 1
- PS
phosphatidylserine
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SUV
small unilamellar vesicle
- THC
Δ9-tetrahydrocannabinol
- TOMM20
translocase of outer mitochondrial membrane 20
- U87MG
human primary glioblastoma cell line
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Isei Tanida (National Institute of Infectious Diseases, Tokyo, Japan) for providing human LC3B, GABARAPL2 and GABARAP plasmids and Dr. Anne Simonsen (Institute of Basic Medical Sciences, Oslo) for providing GABARAPL2-GFP and GABARAP-GFP plasmids. The authors thank Ms Araceli Marcos for technical support.
Funding
This work was supported in part by grants from FEDER/Spanish Ministry of Economy (BFU 2011-28566, BFU 2015-66306-P) and the Basque Government (IT838-13, IT849-13). J.H.H. and Z.A. were predoctoral students supported by the University of the Basque Country. Work in G.V. group was supported by grants from Spanish Ministry of Economy (MINECO)/Acción estratégica en Salud from Instituto de Salud Carlos III and Fondo Europeo de Desarrollo Regional (FEDER) (PI12/02248 and PI15/00339) and Fundació La Marató de TV3 (to G.V.).
References
- [1].Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell 2015; 147:728-41; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- [2].Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res 2014; 24:58-68; PMID:24296784; http://dx.doi.org/ 10.1038/cr.2013.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: More than a nonselective pathway. Int J Cell Biol 2012; 2012:219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/ 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ashrafi G, Schwarz TL The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differentiation 2013; 20(1):31-42; PMID:22743996; http://dx.doi.org/17166847 10.1038/cdd.2012.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem 2007; 282:5617-24; PMID:17166847; http://dx.doi.org/ 10.1074/jbc.M605940200 [DOI] [PubMed] [Google Scholar]
- [7].Kiššová I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem 2004; 279:39068-74; http://dx.doi.org/ 10.1074/jbc.M406960200 [DOI] [PubMed] [Google Scholar]
- [8].Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al.. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 2007; 104:19500-5; PMID:18048346; http://dx.doi.org/ 10.1073/pnas.0708818104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/ 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- [10].Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: The yin and yang of cell death control. Circulation Res 2012; 111:1208-21; PMID:23065344; http://dx.doi.org/ 10.1161/CIRCRESAHA.112.265819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lu H, Li G, Liu L, Feng L, Wang X, Jin H. Regulation and function of mitophagy in development and cancer. Autophagy 2013; 9:1720-36; PMID:24091872; http://dx.doi.org/ 10.4161/auto.26550 [DOI] [PubMed] [Google Scholar]
- [12].Kanki T, Furukawa K, Yamashita S. Mitophagy in yeast: Molecular mechanisms and physiological role. Biochim Biophys Acta - Mol Cell Res 2015; 1853(10 Pt B):2756-65 [DOI] [PubMed] [Google Scholar]
- [13].Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem 2008; 283:32386-93; PMID:18818209; http://dx.doi.org/ 10.1074/jbc.M802403200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012; 8:1462-76; PMID:22889933; http://dx.doi.org/ 10.4161/auto.21211 [DOI] [PubMed] [Google Scholar]
- [15].Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int 2014; 2014:1-7; http://dx.doi.org// 10.1155/2014/238463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cherra SJ, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 2013; 182:474-84; PMID:23231918; http://dx.doi.org/ 10.1016/j.ajpath.2012.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, et al.. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013; 15:1197-205; PMID:24036476; http://dx.doi.org/ 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol 2011; 192:7-16; PMID:21220505; http://dx.doi.org/ 10.1083/jcb.201006159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ren M, Phoon CKL, Schlame M. Metabolism and function of mitochondrial cardiolipin. Progress Lipid Res 2014; 55:1-16; PMID:24769127; http://dx.doi.org/ 10.1016/j.plipres.2014.04.001 [DOI] [PubMed] [Google Scholar]
- [20].Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis 2007; 12:877-85; PMID:17294083; http://dx.doi.org/ 10.1007/s10495-007-0718-8 [DOI] [PubMed] [Google Scholar]
- [21].Marsh D. Lateral pressure in membranes. Biochim Biophys Acta - Rev Biomembr 1996; 1286:183-223; http://dx.doi.org/ 10.1016/S0304-4157(96)00009-3 [DOI] [PubMed] [Google Scholar]
- [22].Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, et al.. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol 2012; 8:831-8; PMID:22922758; http://dx.doi.org/ 10.1038/nchembio.1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Antonny B. Mechanisms of membrane curvature sensing. Annu Rev Biochem 2011; 80:101-23; PMID:21438688; http://dx.doi.org/ 10.1146/annurev-biochem-052809-155121 [DOI] [PubMed] [Google Scholar]
- [24].Fan W, Nassiri A, Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci U. S. A 2011; 108:7769-74; PMID:21518905; http://dx.doi.org/ 10.1073/pnas.1016472108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B, Melia TJ. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol 2014; 16:415-24; PMID:24747438; http://dx.doi.org/ 10.1038/ncb2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kates M, Syz JY, Gosser D, Haines TH. pH-dissociation characteristics of cardiolipin and its 2′-deoxy analogue. Lipids 1993; 28:877-82; PMID:8246687; http://dx.doi.org/ 10.1007/BF02537494 [DOI] [PubMed] [Google Scholar]
- [27].Lewis RNAH, McElhaney RN. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim Et Biophys Acta - Biomembranes 2009; 1788:2069-79; http://dx.doi.org/ 10.1016/j.bbamem.2009.03.014 [DOI] [PubMed] [Google Scholar]
