

Abstract:
Coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide (E/C/F/TAF) has high efficacy and improved renal and bone safety in multiple phase 3 trials; TAF single agent is being studied in 2 phase 3 trials in patients with chronic hepatitis B. We report the results of an open-label, noncomparative switch study evaluating the efficacy and safety of E/C/F/TAF in HIV/hepatitis B virus (HBV)–coinfected adults. At 48 weeks, 91.7% of the 72 participants maintained or achieved virologic suppression (HIV-1 RNA <50 copies/mL; HBV DNA <29 IU/mL). Seroconversion occurred in 2.9% of hepatitis B surface antigen–positive participants and in 3.3% of HBV e antigen–positive participants; 40% of those with abnormal alanine aminotransferase normalized. E/C/F/TAF was associated with improved renal function and reduced bone turnover. These data support the use of E/C/F/TAF in treating HIV/HBV coinfection.
Key Words: tenofovir alafenamide, TAF, elvitegravir, HIV, HBV, coinfection, bone, renal
INTRODUCTION
Tenofovir disoproxil fumarate (TDF)–based regimens are recommended for treatment of HIV/hepatitis B virus (HBV) coinfection by all major treatment guidelines.1–3 Although TDF is potent, well tolerated, and the cornerstone of effective HIV/HBV treatment, it is associated with greater nephrotoxicity4–6 and bone mineral density loss7–9 than other nucleoside/nucleotide reverse transcriptase inhibitors. TDF is a prodrug that is metabolized to tenofovir (TFV), which is diphosphorylated intracellularly to its active metabolite, TFV diphosphate (TFV-DP). Higher circulating plasma TFV levels have been associated with the renal and bone effects of TDF.10 Tenofovir alafenamide (TAF) is a novel prodrug of TFV that is more stable in plasma, allowing for a 10-fold reduction in dose and resulting in a 91% reduction in plasma TFV and a 4-fold increase in intracellular levels of TFV-DP.11
A single tablet coformulation of elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide (E/C/F/TAF) demonstrated high efficacy and improved renal and bone safety in multiple phase 3 trials that have included treatment-naive participants, participants switching from TDF-based regimens, participants with preexisting kidney disease, and adolescents.11–14 It is approved by the US Food and Drug Administration (FDA) and European Medicines Agency for treatment of naive and stably suppressed patients aged 12 years and older and is a recommended initial regimen in the US Department of Health and Human Services guidelines.1 TAF is being studied for the treatment of patients with chronic hepatitis B. In a phase 1b proof-of-concept study, it demonstrated potent anti-HBV activity.15 In vitro and in dogs, TAF had high intrahepatic extraction and enhanced levels of TFV-DP compared to TDF.16 TAF 25 mg compared with TDF 300 mg has been studied in 2 global phase 3 HBV-monoinfected studies—one in treatment-naive or -experienced HBV e antigen (HBeAg)–negative participants (GS-US-320-0108) and one in monoinfected HBeAg-positive participants (GS-US-320-0110). Week 48 results were recently reported.17 Based on these data, TAF has been submitted for regulatory approval for the treatment of HBV in the United States and in the European Union.
This phase 3b, open-label study, GS-US-292-1249 (ClinicalTrials.gov number NCT02071082), conducted in the United States, Canada, and Japan, is the first to evaluate the efficacy and safety of switching to E/C/F/TAF in HIV/HBV-coinfected adults which is currently an unlabeled indication.
METHODS
Study Design and Participants
HIV/HBV-coinfected adults receiving a stable antiretroviral regimen for ≥4 months were eligible. They must have maintained plasma HIV-1 RNA viral load (VL) <50 copies/mL for 6 months before screening and have HBV DNA <9 log10 IU/mL; HBV DNA suppression was not required. Individuals who were currently taking or had previously received regimens concurrently containing 3 active anti-HBV agents (eg, TDF, emtricitabine, and entecavir) were not eligible. Chronic hepatitis B was defined as (1) hepatitis B surface antigen (HBsAg) positive for ≥6 months, (2) screening HBsAg positive and HBeAg or HBV DNA positive for ≥6 months, or (3) positive anti–hepatitis B core (anti-HBc) IgG and negative anti-HBc IgM and positive HBsAg, HBeAg, and/or HBV DNA. All were required to have CD4 count >200 cells per microliter; alanine aminotransferase (ALT) ≤10 × upper limit of normal (ULN); total bilirubin ≤2.5 mg/dL; international normalized ratio ≤1.5; albumin ≥3 g/dL; creatinine clearance by Cockcroft–Gault (CrClCG) ≥50 mL/min; and no cirrhosis, hepatocellular carcinoma, or hepatitis C or D virus infection.
