

Abstract
The contribution of B cells to immunity against many infectious diseases is unquestionably important and well characterized. Here we sought to determine the role of B cells in the induction of T helper 1 (TH1) CD4+ T cells upon vaccination with a TB antigen combined with a TLR4 agonist. We used B cell deficient mice (μMT-/-), tetramer-positive CD4+ T cells, markers of memory ‘precursor’ effector cells (MPEC), and T cell adoptive transfers and demonstrated that the early antigen-specific cytokine-producing TH1 responses are unaffected in the absence of B cells, however MPEC induction is strongly impaired resulting in a deficiency of the memory TH1 response in μMT-/- mice. We further show that antigen-presentation by B cells was necessary for their role in MPEC generation using B cell adoptive transfers from wild type or MHC class II knock-out mice into μMT-/- mice. Our study challenges the view that B cell deficiency exclusively alters the TH1 response at memory time-points. Collectively, our results provide new insightson the multifaceted roles of B cells which will have a high impact on vaccine development against several pathogens including those requiring TH1 cell-mediated immunity.
Introduction
Since the primary objective and hallmark of vaccine design is to generate memory immune responses, a better understanding of the machinery that results in a robust memory response is crucial. Several sequential processes are required for the development of antigen-specific central memory T cell (Tcm) production upon protein-antigen immunization. Primary responses depend on additional components in the vaccine formulation, usually in the form of adjuvants. Adjuvants containing Toll like receptor (TLR) agonists, such as the clinically tested synthetic TLR-4 agonist Glucopyranosyl Lipid Adjuvant (GLA), help promote and influence the fate of a desirable T response through enhanced antigen presentation on dendritic cells (DCs), DC maturation, and production of innate cytokines [1]. Secondary responses require expansion and subsequent contraction of T cells, leaving behind a small percentage of memory T cells that retain proliferative capabilities and are available for future encounters with the specific pathogen. Short lived effector T cells are instead terminally differentiated but provide effector helper functions such as cytokine production or cytotoxic functions that contribute to enhanced magnitude and quality of immunity against subsequent infection (for a review see [2]). It is thought that the long-term fate of antigen experienced T cells can be predicted based on expression of different surface markers and transcription factors. Using expression of two inhibitory surface molecules Woodland and colleagues proposed that memory precursor effector cell (MPEC) could be distinguished as being PD-1+ and KLRG1- [3]. Later, Kaech and colleagues showed that MPEC expressed lower levels of the TH1 committing transcription factor T-bet and the surface marker Ly6C and persist to transition into memory T cells [4]. MPEC cells display enhanced survival during the contraction phase and elicit greater proliferative responses to secondary infection.
Despite the importance of CD4 memory establishment for long-term immunity against pathogens, little is known of the factors influencing the survival of effector T cells and their transition to memory CD4 T cells. Of particular interest is the role of B cells in maintaining long-term T cell memory. The mouse model has provided insight into how B cells affect T cell responses. Diminished T cell memory responses and/or protective immunity generated to several intracellular pathogens including lymphocytic choriomeningitis virus (LCMV)[5, 6], Listeria monocytogenes [7], Francisella tularensis [8], and Mycobacterium tuberculosis [9] have been shown in animals with B cell deficits. Furthermore B cells were shown to be essential for T cell immunity against tumors, where enhanced B16 melanoma growth was observed following anti-CD20 Mab mediated B cell depletion [10].
There are many ways B cells could influence antigen-specific T cell generation and subsequent generation of T cell memory. First, B cells effectively present antigen to T cells through MHC class II (MHC-II) molecules and drive antigen-specific proliferation [11, 12]. Second, B cells produce antibodies that bind antigen and enable the formation of complexes that follicular dendritic cells engulf and use for antigen presentation to circulating T cells, and could additionally be involved in the maintenance of memory T cells [13]. Lastly, cytokine production by B cells and cellular localization are also important factors for shaping CD4 T cell responses [14-16]. B cell toll like receptor (TLR) activation and cytokine production leading to T helper cell differentiation and function, including TH2 [17] and TH1 [18] polarization have also been reported. All of these B cell functions underlie the importance of these cells in protection against a large number of pathogens.
In this study, the mechanistic contribution of B cells to MPEC induction following vaccination was examined. We used the ID93/GLA-SEclinical phase tuberculosis (TB) vaccine, which drives a strong TH1 response, in addition to a vaccine-specific MHC-II tetramer bound to a dominant epitope of Rv3619 (one of the component antigens of the ID93 polyprotein fusion) to follow specific memory T cell responses. In addition to the use of μMT-/- B cell deficient mice, adoptive transfer studies were done with MHC-II knockout mice to determine the role that antigen presentation by B cells play in vaccine-derived T cell immunity.
Results
Memory TH1 responses are severely impaired in B-cell deficient mice
GLA-SE, a TLR4 agonist formulated in a stable nano-emulsion of squalene oil, induces a strong T helper 1 (TH1) response in mice when formulated with the clinically tested TB vaccine polyprotein fusion antigen designated as ID93 [1, 19-21]. Since diminished T cell memory responses have been shown with B cell deficits in several models of infection [5-9] we asked whether B cells were necessary for the induction of TH1 responses to this vaccine. Wild type (wt) and μMT-/- mice were immunized three times at three-week intervals with ID93/GLA-SE. Six weeks after the last immunization, TH1 responses in the spleen and draining lymph node (dLN) were significantly reduced (as indicated by both percentages and CD4+ T cell numbers) in μMT-/- compared to wt mice, including CD4 T cell up-regulation of CD154 and production of the TH1 cytokines IFN-γ, TNF and IL-2 upon stimulation with ID93 [Fig. 1A and B, and for representative flow cytometry plots see Supporting Information (Supp. Inf.) Fig. 2]. The induction of poly-functional antigen-specific TH1 CD4 T cells, expressing IFNγ, TNF and IL-2, was also severely impaired in the μMT-/- mice (Fig. 1C and D, Supp. Inf. Fig. 1C and D).
