

Abstract
Purpose
Recurrent MET exon 14 splicing has been revealed in lung cancers and is a promising therapeutic target. Because we have limited knowledge about the natural history of MET mutant tumors, the current study was aiming to determine the clinical and pathological characteristics in non-small cell lung cancers (NSCLC).
Results
Twenty-three patients (1.3%) were positive for MET exon 14 skipping. Patients with MET exon 14 skipping displayed unique characteristics: female, non-smokers, earlier pathology stage and older age. MET exon 14 skipping indicated an early event as other drivers in lung cancer, while MET copy number gain was more likely a late event in lung cancer. Overall survival (OS) of patients harboring MET exon 14 skipping was longer than patients with KRAS mutation. Almost four-fifths of the lung tumors with MET exon 14 skipping had EGFR and/or HER2 gene copy number gains. EGFR inhibitor showed moderate antitumor activity in treatment of a patient harboring MET exon 14 skipping.
Patients and Methods
From October 2007 to June 2013, we screened 1770 patients with NSCLC and correlated MET status with clinical pathologic characteristics and mutations in EGFR, KRAS, BRAF, HER2, and ALK. Quantitative Real-Time PCR was used to detect MET gene copy number gain. Immunohistochemistry (IHC) was also performed to screen MET exon 14 skipping. Clinicopathological characteristics and survival information were analyzed.
Conclusions
MET exon 14 skipping was detected in 1.3% (23/1770) of the Chinese patients with NSCLC. MET exon 14 skipping defined a new molecular subset of NSCLC with identifiable clinical characteristics. The therapeutic EGFR inhibitors might be an alternative treatment for patients with MET mutant NSCLC.
Keywords: MET, non-small cell lung cancer, surgery, targeted therapy
INTRODUCTION
Lung cancer is the leading cause of cancer deaths worldwide. Non–small cell lung cancer (NSCLC) comprises over 80% of all lung tumors. With better understanding of the major genetic alterations and signaling pathways involved of lung cancer, it has been classified into various subsets with different molecular and clinicopathologic characteristics [1]. Target-based therapeutics designed to specific molecular clusters have revolutionized lung cancer treatment. The most representative examples are gefitinib/erlotinib and crizotinib for lung adenocarcinomas harboring EGFR mutations and ALK/ROS1 fusion, respectively [2, 3]. Even with broad genotyping, there remains 20%–40% of lung adenocarcinoma and 80% of lung squamous cell carcinoma without any known targetable oncogenic mutations [4–7].
MET, encoding the proto-oncogene tyrosine kinase c-MET, is the receptor for hepatocyte growth factor (HGF). The activation of this kinase by amplification and overexpression could promote cancer [8–10]. MET amplification is uncommon in NSCLCs and was observed in 2%–4% of previously untreated patients [8, 9, 11]. MET targeted therapies are currently undergoing early-phase clinical trial evaluations in lung cancer patients [12, 13].
Recently, MET gene exon 14 skipping was identified as a potential driver mutation in lung and colon tumors [14–18]. Strikingly, NSCLC patients harboring these genetic alterations have shown remarkable responses to MET inhibitor (crizotinib or cabozantinib) in several independent early-phase trials, suggesting that MET might be a novel druggable target in lung cancers [15–18]. The clinicopathologic characteristics of lung cancer patients with MET exon 14 skipping have not yet been described. These results provide us with an impetus to understand the natural history of MET mutant tumors.
Here, we demonstrated that MET exon 14 skipping is a novel oncogenic driver in lung cancers by clinical and pathological characteristics. This new molecular subset of patients had distinct features, allowing clinicians to select enriched subpopulations for genotyping and with whom randomized prospective clinical trials of targeted therapies could be efficiently performed.
RESULTS
Clinicopathological characteristics
From October 2007 to June 2013, we screened out 1770 qualified patients in this study cohort, including 1305 adenocarcinomas, 48 adenosquamous carcinomas and 417 squamous cell carcinomas. There were 991 (56.0%) male patients and 779 (44.0%) female patients. Ages below or above 60 years old were almost equally distributed. Seven hundred and seventy-nine patients were current smokers or former smokers; nine hundred and ninety-one patients were non-smokers. Obviously, most adenocarcinomas were female and non-smoking patients. Squamous cell carcinomas predominantly occurred in male and smoking patients. More detailed information was illustrated in Table 1.