The study was conducted in accordance with the Declaration of Helsinki. The protocol was reviewed and approved by central or site-specific institutional review boards or ethics committees. Participants gave written informed consent.
Procedures
Participants were seen at screening, baseline, and at weeks 2, 4, 8, 12, 24, 36, and 48. Laboratory tests included hematological analysis, chemistry tests including liver function tests, fasting lipid parameters, CD4 counts, measures of renal function [CrClCG, urine protein/creatinine (Cr), urine albumin/Cr, retinol binding protein/Cr, β-2-microglobulin (β-2M)/Cr], HBV DNA (Covance Laboratories, Indianapolis, IN), and HIV RNA (Roche TaqMan2.0; Roche Diagnostics, Rotkreuz, Switzerland). Participants with confirmed virologic failure (2 consecutive VL samples >50 copies/mL) and a VL >400 copies/mL at week 8 or later had a second, confirmatory sample sent for resistance analysis by GeneSeq Integrase, PhenoSense GT, and PhenoSense Integrase (Monogram Biosciences, South San Francisco, CA).
HBV resistance surveillance was conducted at baseline by INNO-LiPA Multi-DR v2/v3 (Innogenetics, Ghent, Belgium) in patients with HBV DNA ≥69 IU/mL, which determined the presence of HBV polymerase/reverse transcriptase mutations known to confer resistance to adefovir, lamivudine (3TC), entecavir, clevudine, and/or telbivudine. Population sequencing analysis of HBV polymerase/reverse transcriptase was attempted for all viremic patients (HBV DNA ≥69 IU/mL) at week 48 (or last visit at/after week 24 for patients who discontinued early) and for those with virologic breakthrough defined as either 2 consecutive HBV DNA values ≥69 IU/mL after a result <69 IU/mL or a confirmed ≥1.0 log10 increase in HBV DNA from nadir for those who did not achieve a result <69 IU/mL.
Statistical Analysis
Primary efficacy endpoints were the percentage at week 24 with (1) VL <50 copies/mL (FDA snapshot algorithm) and (2) HBV DNA <29 IU/mL [Missing Failure (M = F)]. Secondary endpoints included the percentage of participants with HIV VL <50 copies/mL at week 48 (FDA snapshot algorithm), with HBV DNA <29 IU/mL at week 48, with HBsAg and HBeAg loss at weeks 24 and 48, with HBsAg-to-HBV surface antibody seroconversion and HBeAg-to-HBV e antibody seroconversion at weeks 24 and 48, with ALT normalization (of those with baseline abnormal ALT), and change from baseline in FibroTest score (using METAVIR18 fibrosis staging classification) at weeks 24 and 48. Participants taking atazanavir at baseline (n = 8) were excluded from analyses of FibroTest scores because the calculation includes serum bilirubin concentration. Safety assessments, including assessment of ALT flare (confirmed serum ALT >2 × baseline value and >10 × ULN, with or without associated symptoms), were summarized using descriptive statistics for the safety analysis set, which included all who received at least 1 dose of study drug. Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities, Version 18.0. P-values reported are from a Wilcoxon signed-rank test.
RESULTS
Of the 100 adults screened, 74 received at least 1 dose of E/C/F/TAF (safety analysis set); 2 who enrolled and took study drug but had no laboratory evidence of chronic HBV were excluded from the efficacy analysis set (n = 72). Baseline characteristics are shown in Table 1. Participants were predominantly male (92%), with median CD4 of 605 cells per microliter, virologically suppressed on ART, with a median duration of HIV infection of 18 years. About 86% had ALT in the normal range, but 60% had moderate to severe fibrosis by FibroTest (METAVIR scores F1–F2, F2, F3, and F4). Almost all (96%) were receiving TDF (and 3TC or FTC)-based regimens before switching. One was receiving an ARV regimen that included only 3TC as an anti-HBV agent, and 2 were receiving regimens with no anti-HBV activity. Seven were evaluated for baseline HBV DNA resistance by INNO-LiPA (HBV DNA ≥ 69 IU/mL): 2 wild type, 4 with 3TC resistance, and 1 with a 3TC compensatory mutation. Through week 48, 6 discontinued study drug: AE (n = 1), lack of HIV efficacy (n = 1), lost to follow-up (n = 1), and withdrawal of consent (n = 3).