Figure 1. Memory TH1 responses in μMT-/- mice versus wt mice.

We immunized wt and μMT-/- mice with ID93/GLA-SE three times, at 3-week intervals, for the analysis of ID93-specific immune responses. CD154 expression and IFN-γ, TNF, and IL-2 production from CD4+ T cells was measured by flow cytometry. We assessed ID93-specific recall immune single (A and B) and polyfunctional (C and D) responses following ID93 stimulation of splenocytes (A and C) or dLN cells (B and D) with 10 μg ml–1 ID93 6 weeks after the final immunization. The gating strategy is shown in Supp. Inf. Fig. 2. Graphs show mean values ± SEM for each group. The p values were determined by two-way ANOVA with Bonferroni correction for multiple comparisons (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Data are representative of three independent experiments with three mice per group.
B cells are necessary for CD4 MPEC induction
To further define the role of B cells in CD4 TH1 response generation we sought to investigate whether early TH1 responses after the first immunization with ID93/GLA-SE were also impaired. Wt and μMT-/- mice were immunized with ID93/GLA-SE and TH1 responses were assessed one week later. At this time-point TH1 responses (percentages) were only modestly affected by B-cell deficiency (Fig. 2) with no discernable difference in poly-functionality (Fig. 2C and D, and for representative flow cytometry plots see Supp. Inf. Fig. 2). In the spleen, TH1 responses were consistently lower in the μMT mice but this response, setting aside CD154 expression, was rarely significant (Fig. 2A). TH1 responses in the dLN were comparable in the two strains, as indicated by both the percentage (Fig. 2) and number of CD4+ T cells (Supp. Inf. Fig. 3B and D). The total numbers of responding TH1 CD4 T cells in the spleens were lower in the μMT-/- mice (Supp. Inf. Fig. 3A and C) but this was expected since μMT-/- have on average a spleen that is six-times smaller than wt mice [22].
Figure 2. TH1 responses following a prime immunization in μMT-/- mice versus wt mice.

We immunized wt and μMT-/- mice once with ID93/GLA-SE for the analysis of ID93-specific immune responses. CD154 expression and IFN-γ, TNF, and IL-2 production from CD4+ T cells was measured by flow cytometry. We assessed ID93-specific recall immune single (A and B) and polyfunctional (C and D) responses following ID93 stimulation of splenocytes (A and C) or dLN cells (B and D) with 10 μg ml–1 ID93 1 week after prime. The gating strategy is shown in Supp. Inf. Fig. 2. Graphs show mean values ± SEM for each group. p values were determined by two-way ANOVA with Bonferroni correction for multiple comparisons (*p<0.05). Data are representative of four independent experiments with five mice per group.
To further characterize the antigen-specific CD4 T cell response to ID93/GLA-SE in B cell deficient mice, we stained the CD4 T cells generated by immunization with a MHC class II tetramer bound to the dominant epitope from Rv3619, one of the component proteins of ID93 [23]. The frequency of tetramer positive CD4 T cells was higher in the μMT-/- mouse strains in the dLN and spleen (Fig. 3A). Again, the total numbers in the spleen were lower in the μMT-/- mice (Supp. Inf. Fig. 4A). Marshall and others [3, 4] have reported a division within the primary CD4 TH1 effector pool similar to the one observed in CD8+ T cells with a population of Memory Precursor Effector Cells (MPEC) that survive contraction and differentiation into competent memory T cells. The MPEC markers PD-1+ and KLRG1- or Ly6CLo and T-betlo were used to evaluate the formation of these particular memory subsets within the tetramer+ CD4 T cells (Fig. 3B and C). B cell deficient μMT-/- mice had significantly lower frequencies of MPEC by these two memory T cell markers in both LN and spleens one week following a prime immunization; the PD-1+ KLRG1- compartment being the most affected (Fig. 3B compared to Fig. 3C). Accordingly, this deficiency was also observable when analyzing the total counts within the PD-1+ KLRG1- compartment (Supp. Inf. Fig. 4B) in both spleen and LN, although this was less prominent within the Ly6CLo and T-betlo compartment (Supp. Inf. Fig. 4C). These results show an impairment of CD4 MPEC induction in ID93/GLA-SE immunized mice in the absence of B cells.
Figure 3. CD4 memory precursor effector T cell (MPEC) induction by ID93/GLA-SE in μMT-/- mice versus wtmice.

We immunized wt and μMT-/- mice once with ID93/GLA-SE for the analysis of ID93-specific immune responses. Peptide-MHCII tetramer staining was used to identify different ID93-specific T cell subsets. Representative flow cytometry plots and graphs are shown for the percentage of CD4 T cells that bind the tetramer (A), the percentage of tetramer-binding CD4 T cells that are PD1+/KLRG1- (B) and the percentage of tetramer-binding CD4 T cells that are T-betLo/Ly6CLo (C). Graphs show mean values ± SEM for each group. The p values were determined by unpaired t-test within each organ (*p<0.05, **p<0.01, ****p<0.0001). Data represents 9 mice pooled from two independent experiments with 4-5 mice per experiment.