Table 1. Clinical characteristics of 1770 patients with NSCLC.
| Adenocarcinoma | Adenosquamous carcinoma | Squamous cell carcinoma | ||||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Total | 1305 | 48 | 417 | |||
| Sex | ||||||
| Male | 573 | 43.9% | 34 | 70.8% | 384 | 92.1% |
| Female | 732 | 56.1% | 14 | 29.2% | 33 | 7.9% |
| Age | ||||||
| > 60 y | 657 | 50.3% | 29 | 60.4% | 228 | 54.7% |
| < 60 y | 648 | 49.7% | 19 | 39.6% | 189 | 45.3% |
| Smoking status | ||||||
| Smoker | 401 | 30.7% | 27 | 56.3% | 351 | 84.2% |
| Never-smoker | 904 | 69.3% | 21 | 43.8% | 66 | 15.8% |
| Pathologic stage | ||||||
| 0 | 32 | 2.5% | 0 | 0.0% | 0 | 0.0% |
| IA | 510 | 39.1% | 7 | 14.6% | 70 | 16.8% |
| IB | 171 | 13.1% | 11 | 22.9% | 104 | 24.9% |
| IIA | 113 | 8.7% | 3 | 6.3% | 65 | 15.6% |
| IIB | 43 | 3.3% | 5 | 10.4% | 47 | 11.3% |
| IIIA | 352 | 27.0% | 21 | 43.8% | 125 | 30.0% |
| IIIB | 30 | 2.3% | 1 | 2.1% | 4 | 1.0% |
| IV | 54 | 4.1% | 0 | 0.0% | 2 | 0.5% |
| Differentiation | ||||||
| Well | 197 | 15.1% | 0 | 0.0% | 8 | 1.9% |
| Moderate | 734 | 56.2% | 16 | 33.3% | 188 | 45.1% |
| Poor | 374 | 28.7% | 32 | 66.7% | 221 | 53.0% |
| EGFR Mutation | ||||||
| Present | 855 | 65.5% | 20 | 41.7% | 17 | 4.1% |
| Absent | 450 | 34.5% | 28 | 58.3% | 400 | 95.9% |
| KRAS Mutation | ||||||
| Present | 107 | 8.2% | 6 | 12.5% | 6 | 1.4% |
| Absent | 1198 | 91.8% | 42 | 87.5% | 411 | 98.6% |
| HER2 Insertion | ||||||
| Present | 32 | 2.5% | 1 | 2.1% | 4 | 1.0% |
| Absent | 1273 | 97.5% | 47 | 97.9% | 413 | 99.0% |
| BRAF Mutation | ||||||
| Present | 20 | 1.5% | 0 | 0.0% | 0 | 0.0% |
| Absent | 1285 | 98.5% | 4 | 8.3% | 417 | 100.0% |
| ALK Fusion | ||||||
| Present | 70 | 5.4% | 4 | 8.3% | 2 | 0.5% |
| Absent | 1235 | 94.6% | 44 | 91.7% | 415 | 99.5% |
| RET Fusion | ||||||
| Present | 18 | 1.4% | 2 | 4.2% | 0 | 0.0% |
| Absent | 1287 | 98.6% | 46 | 95.8% | 417 | 100.0% |
| ROS1 Fusion | ||||||
| Present | 11 | 0.8% | 0 | 0.0% | 0 | 0.0% |
| Absent | 1294 | 99.2% | 48 | 100.0% | 417 | 100.0% |
| FGFR1/3 Fusion | ||||||
| Present | 6 | 0.5% | 0 | 0.0% | 12 | 2.9% |
| Absent | 1299 | 99.5% | 48 | 100.0% | 405 | 97.1% |
| MET Skipping | ||||||
| Present | 21 | 1.6% | 2 | 4.2% | 0 | 0.0% |
| Absent | 1284 | 98.4% | 46 | 95.8% | 417 | 100.0% |
| PIK3CA Mutation | ||||||
| Present | 22 | 1.7% | 1 | 2.4% | 12 | 7.5% |
| Absent | 1270 | 98.3% | 40 | 97.6% | 147 | 92.5% |
| CTNNB1 Mutation | ||||||
| Present | 32 | 3.8% | 1 | 2.4% | 2 | 0.5% |
| Absent | 815 | 96.2% | 40 | 97.6% | 371 | 99.5% |
Screening of MET exon 14 skipping in 1770 NSCLCs
The genetic alterations of 14 genes were screened in all NSCLCs (Table 1, Figure 1). Of 1770 patients, MET exon 14 skipping was detected in 23 patients (1.3%), which was the fourth most frequently identified driver in NSCLCs (Figure 1A) (Detailed data was shown in Supplementary Table S2). It was found in 21 (1.6%) of 1305 adenocarcinomas, and in two (4.2%) of 48 adenosquamous cell carcinomas. No mutant MET was detected in squamous carcinomas. We further revealed that 1.9% (17 of 904) of non-smoking lung adenocarcinomas (NSLAD) harbored MET exon 14 skipping (Figure 1B). Among 80 NSLADs that were negative for known oncogenic drivers, 21.25% of them harbored mutant MET.