- [28].Salazar M, Carracedo A, Salanueva ÍJ, Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C, Torres S, García S, et al.. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 2009; 119:1359-72; PMID:19425170; http://dx.doi.org/ 10.1172/JCI37948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011; 80:125-56; PMID:21548784; http://dx.doi.org/ 10.1146/annurev-biochem-052709-094552 [DOI] [PubMed] [Google Scholar]
- [30].Schreiber A, Peter M. Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochim Et Biophys Acta - Mol Cell Res 2014; 1843:163-81; http://dx.doi.org/ 10.1016/j.bbamcr.2013.03.019 [DOI] [PubMed] [Google Scholar]
- [31].Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: Sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 2012; 287:39275-90; PMID:23043107; http://dx.doi.org/ 10.1074/jbc.M112.378109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end - Directed vesicle transport. J Cell Biol 2010; 188:253-69; PMID:20100911; http://dx.doi.org/ 10.1083/jcb.200907015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al.. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505-16; PMID:19250911; http://dx.doi.org/ 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- [34].Sugawara K, Suzuki NN, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J Biol Chem 2005; 280:40058-65; PMID:16183633; http://dx.doi.org/ 10.1074/jbc.M509158200 [DOI] [PubMed] [Google Scholar]
- [35].Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al.. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333:228-33; PMID:21617041; http://dx.doi.org/ 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein Á, Bloor S, Rutherford TJ, Freund SM, Komander D, Randow F. LC3C, bound selectively by a Noncanonical LIR Motif in NDP52, is required for antibacterial autophagy. Mol Cell 2012; 48:329-42; PMID:23022382; http://dx.doi.org/ 10.1016/j.molcel.2012.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, et al.. VHL-Regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012; 21:532-46; PMID:22516261; http://dx.doi.org/ 10.1016/j.ccr.2012.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-Anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87-97; PMID:19619494; http://dx.doi.org/ 10.1016/j.devcel.2009.06.013 [DOI] [PubMed] [Google Scholar]
- [39].Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/ 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- [40].Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al.. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45-51; PMID:20010802; http://dx.doi.org/ 10.1038/embor.2009.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al.. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 2010; 107:378-83; PMID:19966284; http://dx.doi.org/ 10.1073/pnas.0911187107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al.. Hereditary early-onset Parkinson disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/ 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- [43].Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; PMID:20126261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basañez G, Terrones O, Martinou JC. Specific interaction with cardiolipin triggers functional activation of Dynamin-Related Protein 1. PLoS One 2014; 9:e102738; PMID:25036098; http://dx.doi.org/ 10.1371/journal.pone.0102738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, et al.. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 2010; 12:1154-65; PMID:21102437; http://dx.doi.org/ 10.1038/ncb2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M, Liu M, Tian Y, Lu P, Wang FL, et al.. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res 2012; 22:473-89; PMID:22310240; http://dx.doi.org/ 10.1038/cr.2012.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Landajuela A, Hervás JH, Antón Z, Montes LR, Gil D, Valle M, Rodriguez JF, Goñi FM, Alonso A. Lipid geometry and bilayer curvature modulate LC3/GABARAP-mediated model autophagosomal elongation. Biophys J 2016; 110(2):411-22; PMID:26789764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen JJ, Yu BP. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic Biol Med 1994; 17:411-8; PMID:7835747; http://dx.doi.org/ 10.1016/0891-5849(94)90167-8 [DOI] [PubMed] [Google Scholar]
- [49].Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al.. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433-46; PMID:18200046; http://dx.doi.org/ 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Goñi FM. The basic structure and dynamics of cell membranes: an update of the Singer-Nicolson model. Biochim Biophys Acta 2014; 1838:1467-76 [DOI] [PubMed] [Google Scholar]
- [51].Lemmin T, Bovigny C, Lançon D, Dal Peraro M. Cardiolipin models for molecular simulations of bacterial and mitochondrial membranes. J Chem Theory Comput 2013; 9:670-8; PMID:26589063; http://dx.doi.org/ 10.1021/ct300590v [DOI] [PubMed] [Google Scholar]
- [52].Khalifat N, Fournier JB, Angelova MI, Puff N. Lipid packing variations induced by pH in cardiolipin-containing bilayers: The driving force for the cristae-like shape instability. Biochim Biophys Acta - Biomembr 2011; 1808:2724-33; http://dx.doi.org/ 10.1016/j.bbamem.2011.07.013 [DOI] [PubMed] [Google Scholar]
- [53].Khalifat N, Puff N, Bonneau S, Fournier JB, Angelova MI. Membrane deformation under local pH gradient: mimicking mitochondrial cristae dynamics. Biophys J 2008; 95:4924-33; PMID:18689447; http://dx.doi.org/ 10.1529/biophysj.108.136077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kiššova I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 2007; 3:329-36; http://dx.doi.org/ 10.4161/auto.4034 [DOI] [PubMed] [Google Scholar]
- [55].Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792-802; PMID:20418806; http://dx.doi.org/ 10.1038/emboj.2010.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mayer LD, Hope MJ, Cullis PR. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta 1986; 858:161-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.