B cells are necessary for memory CD4 T cell programming
Next we wanted to further characterize the phenotype of the TH1 T cells that are induced in the absence of B cells by distinguishing between the role of B cells in the induction and maintenance of antigen-specific MPEC. To determine this, we adoptively transferred T cells from μMT-/- mice (generated following a prime with ID93/GLA-SE) into B cell competent wt mice, and investigated the survival and stimulation/expansion of TH1 CD4 T cells following a boost in the recipient mice. We immunized wt (CD90.2), μMT-/- (CD90.2) and wt CD90.1 mice with ID93/GLA-SE and isolated their CD4 T cells one week later. CD45.1 recipient mice received either a mix of wt CD90.2 and wt CD90.1 cells (CD90.1 cells are included as an internal control) or a mix of μMT-/- (CD90.2) and wt CD90.1 cells. In this experiment we expected that because CD90.1 and CD90.2 CD4 T cells are generated in the same background (in wt mice) that the antigen-specific CD4 T cells would have equal survival and secondary proliferation capacities, i.e 100% of recovery of antigen specific CD90.2 T cells compared to the internal control CD90.1 (Fig. 4).
Figure 4. Functionality of adoptively-transferred TH1 CD4+ T cells generated in μMT-/- mice versus wt mice.

We isolated CD4 T cells from spleens and dLN of either wt (CD90.2) mice, μMT-/- (CD90.2) or CD90.1 mice. We performed adoptive transfer into CD45.1 naïve mice using either a mix of wt (CD90.2) and CD90.1 CD4 T cells (control group, black histogram) or a mix of μMT-/- (CD90.2) and CD90.1 CD4 T cells (white histogram) for the analysis of ID93-specific immune responses. CD154 expression and IFN-γ, TNF, and IL-2 production from CD4+ T cells was measured by flow cytometry. We assessed ID93-specific immune responses by recall of splenocytes with ID93. (A) Schematic representation of the experimental design. (B-C) Percentage recovery of tetramer + or responding CD90.2 cells when compared to the internal control CD90.1 (see Material and Methods for calculation) (B) two weeks after transfer or (C) one week after boost. Graphs show mean values ± SEM for each group. The p values were determined by two-way ANOVA with Bonferroni correction for multiple comparisons (*p<0.05, *****p<0.00001). Data represents 9-10 mice pooled from two independent experiments with 4-5 mice per experiment.
To analyze the survival capacities of the ID93/GLA-SE generated CD4 T cells from μMT-/- mice and wt mice, we compared the percent recovery of CD45.2 tetramer +/CD90.2 T cells relative to the internal control CD90.1 T cells two weeks following adoptive transfer (for representative flow cytometry plots see Supp. Inf. Fig. 5). No significant difference was found in the percentage recovery of CD90.2 tetramer + CD4 T cells from μMT-/- compared to CD90.2 tetramer + CD4 T cells from wt mice (Fig. 4B, left graph). Next we looked at the differences in the percent recovery of CD45.2/CD90.2 TH1 cytokine-producing CD4 T cells (relative to the internal control CD90.1 T cells), 2 weeks after adoptive transfer (Fig. 4B and Supplemental Fig. 5). Surprisingly, even though total number of tetramer + CD4 T cells is unaffected, the percent recovery of TH1 cytokine-producing cells was disparate. Two weeks after transfer into wt mice, we observed a consistent reduction of approximately 30% in the recovered percentage of CD90.2 TH1 cytokine-producing CD4 T cells from μMT-/- mice relative to the internal control of CD90.1 T cells (Fig. 4B, right graph).
In order to assess the secondary proliferation capacities of the antigen specific-CD4 T cells from wt and μMT-/- mice, we then compared the percent recovery of tetramer +/CD90.2 T cells (relative to the internal CD90.1 T cells) found in the adoptively transferred mice one week following a boost with ID93/GLA-SE (Fig. 4A and C). Again, no significant difference was found in the percentage recovery of CD90.2 tetramer + CD4 T cells from μMT-/- compared to wt mice (Fig. 4C, left graph). However the percent recovery of cytokine-producing CD90.2 CD4 T cells (relative to the CD90.1 internal control) was again reduced when the CD90.2 CD4 T cells were generated in the μMT-/- compared to the CD90.1 CD4 T cells generated in wt mice (Fig. 4C, right graph). This reduction of approximately 30%in the CD90.2 TH1 cytokine-producing cells from μMT-/- mice (relative to the internal control CD90.1) is similar to the one observed at the survival time-point (2 weeks after adoptive transfer), suggesting that the cells that were not able to produce cytokines at 2 weeks after transfer were also not able to undergo a secondary proliferation upon boost at this time point (Fig. 4C compared to Fig. 4B, right graphs).
These results (in addition to TH1 responses as shown in Fig. 1, 6 weeks following a boost in μMT-/- mice versus wt mice, suggest that whenantigen-specific TH1 CD4 T cells are generated in B cell deficient mice they lose their capacity to respond to antigen recall, resulting in the loss of TH1 cytokine production, as well as their proliferative capacities over time, even when transferred in B-cell sufficient mice.