Figure 1. Mutational profiles in non-small cell lung cancer.
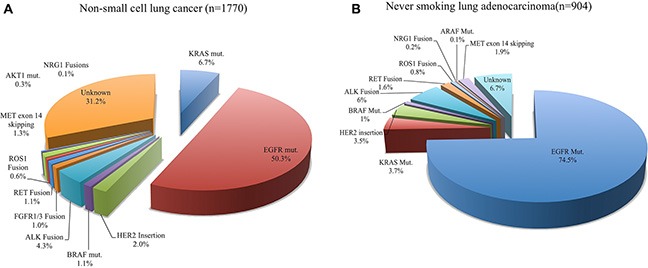
(A) Oncogenic driver mutations in 1770 non-small cell lung cancers; (B) Oncogenic driver mutations in 904 non-smoking lung adenocarcinomas.
IHC analysis of MET exon 14 skipping positive NSCLCs
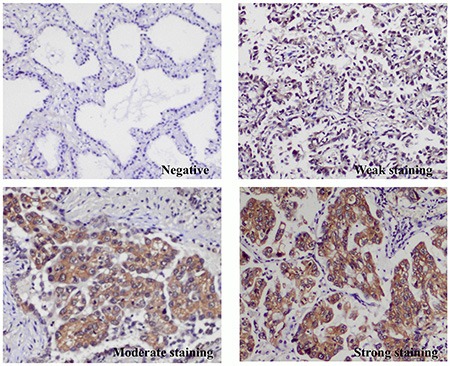
MET exon 14 skipping can only be detected using mRNA isolated from frozen tissues, which are commonly not available in clinical practice. Thus, we examined whether IHC could be utilized as a screening tool for detecting mutant MET. Of the 23 MET positive samples subjected to IHC, four scored as negative, nine as weak, eight as moderate, and two as strong. Another 64 pan-negative lung adenocarcinomas were also stained for MET protein; 42 scored as negative, 12 scored as weak, seven as moderate, and three as strong (Figure 3). The frequency of samples with MET positive in MET exon 14 skipping tumors was significantly higher than that in pan-negative samples (82.6% VS. 34.4%, Chi-Square test, p < 0.0001).
Figure 3. Representation of immunohistochemistry (IHC) staining of MET protein in pan-negative lung adenocarcinomas and MET exon 14 skipping positive non–small-cell lung cancers.
EGFR and/or HER2 copy number gains were common in MET Exon 14 skipping lung tumors
We showed that 18 of 23 (78.3%) lung tumors with MET exon 14 skipping had EGFR and/or HER2 gene copy number gains. Among 19 invasive lung cancers, 94.7% of MET mutant tumors showed EGFR and/or HER2 gene copy gains, but none of the cases with AAH/AIS harbored EGFR or HER2 gene copy alterations (Table 3), indicating the pivotal role of EGFR or HER2 in formation of invasive lung carcinoma in MET positive tumors. We also found that EGFR copy number gain was seen in all lung adenocarcinoma patients whose tumor harbored MET exon 14 skipping in TCGA (The Cancer Genome Atlas) cohort [6].