TABLE 1.
Baseline Demographic and Disease Characteristics

HIV Efficacy
E/C/F/TAF maintained a high rate of virologic success (VL <50 copies/mL by FDA snapshot algorithm) at week 24 [94.4%; 95% confidence interval (CI): 86.4% to 98.5%] and at week 48 [91.7%; 95% CI: 82.7% to 96.9%] (Fig. 1A). Two met criteria for HIV virologic failure but had VLs <400 copies/mL, below the VL required for resistance testing.
FIGURE 1.
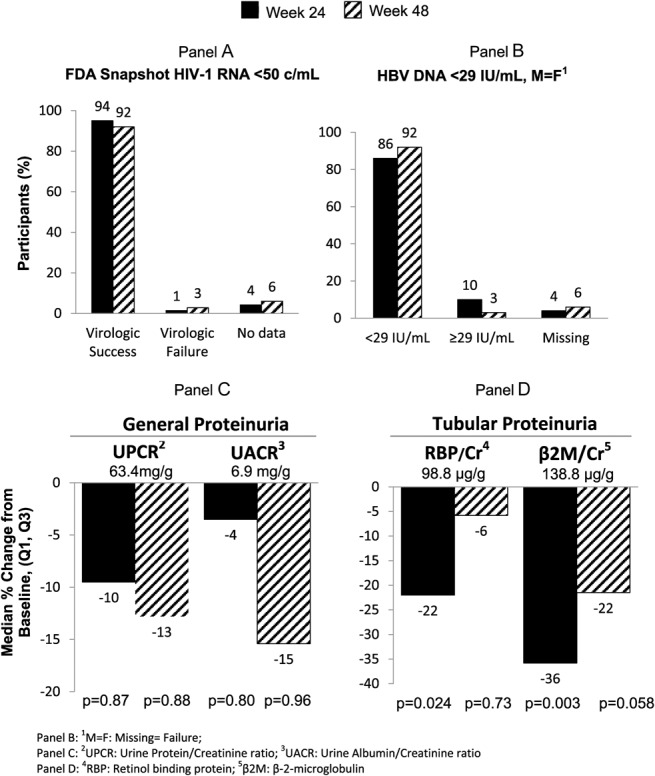
Virologic results and proteinuria at weeks 24 and 48.
HBV Efficacy
The percentage of participants with HBV DNA <29 IU/mL (M = F) was 86.1% at baseline, 86.1% (95% CI: 75.9% to 93.1%) at week 24, and 91.7% (95% CI: 82.7% to 96.9%) at week 48 (Fig. 1B). Of the 10 with baseline HBV DNA >29 IU/mL, 7 were undetectable by week 48, 2 had declining HBV DNA, and 1 withdrew consent. No individual with baseline undetectable HBV DNA at baseline became detectable at week 48; 3 were missing data at week 48—1 discontinued because of AE, 1 withdrew consent, and 1 had missing data at week 48 but was undetectable at weeks 36 and 60. Two qualified for HBV resistance analyses: 1 with baseline 3TC resistance who had HBV DNA of 3580 IU/mL at baseline and experienced a 0.5 log drop by week 48 to 170 IU/mL but was still >69 IU/mL, and 1 with baseline 3TC resistance who discontinued at week 36 with HBV DNA ≥69 IU/mL. No mutations associated with resistance to tenofovir were observed.
Serologic Response: HBsAg and HBeAg Loss and Seroconversion
Of the 70 participants who were HBsAg positive and HBsAb negative at baseline, 1 (1.4%) experienced HBsAg-to-HBsAb seroconversion at week 24, 2 (2.9%) lost HBsAg with 1 also gaining HBsAb at week 48. Of the 30 who were HBeAg positive and HBeAb negative at baseline, 1 (3.3%) experienced HBeAg-to-HBeAb seroconversion at week 24 and 1 (3.3%) experienced HBeAg loss without gaining HBeAb at week 48.