MPEC impairment can be partially restored by B cell transfer and is MHC-II dependent
Even though the capacity of B cells to present antigens to the CD4 T cells is well established and a long-held view, their contribution to T cell priming and maintenance in vivo is still debated[11, 18, 24-28]. We sought to investigate whether their APC function is implicated in this process. To test this, we transferred B cells from wt or MHCII-/- mice nto μMT-/- mice and compared the MPEC generation in the dLN, where the MPEC deficiency phenotype observed was strongest. Three days after adoptive transfer, mice were immunized with ID93/GLA-SE and the responses in the dLN assessed 1 week later to determine ID93-specific immune responses. As shown above, there was no significant difference in the frequency (or total counts) of tetramer+ CD4 T cells (Fig. 5A and Supp. Inf. Fig. 6A) and the PD1+ KLRG1- was strongly reduced in μMT-/- compared to wt (Fig. 5B, and Supp. Inf. Fig. 6B). This impairment was partially reversed when wt B cells but not MHCII-/- B cells were transferred to μMT-/- mice (Fig. 5B and Supp. Inf. Fig. 6B). A similar tendency was observed for the Ly6CLo T-betLo subpopulation (Fig. 5C and Supp. Inf. Fig. 6C) pointing to a role of B cells as APC for the induction of MPEC by ID93/GLA-SE immunization.
Figure 5. MPEC generation in μMT-/- mice following adoptive transfer of B cells from either wt or MHCII-/- mice.
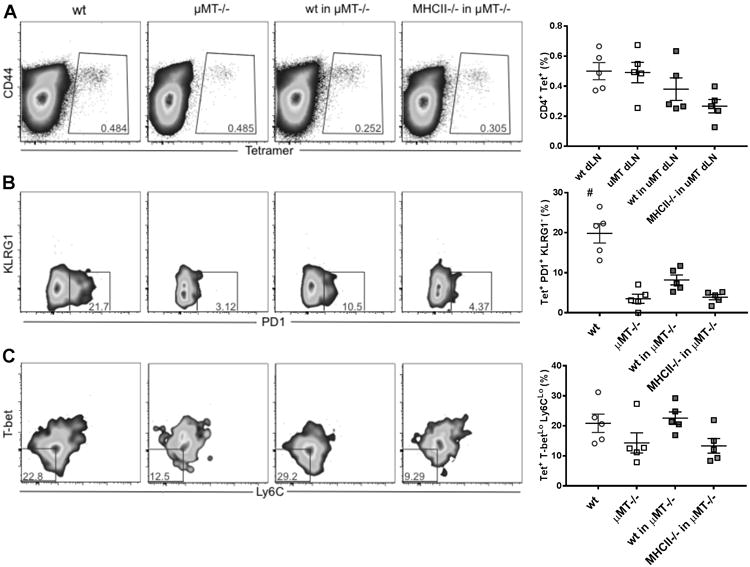
We adoptively transferred B cells from wt or MHCII-/- mice into μMT-/- mice. Three days later, we immunized wt, μMT-/- or μMT-/- adoptively transferred mice once with ID93/GLA-SE for the analysis of ID93-specific immune responses. Peptide-MHCII tetramer staining was used to identify different ID93-specific T cell subsets. Representative flow cytometry plots and graphs are shown for the percentage of CD4 T cells that bind the tetramer (A), the percentage of tetramer-binding CD4 T cells that are PD1+/KLRG1- (B) and the percentage of tetramer-binding CD4 T cells that are T-betLo/Ly6CLo (C). The gating strategy is shown in Supp. Inf. Fig. 5. Graphs show mean values ± SEM for each group. The p values were determined by one-way ANOVA with Bonferroni correction for multiple comparisons (#p<0.001compared to all other groups). Data are representative of two independent experiments with five mice per group.
Discussion
B cell-T cell cross talk is important for generation of effector adaptive immunity in several systems including in infectious disease settings and in cancer models. Indeed impairment of memory T cell responses to infectious diseases has been observed in the absence of B cells (μMT-/- mice)[5-9].
The rapeutic use of B cell-depleting monoclonal antibodies in patients diagnosed with B cell lymphomas [29] or autoimmune disorders [30-33] has brought new evidence of B cell involvement in affecting the T cell compartment. For instance, these therapies are well tolerated and adverse problems are relatively rare, but there have been reports of recurrent infections that depend on cellular immunity in these patients [34-38]. Furthermore, B cell depleting monoclonal antibody therapies also ameliorate the disease course in patients with CD4+ T cell-mediated autoimmunity [39, 40].
Consistent with these previous findings in mice and humans, we show that MPEC CD4 T cell generation upon immunization with a clinical TB vaccine is impaired in B cell deficient mice and results in a dramatically diminished memory T cell recall response.
Our results suggest that while vaccine-specific memory TH1 responses are affected by the lack of B cells, they are not defective at earlier time points. At 1 week after immunization functional TH1 responses are mostly unaffected. This phenomenon following acute LCMV infection in mice has been observed and similarly, deficiency was shown only in the memory T cell population and not in the primary response [5]. Interestingly, even though these early (1 week after prime) antigen-specific cells seem functionally similar in their capacity to produce TH1 cytokines when generated in either wt or B cell deficient mice, they are phenotypically different. B-cell deficient mice fail to generate MPEC, which are defined and characterized by others as either PD1+ KLRG1- or T-betlo Ly6CLo [3, 4]. These data demonstrate that B cells might be needed for priming a robust long-lasting TH1 response characterized by the generation of antigen-specific MPEC.