Table 3. Characteristics of non-small cell lung cancer patients with MET exon 14 skipping.
| Cases | Sex | Age | Smoking | P. | T. | Subtype | Stage | MET IHC | EGFR CN | HER2 CN | TTF1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 67 | Never | A | 3.5 | Acinar | IB | ++ | > 8 | > 6 | – |
| 2 | F | 77 | Never | A | 2 | Lepidic+Acinar | IB | + | > 4 | 2 | + |
| 3 | M | 66 | Smoker | A | 4 | Papillary | IIA | ++ | > 4 | 2 | + |
| 4 | F | 74 | Never | A | 2.3 | Lepidic | IA | ++ | > 6 | 2 | + |
| 5 | F | 54 | Never | A | 2 | Acinar+Papillary | IB | ++ | > 10 | 2 | – |
| 6 | M | 60 | Smoker | A | 4.5 | Solid | IB | ++ | > 7 | 2 | + |
| 7 | F | 75 | Never | A | 2.5 | Solid | IB | +++ | > 5 | 2 | + |
| 8 | F | 60 | Never | A | 4 | Acinar+Lepidic | IIB | + | > 8 | 2 | + |
| 9 | F | 62 | Never | A | 3 | Acinar+Solid | IA | ++ | > 5 | 2 | + |
| 10 | F | 49 | Never | A | 1 | AAH | 0 | – | 2 | 2 | + |
| 11 | F | 60 | Never | A | 2.8 | Lepidic | IA | + | 2 | 4 | + |
| 12 | F | 75 | Never | A | 2.5 | IMA+Acinar | IA | + | > 5 | 2 | – |
| 13 | M | 76 | Never | A | 0.5 | Acinar | IA | ++ | > 5 | > 5 | + |
| 14 | M | 76 | Smoker | A | 2.8 | Lepidic+Acinar | IA | – | > 5 | 3 | + |
| 15 | M | 67 | Never | A | 1.5 | AIS | 0 | – | 2 | 2 | + |
| 16 | F | 57 | Never | A | 0.8 | AIS | 0 | + | 2 | 2 | + |
| 17 | F | 73 | Never | A | 4 | Papillary | IIA | + | 2 | 2 | + |
| 18 | F | 47 | Never | A | 0.9 | AIS | 0 | – | 2 | 2 | + |
| 19 | F | 63 | Never | A | 2 | Papillary+Micropapillary | IIA | + | > 7 | 2 | + |
| 20 | M | 68 | Smoker | A | 3.2 | Solid | IIA | ++ | > 8 | > 3 | + |
| 21 | M | 68 | Never | A | 3.5 | Acinar+Solid | IIIA | +++ | > 5 | 3 | + |
| 22 | M | 65 | Smoker | AS | 10 | N.A. | IIB | + | > 4 | 2 | + |
| 23 | F | 77 | Never | AS | 1.5 | N.A. | IA | + | > 5 | > 4 | + |
Abbreviations: F, female, M, male; P, pathology; T. tumor size (cm); AAH, atypical adenomatous hyperplasia; AIS, adenocarcinoma in situ; IHC, MET immunohistochemistry; EGFR CN, EGFR gene copy by EGFR-FISH analysis; HER2 CN, HER2 gene copy by HER2-FISH analysis; N.A., not available; AAH, Atypical adenomatous hyperplasia; AIS, adenocarcinoma in situ (AIS).
Clinicopathologic characteristics of patients with MET exon 14 skipping
Here, we demonstrated that patients harboring MET exon 14 skipping defined a novel genetic subset of NSCLC. Patients with MET exon 14 skipping displayed unique characteristics: female, non-smokers, earlier pathology stage and older age (Table 2). Lepidic/acinar component were present in 58.8% of invasive lung adenocarcinomas with MET exon 14 skipping (Table 3). Atypical adenomatous hyperplasia (AAH)/ adenocarcinoma in situ (AIS) were observed in 17.4% (4 of 23) of MET positive tumors, indicating that it was an early event as other drivers in lung cancer. In contrast to patients with MET exon 14 skipping, patients with MET copy number gain were more prone to have late stage disease, poor differentiation, and acinar/solid/invasive mucinous adenocarcinoma (Table 4), indicating that MET copy number gain is a late event in lung cancer.