Biochemical Response: ALT Normalization
Of the 10 participants (13.9%) with ALT values >ULN at baseline, 5 (50.0%) achieved ALT normalization at week 24 and 4 (40.0%) achieved ALT normalization at week 48. Of the 62 (86.1%) with normal ALT values at baseline, 54 (87.1%) and 57 (91.9%) remained normal at weeks 24 and 48, respectively. There were no ALT flares. Two (2.7%) had elevated AST or ALT levels >3 × ULN during this study; 1 (1.4%) had AST and ALT levels >5 × ULN (during acute HCV infection).
Hepatic Fibrosis Response
Median (Q1, Q3) FibroTest scores were 0.35 (0.21, 0.51) at baseline; the change from baseline was −0.02 (−0.11, 0.05; P = 0.064) at week 24 and −0.04 (−0.11, 0.03; P = 0.018) at week 48. Of the 60 with paired baseline and week 48 data, 9 improved, 45 had no change, and 6 worsened in fibrosis stage.
General Safety
The most frequently reported study drug–related AEs were diarrhea (4.1%) and increased appetite (2.7%). One participant (1.4%) discontinued E/C/F/TAF because of AEs of increased weight and appetite. Serious AEs were infrequent (8.1%); none were reported as study drug related: acute myocardial infarction; appendicitis; benign prostatic hyperplasia and prostatitis; diabetes mellitus and limb abscess; pneumonia; pneumococcal bacteremia, meningitis, and pneumonia (n = 1 each). There were no deaths. There were small increases in total cholesterol, direct LDL cholesterol (both P < 0.001), and HDL cholesterol (P = 0.054) but no changes in the total cholesterol-to-HDL ratio (P = 0.12).
Renal and Bone Outcomes
Renal and bone outcomes were consistent with those seen in other TAF studies. Participants switching from TDF-based regimens experienced improvements in CrClCG. There was no proximal tubulopathy or drug discontinuation because of renal AEs. There were declines in markers of proximal tubular proteinuria (retinol binding protein/Cr and β-2M/Cr) (Fig. 1) and in clinically significant proteinuria (UPCR ≥ 200 mg/g) and albuminuria (UACR ≥ 30 mg/g).
Statistically significant declines in markers of bone turnover (serum CTX and P1NP) were observed. One participant had a traumatic calcaneus fracture classified as not study drug related by the investigator.
DISCUSSION
This is the first study to evaluate the efficacy and safety of switching to E/C/F/TAF in HIV/HBV-coinfected adults. One year after switching from predominantly TDF-based regimens to E/C/F/TAF, participants maintained high rates of HIV and HBV suppression, had improved renal function, and reduced biomarkers of bone turnover, consistent with other E/C/F/TAF studies.11–14 E/C/F/TAF was well tolerated with no discontinuations because of renal events. Seroconversion occurred in 2.9% of HBsAg-positive participants and 3.3% of HBeAg-positive participants; 40% of those with abnormal ALT normalized by week 48; which is lower than the percentage seen in naive HBV-monoinfected populations and similar to treatment-experienced coinfected populations.19–21 There were no ALT flares, and assessments of other liver-related parameters did not suggest increased hepatic risk.
This study was open-label, with a small sample size and no comparator group. Most participants had suppressed HBV infection at baseline. Despite these limitations, this study provides the first comprehensive assessment of the efficacy of E/C/F/TAF against both HIV and HBV and a detailed examination of both liver and renal endpoints relevant to the safety of TAF-based regimens in HIV/HBV-coinfected adults.
In this first study in HIV/HBV-coinfected participants with suppressed HIV infection, E/C/F/TAF was effective against HIV and HBV, well tolerated, and demonstrated improvements in renal and bone safety consistent with the clinical profile of TAF. These data support the use of E/C/F/TAF in treating HIV/HBV coinfections.
ACKNOWLEDGMENTS
We thank the participants, their partners, and families and all Principal Investigators and their Study Staff.