To further explore whether there was an impairment in the survival of TH1 memory T cells in μMT-/- mice, we performed a mixed-cell adoptive transfer experiment using wt CD90.1 T cells combined with either wt CD90.2 or μMT-/- CD90.2 T cells, where the survival of CD4 T cells derived from either B cell deficient or wtID93/GLA-SE immunized mice could be followed relative to the internal CD90.1 T cell control. We also examined whether transferred cells could be boosted with a subsequent immunization. While we observed a significant reduction in the recovery of responding TH1 CD4 T cells from B cell deficient donors compared to the internal control, there was no significant further reduction (indicative of cell death)or expansion (indicative of cell proliferation) in the percentage of cells when those surviving cells were boosted (at week 3, as shown in Fig. 4A). This suggests that the main impairment in of TH1 cells generated in B cell deficient mice is in their lack of capacity to respond to antigen stimulation and in their subsequent failure to proliferate; an impairment that is not rescued by the presence of B cells after the priming event. When looking strictly at tetramer numbers there was no apparent difference in wt and B cell deficient mice, similar to that observed in prior studies [5]. This suggests that the responding TH1 CD4 T cells generated in μMT-/- mice might be impaired rather than dying at the time point observed in this study (2 weeks after adoptive transfer and 3 weeks after the prime immunization). These data confirm our results above showing that the MPEC generation is impaired in B cell deficient mice and thus, B cells are required upon priming. Further experiments will be needed to investigate whether B cells are also required for TH1 CD4+ T cell maintenance.
To investigate antigen-specific CD4 T cells further, the MHC-II tetramer-positive T cells that recognize an epitope of the ID93/GLA-SE vaccine were used to determine whether CD4 MPEC could be restored following transfer of wt B cells into μMT-/- recipient mice. We show that MPEC sarepartially restored as determined by T-betlo Ly6Clo or PD1+ KLRG1- stained cells (memory phenotype). B cell transfers do not restore B cell numbers to similar levels observed in wt mice, which could explain why only a partial restoration of the phenotype was observed. Interestingly, MHCII-/- B cells from wt mice were not capable of restoring the memory phenotype, suggesting that antigen presentation by B cells is required for MPEC generation. While we indeed show that wt B cells but not MHCII-/- B cells were capable of restoring the B cell deficient phenotype, another possibility that cannot be overlooked is the potential role of B7/1 and B7/2 during antigen presentation [41, 42]. We plan to address this possibility in future studies using B cells deficient in B7/1, B7/2 or both molecules. The requirement for antigen presentation by B cells is strongly debated [11, 18, 24-28]. Gray et al showed that antigen presentation by B cells is not necessary for inducing primary T cell responses to Salmonella enterica infection and instead is needed later (7-14 days) for effector T cell response sustenance [18], which disagrees with the results presented here where B cells are required earlier. In their study, however, survival of the T cells generated at day 7 in mixed bone marrow chimeras, in which B cells could not present antigenvia MHC-II, was not tested. As shown in our study those TH1 T cells might have normal cytokine producing capacities at that time point, but may already be poised to become impaired later on. In additional studies this function was shown to be needed much earlier (at days 3-4) upon immunization with a model vaccination regimen [11]. One caveat of this study is that they used a TCR transgenic model in which the frequency of naïve antigen-specific T cells is artificially increased; they hypothesized that the APC capacity of DCs might be insufficient in this model. Another hypothesis which is in agreement with the results presented here is that the antigen presenting capacity of B cells may be needed upon immunization, where APC activation is less robust than that observed following infection. Antigen-specific antibodies have been shown to enhance antigen uptake and presentation by non-B cell APC [43, 44]. It should then be pointed out that, in our model, the inability of MHCII-/- B cells to resolve the memory T cell phenotype could be due to their inability to receive T cell help, and thus may be unable to produce Ag-specific antibodies.
Another cave at of our studies is that they were performed in μMT-/- mice which have altered splenic architecture (accounting for the differences observed between frequencies and total numbers of cells in the spleens) and which could potentially influence our results [22, 45]. However, the B cell deficient phenotype was partially restored when wt and not MHC-II-/- B cells were transferred in the μMT-/- mice 3 days prior to immunization, suggesting that the CD4 MPEC generation impairment observed in the μMT-/- mice is due to a lack of antigen presentation by B cells rather than a defect in the splenic architecture of those mice. Taken together these data show that B cell antigen presentation is necessary, from the earliest stages of the response, to drive T cells down the memory pathway in the context of vaccination. Previous studies, done with another model antigen, have shown that dendritic cells and their antigen presenting capacity are essential for the generation of a T cell response influenced by GLA-SE [46]. Consequently both DC and B cells MHC-II presentation are necessary for the generation of a robust long-lasting TH1 responses to antigens combined with GLA-SE. Further studies will be necessary to determine why the antigen presentation by DC's and B cells is not redundant in this system and in particular what kind of extra and essential antigen presentation capacity is brought about by B cells.
The data presented show that there is failure to develop a strong TH1 memory response in the absence of B cells or in the absence of their antigen-presenting capacity. These results highlight the importance of a multi-faceted immune system working in concert for cell-mediated immunity against a number of important pathogens. Collectively, our studies and recent advances in understanding the role B cells play in both humoral and cellular immunity will have a high impact on vaccine development against several pathogens especially those requiring TH1 cell-dependent immune responses, and will be useful to clinicians that utilize B cell depleting monoclonal antibody therapy in patients with B cell disorders.
Materials and Methods
Animals and immunizations
Female wt C57BL/6 mice, μMT-/- (B cell deficient), CD45.1+, CD90.1+, and MHC-II-/- mice, 6-10 weeks of age, were purchased from The Jackson Laboratory. All strains were on the C57BL/6 background. Mice were immunized by i.m. injection with either saline or ID93 (a recombinant fusion protein comprised of Rv3619, Rv1813, Rv3620, and Rv2608) formulated with the adjuvant GLA-SE to provide a final vaccine dose of 0.5μg ID93 and 5μg GLA-SE [21]. All animals were housed in the IDRI animal care facility (Seattle, WA) under specific pathogen-free conditions. All animal experiments and protocols used in this study were approved by IDRI's Animal Care and Use Committee (ACUC).