Table 2. Clinicopathologic characteristics of patients with MET exon 14 skipping in lung adenocarcinomas.
| MET skipping | EGFR | P | KRAS | P | ALK | P | *Pan-negative | P | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | 21 | 855 | % | 107 | % | 70 | % | 165 | % | |||||
| Gender | ||||||||||||||
| Male | 7 | 33.3 | 312 | 36.5 | 85 | 79.4 | 26 | 37.1 | 114 | 69.1 | ||||
| Female | 14 | 66.7 | 543 | 63.5 | .823 | 22 | 20.6 | .001 | 44 | 62.9 | .802 | 51 | 30.9 | .003 |
| Age | ||||||||||||||
| ≥ 60 y | 17 | 81.0 | 440 | 51.5 | 48 | 44.9 | 23 | 32.9 | 93 | 56.4 | ||||
| < 60 y | 4 | 19.0 | 415 | 48.5 | .008 | 59 | 55.1 | .003 | 47 | 67.1 | .001 | 72 | 43.6 | .035 |
| Smoking status | ||||||||||||||
| Smoking | 4 | 19 | 182 | 21.3 | 73 | 68.2 | 16 | 22.9 | 102 | 61.8 | ||||
| Never | 17 | 81 | 673 | 78.7 | .529 | 34 | 31.8 | .001 | 54 | 77.1 | .485 | 63 | 38.2 | .001 |
| Tumor size | ||||||||||||||
| < 3 cm | 13 | 61.9 | 560 | 65.5 | 52 | 48.6 | 38 | 54.3 | 75 | 45.5 | ||||
| ≥ 3 cm | 8 | 38.1 | 289 | 33.8 | .816 | 55 | 51.4 | .341 | 32 | 45.7 | .621 | 90 | 54.5 | .171 |
| LN | ||||||||||||||
| 0 | 15 | 71.4 | 553 | 64.7 | 68 | 63.6 | 31 | 44.3 | 91 | 55.2 | ||||
| 1 | 5 | 23.8 | 78 | 9.1 | 12 | 11.2 | 15 | 21.4 | 11 | 6.7 | ||||
| 2 | 1 | 4.8 | 219 | 25.6 | 26 | 24.3 | 19 | 27.1 | 60 | 36.4 | ||||
| 3 | 0 | 0 | 5 | 0.6 | .037 | 1 | 0.9 | .131 | 5 | 7.1 | .056 | 3 | 1.8 | .004 |
| Stage | ||||||||||||||
| 0 | 4 | 19 | 16 | 1.9 | 0 | 0 | 1 | 1.4 | 5 | 3 | ||||
| I | 11 | 52.4 | 482 | 56.4 | 53 | 49.5 | 26 | 37.1 | 68 | 41.2 | ||||
| II | 5 | 23.8 | 88 | 10.3 | 21 | 19.6 | 12 | 17.1 | 25 | 15.2 | ||||
| III | 1 | 4.8 | 230 | 26.9 | 30 | 28.0 | 28 | 40.0 | 61 | 37.0 | ||||
| IV | 0 | 0 | 39 | 4.6 | .001 | 3 | 2.8 | .001 | 3 | 4.3 | .001 | 6 | 3.6 | .001 |
| Differentiation | ||||||||||||||
| Well | 6 | 28.6 | 139 | 16.3 | 11 | 10.3 | 6 | 8.6 | 19 | 11.5 | ||||
| Moderate | 8 | 38.1 | 519 | 60.7 | 53 | 49.5 | 43 | 61.4 | 74 | 44.8 | ||||
| Poor | 7 | 33.3 | 197 | 23.0 | .103 | 43 | 40.2 | .077 | 21 | 30. | .038 | 72 | 43.6 | .096 |
| Histology | ||||||||||||||
| AAH/AIS | 4 | 19 | 43 | 5.0 | 1 | 0.9 | 1 | 1.4 | 9 | 5.5 | ||||
| Lepidic | 4 | 19 | 93 | 10.9 | 5 | 4.7 | 1 | 1.4 | 8 | 4.8 | ||||
| Acinar | 6 | 28.6 | 455 | 53.2 | 34 | 31.8 | 26 | 37.1 | 58 | 35.2 | ||||
| Papillary | 3 | 14.3 | 132 | 15.4 | 10 | 9.3 | 6 | 8.6 | 15 | 9.1 | ||||
| Solid | 3 | 14.3 | 85 | 9.9 | 33 | 30.8 | 20 | 28.6 | 58 | 35.2 | ||||
| M-P | 0 | 0.0 | 18 | 2.1 | 0 | 0 | 0 | 0 | 3 | 1.8 | ||||
| IMA | 1 | 4.8 | 14 | 1.6 | 22 | 20.6 | 16 | 22.9 | 10 | 6.1 | ||||
| Enteric | 0 | 0.0 | 0 | 0.0 | .045 | 0 | 0 | .001 | 0 | 0 | .001 | 2 | 1.2 | .028 |
Abbreviations: LN, lymph nodes; AAH, Atypical adenomatous hyperplasia; AIS, adenocarcinoma in situ (AIS); M-P, micropapillary; IMA, Invasive mucinous adenocarcinoma
Table 4. Comparison of clinicopathologic characteristics of patients with MET exon 14 skipping and with MET copy number gain in lung adenocarcinomas.