GS US 292–1249 Study Investigators: P.B. (Be Well Medical Center), L. Bhatti (AHF Research Center), C.B. (Central TX Clinical Research), J.B. (Maple Leaf Research), G.C. (Crofoot Research Center), C. Dietz (Kansas City Free Clinic), E. Elion (Whitman Walker Clinic), J.G. (Southwest CARE Center), J. Gathe (Therapeutic Concepts), Anthony Mills, MD, S.O. (National Center for Global Health and Barts & The London NHS Trust), O. Osiyemi (Triple O Medical Services, Inc.), G. Pierone (Treasure Coast Infectious Disease Consultants), D. Prelutsky (Southampton Healthcare), M. Ramgopal (Midway Immunology & Research Center), Gary Richmond, MD, PA, Barry M. Rodwick, MD, Peter J. Ruane, MD, Peter Shalit, MD, L. Sloan (North TX Infectious Diseases Consultants), T. Vanig (Spectrum Medical Group), S. Walmsley (University Health Network), and M. Wohlfeiler (AIDS Healthcare Foundation).
Footnotes
Supported by Gilead Sciences, Inc.
The 48-week results have been presented at the 8th IAS Conference on HIV Pathogenesis, Treatment and Prevention, July 19–22, 2015, Vancouver, BC, Canada.
J.G. reports receiving consulting/advisory fees from Bristol Myers Squibb (BMS), Gilead, Janssen Therapeutics, Merck, ViiV/GlaskoSmithKilne (GSK) and institutional grant support from AbbVie, BMS, Gilead, Janssen, Merck, Sangamo BioSciences, and GSK/ViiV outside the submitted work. J.B. has received study-related reimbursement from Gilead during the conduct of the study; conference sponsorship from Gilead Canada, Merck Canada, ViiV Canada, and Janssen Canada; speaker fees from Gilead Canada and Merck Canada; and consultant fees from Gilead Canada, Merck Canada, ViiV Canada, and Abbvie Canada outside the submitted work. G.C. reports grants from Gilead during the conduct of the study; grants and personal fees from Gilead, ViiV, Janssen, and Merck outside of the submitted work; and personal fees from Gilead and ViiV outside the submitted work. P.B. reports receiving other fees (Speakers Bureau, study-related reimbursement, advisory boards) from Gilead outside the submitted work. C.B. has received grants Gilead, Theratech, BMS, SlieaGen, ViiV/GSK, Daiichi Sankyo, Vertex, Novo Nordisk, Sanofi, Shionogi; advisory board fees from Gilead, Theratech, VMS, and ViiV/GSK; speaker fees from Gilead; and consultant fees from ViiV/GSK during the conduct of the study; and grants from Theratech, BMS, SlieaGen, ViiV/GSK, Daiichi Sankyo, Vertex, Novo Nordisk, Sanofi, Shionogi, Elcelyx; advisory board fees from Theratch and BMS; and speaker fees from Theratch outside the submitted work. S.O. has received research grants from Merck Sharp & Dohme (MSD) and honoraria from MSD, ViiV, Trii Pharmaceuticals, AbbVie, Japan Tobacco, and Janssen outside the submitted work. A.C., W.G., M.F., M.D., and S.M. are employees of Gilead and hold stock interest in the company. J.G., J.B., G.C., P.B., A.M., C.B. were principal investigators and A.C., W.G., M.F., M.D., and S.M. were employees of Gilead Sciences and were the scientific, medical, and operational leaders responsible for this study's design, conduct, oversight, and analyses. All authors have reviewed the results of this study and approved the manuscript.