Spleen and LN processing
Single-cell suspensions of splenocytes or draining LN (dLN) cells were obtained by dissociating the whole organ through a 100-μm nylon cell strainer (BD Falcon). Red blood cells from the spleens were lysed with Red Blood Cell Lysis Buffer (eBioscience) and resuspended in RPMI 1640/10%FBS with pen/strep and glutamine (cRPMI) for further analysis.
Intracellular Cytokine Staining and tetramer staining
Processed splenocytes or dLN (inguinal) cells were stimulated for 2 hours with either ID93 (10 μg/ml) in cRPMI, or cRPMI alone. Stimulated cells were then treated with Brefeld in A (Golgi Plug, BD Biosciences) and incubated for 8 hours at 37°C, then transferred to 4°C. Cells were surface stained with fluorochrome labeled antibodies to CD4 (clone GK1.5), CD8 (clone 53-6.7), CD90.1 (clone OX-7), CD90.2 (clone 53-2.1), CD45.1 (clone A20) and CD45.2 (clone 104) in the presence of 20% normal mouse serum for 20 min at 4°C. Cells were washed and permeabilized with Cytofix/Cytoperm (BD Biosciences) for 20 min at room temperature. Cells were washed twice with Perm/Wash (BD Biosciences) and stained intracellularly with fluorochrome labeled antibodies to IFN-γ (clone XMG-1.2), IL-2 (JES6-5H4), TNF (MP6-XT22) and CD154 (clone MR1) for one hour at 4°C. Cells were washed and resuspended in PBS+1% BSA. Up to 107 events were collected on a four laser LSRF or tessa flow cytometer (BD Biosciences). Data were analyzed with Flow Jo. Cells were gated as singlets > lymphocytes > CD4+ CD8 – > cytokine positive; except for the T cell survival analysis where cells were gated as singlets > lymphocytes > CD4+ CD8- CD45.1- CD45.2+ > cytokine positive or tetramer + > CD90.2+. ID93-specific response frequencies were determined by subtracting the frequency of unstimulated cells with positive responses from ID93-stimulated cells in matched samples.
Percentage recovery of CD90.2 cells for each category was calculated as follow: . Alternatively CD4 cells were positively selected with the mouse CD4 (L3T4) microbeads from Miltenyi Biotec and stained, in the presence of 20% normal mouse sera, for 1h at 37°C with an I-Ab tetramer presenting the dominant epitope of Rv3619 (VIYEQANAHGQ), one of the components of ID93 [23]. APC-labeled tetramers were provided by the National Institutes of Health Tetramer Core Facility. Cells were washed and stained for surface CD4 (clone RM4-5), CD8 (clone 53-6.7), Ly6G (clone 1A8), CD11b (clone M1/70), CD19 (clone 1D3), CD11c (clone N418), F4/80 (clone BM8), ter119 (clone TER-119), CD44 (clone IM7), Ly6C (clone HK1.4), PD-1 (clone 29F.1A12) and KLRG1 (clone 2F1). Cells were washed and permeabilized with Cytofix/Cytoperm (BD Biosciences) for 20 min at room temperature. Cells were washed twice with Perm/Wash (BD Biosciences) and stained intracellularly with fluorochrome labeled antibodies to T-bet (clone 4B100) overnight at 4°C. Cells were washed and resuspended in PBS+1% BSA. Up to 107 events were collected on a four laser LSRF or tess a flow cytometer (BD Biosciences). Data were analyzed with Flow Jo. Cells were gated as singlets > lymphocytes > CD4+ Dump- (Ly6G, CD11b, CD19, CD11c, CD8, F4/80, ter119) > tetramer+ > PD1+ KLRG1- or T-betLo Ly6CLo.
B and T cell adoptive transfers
For the B cell adoptive transfer experiments, spleens and LNs (inguinal, popliteal, iliac, axillar and brachial) from 5 naïve donor mice per strain (wt or MHCII-/-) were harvested and processed as described above. Cells were pooled for each strain and B cells were isolated by positive selection with the mouse Pan B Cell Isolation Kit II (Miltenyi Biotec) according to the manufacturer's instructions. Approximately 2.5×107 B cells/mouse in 300μl of PBS were transferred into naïve recipient μMT-/- mice by i.v. injection.
For the CD4 T cell adoptive transfer experiments, 5 wt (CD90.2), 5 μMT-/- (CD90.2) and 10 CD90.1 mice were immunized with ID93/GLA-SE. One week later their spleens and dLN (inguinal, popliteal, iliac and axillar) were harvested and processed as described above. Total cells from the 5 wt (CD90.2) and 5 CD90.1 constituting the first combined donor population (control) and 5 μMT-/- (CD90.2) and 5 CD90.1 mice constituting the second combined donor population (experimental) were pooled and CD4 T cells were isolated with the mouse CD4 T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer's instructions. Approximately 3.5×107 CD4 T cells/mouse in 300μl of PBS were transferred into naïve CD45.1 recipient mice by i.v. injection (see Fig. 5a for a schematic representation of the experimental design).