| MET E14 skipping | % | MET copy number gain | % | P | |
|---|---|---|---|---|---|
| Total | 21 | 47 | |||
| Gender | |||||
| Male | 7 | 33.33% | 22 | 46.81% | |
| Female | 14 | 66.67% | 25 | 53.19% | 0.427 |
| Age | |||||
| ≥ 60 y | 17 | 80.95% | 27 | 57.45% | |
| < 60 y | 4 | 19.05% | 20 | 42.55% | 0.052 |
| Smoking status | |||||
| Smoking | 4 | 19.05% | 14 | 29.79% | |
| Never-smoking | 17 | 80.95% | 33 | 70.21% | 0.269 |
| Tumor size | |||||
| < 3 | 13 | 61.90% | 29 | 61.70% | |
| ≥ 3 | 8 | 38.10% | 18 | 38.30% | 1.000 |
| Lymph Node status | |||||
| 0 | 15 | 71.43% | 28 | 59.57% | |
| 1 | 5 | 23.81% | 5 | 10.64% | |
| 2 | 1 | 4.76% | 13 | 27.66% | |
| 3 | 0 | 0.00% | 1 | 2.13% | 0.103 |
| Pathologic stage | |||||
| 0 | 4 | 19.05% | 1 | 2.13% | |
| I | 11 | 52.38% | 23 | 48.94% | |
| II | 5 | 23.81% | 6 | 12.77% | |
| III | 1 | 4.76% | 16 | 34.04% | |
| IV | 0 | 0.00% | 1 | 2.13% | 0.016 |
| Differentiation | |||||
| Well | 6 | 28.57% | 3 | 6.38% | |
| Moderate | 8 | 38.10% | 25 | 53.19% | |
| Poor | 7 | 33.33% | 19 | 40.43% | 0.043 |
| Predominant histology subtype | |||||
| AAH/AIS | 4 | 19.05% | 1 | 2.13% | |
| Lepidic | 4 | 19.05% | 2 | 4.26% | |
| Acinar | 6 | 28.57% | 22 | 46.81% | |
| Papillary | 3 | 14.29% | 4 | 8.51% | |
| Solid | 3 | 14.29% | 11 | 23.40% | |
| Micropapillary | 0 | 0.00% | 1 | 2.13% | |
| IMA | 1 | 4.76% | 6 | 12.77% | 0.046 |
Abbreviations: AAH, Atypical adenomatous hyperplasia; AIS, adenocarcinoma in situ (AIS); IMA, Invasive mucinous adenocarcinoma.
The proportion of precancerous or early lesions (AAH/AIS/Lepidic) (38.1%) in MET exon 14 skipping tumors was significantly higher than that of EGFR (15.9%), KRAS (5.6%), HER2 (21.9%), BRAF (15%), ALK (2.9%), RET (5.6%), and pan-negative (10.3%) lung adenocarcinomas, indicating that mutant MET alone might be insufficient to transform normal cells but depend on other genetic alterations progressing into more aggressive carcinoma subtypes.
Survival analyses
The median relapse-free survival (RFS) was 46.2 months for MET compared with 48.2 months for EGFR (P = 0.152), 41.2 months for KRAS (P = 0.088), 39.4 months for ALK (P = 0.462), and 35 months for HER2 (P = 0.13). The median overall survival (OS) was 70 months for MET compared with 67.2 months for EGFR (P = 0.841), 60 months for KRAS (P = 0.016), 52.7 months for ALK (P = 0.95), and 49.4 months for HER2 (P = 0.321) (Figure 2). Multivariate analysis was not feasible because of the small number of deaths in the MET mutation group.
Figure 2.