REFERENCES
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-infected Adults and Adolescents; 2015:1–166. Available at: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed January 23, 2016. [Google Scholar]
- 2.Huldrych F, Gunthard HF, Aberg JA, et al. Antiretroviral treatment of adult HIV infection: 2014 recommendations of the International Antiviral Society-USA Panel. JAMA. 2014;312:410–425. [DOI] [PubMed] [Google Scholar]
- 3.(EACS) EACS. EACS Guideline Version 8.0; 2015. European AIDS Clinical Society. Available at http://www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. Accessed January 23, 2016. [Google Scholar]
- 4.Hall AM, Hendry BM, Nitsch D, et al. Tenofovir-associated kidney toxicity in HIV-infected patients: a review of the evidence. Am J Kidney Dis. 2011;57:773–780. [DOI] [PubMed] [Google Scholar]
- 5.Gupta SK. Tenofovir-associated Fanconi syndrome: review of the FDA adverse event reporting system. AIDS Patient Care and STDS. 2008;22:99–103. [DOI] [PubMed] [Google Scholar]
- 6.Post FA, Moyle GJ, Stellbrink HJ, et al. Randomized comparison of renal effects, efficacy, and safety with once-daily abacavir/lamivudine versus tenofovir/emtricitabine, administered with efavirenz, in antiretroviral-naive, hiv-1-infected adults: 48-week results from the ASSERT Study. J Acquir Immune Defic Syndr. 2010;55:49–57. [DOI] [PubMed] [Google Scholar]
- 7.Stellbrink HJ, Orkin C, Arribas JR, et al. Comparison of changes in bone density and turnover with abacavir-lamivudine versus tenofovir-emtricitabine in HIV-infected adults: 48-week results from the ASSERT study. Clin Infect Dis. 2010;51:963–972. [DOI] [PubMed] [Google Scholar]
- 8.Schafer JJ, Manlangit K, Squires KE. Bone health and human immunodeficiency virus infection. Pharmacotherapy. 2013;33:665–682. [DOI] [PubMed] [Google Scholar]
- 9.McComsey GA, Kitch D, Daar ES, et al. Bone mineral density and fractures in antiretroviral-naive persons randomized to receive abacavir-lamivudine or tenofovir disoproxil fumarate-emtricitabine along with efavirenz or atazanavir-ritonavir: Aids Clinical Trials Group A5224s, a substudy of ACTG A5202. J Infect Dis. 2011;203:1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Rompay KKA, Durand-Gasselin L, Brignolo LL, et al. Chronic administration of tenofovir to rhesus macaques from infancy through adulthood and pregnancy: summary of pharmacokinetics and biological and virological effects. Antimicrob Agents Chemother. 2008;52:3144–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. 2015;385:2606–2615. [DOI] [PubMed] [Google Scholar]
- 12.Mills A, Arribas JR, Andrade-Villanueva J, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in antiretroviral regimens for virologically suppressed adults with HIV-1 infection: a randomised, active-controlled, multicentre, open-label, phase 3, non-inferiority study. Lancet. 2015;16:43–52. [DOI] [PubMed] [Google Scholar]
- 13.Pozniak A, Arribas JR, Gathe J, et al. Switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected patients with renal impairment: 48 week results from a single-arm, multi-center, open-label, Phase 3 study. J Acquir Immune Defic Syndr. 2015;71:530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaur A, Fourie J, Chokephaibulkit K, et al. Pharmacokinetics, efficacy and safety of an integrase inhibitor-based single-tablet regimen in HIV-infected treatment-naive adolescents [Poster 909]. Paper presented at: 21st Conference on Retroviruses and Opportunistic Infections; March 3–6, 2014; Boston, MA.
- 15.Agarwal K, Fung SK, Nguyen TT, et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol. 2015;62:533–540. [DOI] [PubMed] [Google Scholar]
- 16.Murakami E, Wang T, Park Y, et al. Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis B virus therapy. Antimicrob Agents Chemother. 2015;59:3563–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilead Sciences Inc., Foster City, CA. Gilead Submits New Drug Application to U.S. Food and Drug Administration for Tenofovir Alafenamide (TAF) for the Treatment of Chronic Hepatitis B. Foster City, CA; 2016. [Google Scholar]
- 18.METAVIR Cooperative Study Group. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology. 1994;20(1 pt 1):15–20. [PubMed] [Google Scholar]
- 19.Fung S, Kwan P, Fabri M, et al. Randomized comparison of tenofovir disoproxil fumarate vs emtricitabine and tenofovir disoproxil fumarate in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology. 2014;146:980–988. [DOI] [PubMed] [Google Scholar]
- 20.Marcellin P, Heathcote EJ, Buti M, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359:2442–2455. [DOI] [PubMed] [Google Scholar]
- 21.Martin-Carbonero L, Teixeira T, Poveda E, et al. Clinical and virological outcomes in HIV-infected patients with chronic hepatitis B on long-term nucleos(t)ide analogues. AIDS. 2011;25:73–79. [DOI] [PubMed] [Google Scholar]