Statistical Analysis
Statistical analysis was determined by one-way or two-way ANOVA with Bonferroni correction for multiple comparisons, or by student's t-tests as indicated for each experiment. Graphs and statistical analyses were performed using Graph Pad Prism 5 (Graph Pad Software, San Diego, CA). p<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We would like to thank Dave Argilla, Ian Bishop, Valerie Reese, Hillarie Windish, Tara Evers and Elyse Beebe for their technical assistance and helpful discussions, as well as the Formulation team and Vivarium team at the Infectious Disease Research Institute.
This work was funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200800045C and Grant Number U01AI078054 (under the Principal Investigator Dr. Rhea Coler).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, National Institute of Allergy And Infectious Diseases, or the National Institutes of Health nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Conflict of Interest: The authors declare no financial or commercial conflicts of interest.
References
- 1.Coler RN, Baldwin SL, Shaverdian N, Bertholet S, Reed SJ, Raman VS, Lu X, et al. A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PloS one. 2010;5:e13677. doi: 10.1371/journal.pone.0013677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiley WW, Shafiani S, Wittmer ST, Tucker-Heard Gs, Moon JJ, Jenkins MK, Urdahl KB, et al. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19408–19413. doi: 10.1073/pnas.1006298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish Ia, Rutishauser R, et al. Differential Expression of Ly6C and T-bet Distinguish Effector and Memory Th1 CD4 + Cell Properties during Viral Infection. Immunity. 2011;35:633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misumi I, Whitmire JK. B Cell Depletion Curtails CD4+ T Cell Memory and Reduces Protection against Disseminating Virus Infection. Journal of immunology (Baltimore, Md : 1950) 2014;192:1597–1608. doi: 10.4049/jimmunol.1302661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitmire JK, Asano MS, Kaech SM, Sarkar S, Hannum LG, Shlomchik MJ, Ahmed R. Requirement of B cells for generating CD4+ T cell memory. J Immunol. 2009;182:1868–1876. doi: 10.4049/jimmunol.0802501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuzaki G, Vordermeier HM, Hashimoto A, Nomoto K, Ivanyi J. The role of B cells in the establishment of T cell response in mice infected with an intracellular bacteria, Listeria monocytogenes. Cell Immunol. 1999;194:178–185. doi: 10.1006/cimm.1999.1503. [DOI] [PubMed] [Google Scholar]
- 8.Elkins KL, Bosio CM, Rhinehart-Jones TR. Importance of B cells, but not specific antibodies, in primary and secondary protective immunity to the intracellular bacterium Francisella tularensis live vaccine strain. Infect Immun. 1999;67:6002–6007. doi: 10.1128/iai.67.11.6002-6007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vordermeier HM, Venkataprasad N, Harris DP, Ivanyi J. Increase of tuberculous infection in the organs of B cell-deficient mice. Clinical and experimental immunology. 1996;106:312–316. doi: 10.1046/j.1365-2249.1996.d01-845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–4016. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol. 2006;176:3498–3506. doi: 10.4049/jimmunol.176.6.3498. [DOI] [PubMed] [Google Scholar]
- 12.Morales-Aza B, Glennie SJ, Garcez TP, Davenport V, Johnston SL, Williams NA, Heyderman RS. Impaired maintenance of naturally acquired T-cell memory to the meningococcus in patients with B-cell immunodeficiency. Blood. 2009;113:4206–4212. doi: 10.1182/blood-2008-08-171587. [DOI] [PubMed] [Google Scholar]
- 13.Nayak R, Lal G, Shaila MS. Perpetuation of immunological memory: role of serum antibodies and accessory cells. Microbes Infect. 2005;7:1276–1283. doi: 10.1016/j.micinf.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojciechowski W, Harris DP, Sprague F, Mousseau B, Makris M, Kusser K, Honjo T, et al. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity. 2009;30:421–433. doi: 10.1016/j.immuni.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol. 2012;13:681–690. doi: 10.1038/ni.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horsnell WG, Darby MG, Hoving JC, Nieuwenhuizen N, McSorley HJ, Ndlovu H, Bobat S, et al. IL-4Ralpha-associated antigen processing by B cells promotes immunity in Nippostrongylus brasiliensis infection. PLoS Pathog. 2013;9:e1003662. doi: 10.1371/journal.ppat.1003662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barr TA, Brown S, Mastroeni P, Gray D. TLR and B cell receptor signals to B cells differentially program primary and memory Th1 responses to Salmonella enterica. J Immunol. 2010;185:2783–2789. doi: 10.4049/jimmunol.1001431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orr MT, Duthie MS, Windish HP, Lucas EA, Guderian JA, Hudson TE, Shaverdian N, et al. MyD88 and TRIF synergistic interaction is required for TH1-cell polarization with a synthetic TLR4 agonist adjuvant. European journal of immunology. 2013;43:2398–2408. doi: 10.1002/eji.201243124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orr MT, Kramer RM, Barnes Lt, Dowling QM, Desbien AL, Beebe EA, Laurance JD, et al. Elimination of the cold-chain dependence of a nanoemulsion adjuvanted vaccine against tuberculosis by lyophilization. Journal of controlled release : official journal of the Controlled Release Society. 2013;177C:20–26. doi: 10.1016/j.jconrel.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertholet S, Ireton GC, Ordway DJ, Windish HP, Pine SO, Kahn M, Phan T, et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Science translational medicine. 2010;2:53ra74–53ra74. doi: 10.1126/scitranslmed.3001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asano MS, Ahmed R. CD8 T cell memory in B cell-deficient mice. J Exp Med. 1996;183:2165–2174. doi: 10.1084/jem.183.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orr MT, Fox CB, Baldwin SL, Sivananthan SJ, Lucas E, Lin S, Phan T, et al. Adjuvant formulation structure and composition are critical for the development of an effective vaccine against tuberculosis. Journal of controlled release : official journal of the Controlled Release Society. 2013;172:190–200. doi: 10.1016/j.jconrel.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J Immunol. 1999;162:5695–5703. [PubMed] [Google Scholar]
- 25.Epstein MM, Di Rosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell-deficient mice. J Exp Med. 1995;182:915–922. doi: 10.1084/jem.182.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams GS, Oxenius A, Hengartner H, Benoist C, Mathis D. CD4+ T cell responses in mice lacking MHC class II molecules specifically on B cells. Eur J Immunol. 1998;28:3763–3772. doi: 10.1002/(SICI)1521-4141(199811)28:11<3763::AID-IMMU3763>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 27.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 28.Mollo SB, Zajac AJ, Harrington LE. Temporal requirements for B cells in the establishment of CD4 T cell memory. Journal of immunology (Baltimore, Md : 1950) 2013;191:6052–6059. doi: 10.4049/jimmunol.1302033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foa R. Changes in the treatment landscape for chronic lymphoid leukemia. N Engl J Med. 2014;371:273–274. doi: 10.1056/NEJMe1405766. [DOI] [PubMed] [Google Scholar]
- 30.Stasi R, Pagano A, Stipa E, Amadori S. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood. 2001;98:952–957. doi: 10.1182/blood.v98.4.952. [DOI] [PubMed] [Google Scholar]
- 31.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 32.Naismith RT, Piccio L, Lyons JA, Lauber J, Tutlam NT, Parks BJ, Trinkaus K, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology. 2010;74:1860–1867. doi: 10.1212/WNL.0b013e3181e24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez-Puerta JA, Quintana LF, Stone JH, Ramos-Casals M, Bosch X. B-cell depleting agents for ANCA vasculitides: a new therapeutic approach. Autoimmun Rev. 2012;11:646–652. doi: 10.1016/j.autrev.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Goldberg SL, Pecora AL, Alter RS, Kroll MS, Rowley SD, Waintraub SE, Imrit K, et al. Unusual viral infections (progressive multifocal leukoencephalopathy and cytomegalovirus disease) after high-dose chemotherapy with autologous blood stem cell rescue and peritransplantation rituximab. Blood. 2002;99:1486–1488. doi: 10.1182/blood.v99.4.1486. [DOI] [PubMed] [Google Scholar]
- 35.Suzan F, Ammor M, Ribrag V. Fatal reactivation of cytomegalovirus infection after use of rituximab for a post-transplantation lymphoproliferative disorder. N Engl J Med. 2001;345:1000. doi: 10.1056/NEJM200109273451315. [DOI] [PubMed] [Google Scholar]
- 36.Tsutsumi Y, Yamamoto Y, Ito S, Ohigashi H, Shiratori S, Naruse H, Teshima T. Hepatitis B virus reactivation with a rituximab-containing regimen. World J Hepatol. 2015;7:2344–2351. doi: 10.4254/wjh.v7.i21.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salvana EM, Salata RA. Infectious complications associated with monoclonal antibodies and related small molecules. Clin Microbiol Rev. 2009;22:274–290. doi: 10.1128/CMR.00040-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gea-Banacloche JC, Weinberg GA. Monoclonal antibody therapeutics and risk for infection. Pediatr Infect Dis J. 2007;26:1049–1052. doi: 10.1097/INF.0b013e31815a044f. [DOI] [PubMed] [Google Scholar]
- 39.Liossis SN, Sfikakis PP. Rituximab-induced B cell depletion in autoimmune diseases: potential effects on T cells. Clin Immunol. 2008;127:280–285. doi: 10.1016/j.clim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Leandro MJ, de la Torre I. Translational Mini-Review Series on B Cell-Directed Therapies: The pathogenic role of B cells in autoantibody-associated autoimmune diseases--lessons from B cell-depletion therapy. Clin Exp Immunol. 2009;157:191–197. doi: 10.1111/j.1365-2249.2009.03978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris NL, Ronchese F. The role of B7 costimulation in T-cell immunity. Immunol Cell Biol. 1999;77:304–311. doi: 10.1046/j.1440-1711.1999.00835.x. [DOI] [PubMed] [Google Scholar]
- 42.Gimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, Nadler LM. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci U S A. 1991;88:6575–6579. doi: 10.1073/pnas.88.15.6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coughlan S, Harkiss GD, Hopkins J. Enhanced proliferation of CD4+ T cells induced by dendritic cells following antigen uptake in the presence of specific antibody. Vet Immunol Immunopathol. 1996;49:321–330. doi: 10.1016/0165-2427(95)05478-2. [DOI] [PubMed] [Google Scholar]
- 44.Manca F, Fenoglio D, Li Pira G, Kunkl A, Celada F. Effect of antigen/antibody ratio on macrophage uptake, processing, and presentation to T cells of antigen complexed with polyclonal antibodies. J Exp Med. 1991;173:37–48. doi: 10.1084/jem.173.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crowley MT, Reilly CR, Lo D. Influence of lymphocytes on the presence and organization of dendritic cell subsets in the spleen. J Immunol. 1999;163:4894–4900. [PubMed] [Google Scholar]
- 46.Pantel A, Cheong C, Dandamudi D, Shrestha E, Mehandru S, Brane L, Ruane D, et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T-cell immunity in vivo. European journal of immunology. 2012;42:101–109. doi: 10.1002/eji.201141855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.