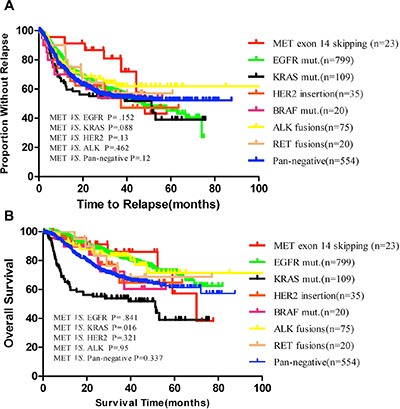
(A) Kaplan–Meier survival curves for relapse free according to MET exon 14 skipping or other known oncogenic drivers in 1635 NSCLC patients; (B) Overall survival according to MET exon 14 skipping or other known oncogenic drivers in 1393 NSCLC patients. Pan-negative indicates EGFR, KRAS, HER2, BRAF, ALK, RET, and MET negative.
EGFR inhibitor demonstrates durable clinical response in a patient with advanced MET exon 14 skipping positive NSCLC
Among the 23 NSCLC patients with MET exon 14 skipping in our cohort, a 68 year-old non-smoker with MET, EGFR, and HER2 gene copy number gains in his tumor had relapsed. The patient showed a stable disease response to EGFR inhibitor Iressa, and the status had been maintained for 12 months (Figure 4). This modest clinical activity of Iressa might indicate that blockage of EGFR might serve as a salvage therapy for treatment of advanced MET exon 14 skipping positive patients if MET inhibitor is not available.
Figure 4. EGFR inhibitor shows durable clinical response in a patient harboring MET exon 14 skipping.
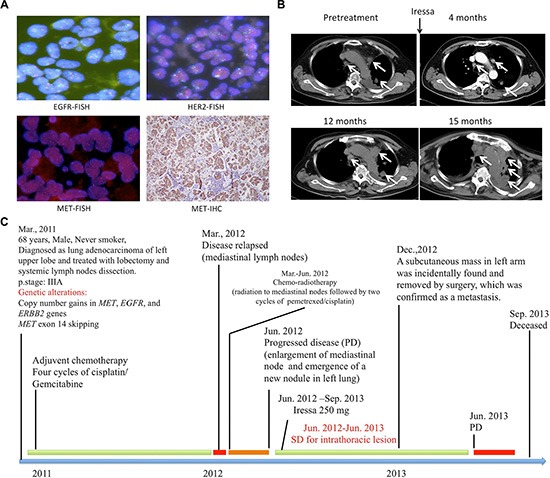
(A) FISH indicated that a MET exon 14 skipping positive non-smoker harbored EGFR, HER2, and MET copy number gains; IHC showed that this patient's tumor was positive for MET expression; (B) CT scan of the thorax demonstrating mediastinal lymph nodes enlargement and a left lobe mass before and after treatment with oral Iressa daily; (C) Timeline of the patient with MET exon 14 skipping lung.
DISCUSSION
In this study, we identified a subset of patients harboring MET exon 14 skipping as a novel oncogenic driver in lung cancers. As we previous reported [19], 90% of non-smoking lung adenocarcinomas (NSLAD) of East Asian populations were found to harbor known drivers that could be exploited as therapeutic targets. In this study, we examined 904 non-smoking NSLADs for all known oncogenic drivers. To the best of our knowledge, this study represents the most comprehensive analysis of mutation profiles in NSLADs. Of all non-smokers, 93.3% were found to carrying oncogenic driver mutations and 1.9% harbored MET exon 14 skipping. Given that MET exon 14 skipping defines a distinct subset of NSCLC patients characterized by female, non-smoker, and older age, further prospective clinical trials could exploit these clinical profiles to help clinicians select patients most likely to carry mutant MET and evaluate the clinical efficacy of MET inhibitor.
MET exon 14 skipping was not reckoned as a driver in lung cancer until the high-throughput sequencing revealed that this mutant kinase was mutually exclusive with known driver events [6, 7]. In TCGA's cohort, nine of 230 (3.9%) lung adenocarcinomas harbored MET exon 14 skipping mutation [6]. Seo JS, et al. reported three patients with MET exon 14 skipping mutation among a total of 200 patients, accounting for 1.5% in Asian populations [7]. Combined with our frequency of 1.6%, it seemed that Western populations had a slightly higher prevalence of the MET exon 14 skipping mutation than that of Asian populations. With the wide application of large-scale genome-sequencing technologies, many novel cancer-associated exon-skipping events have been identified [6, 7]. The copy number of known driver oncogenes, including MET, remains unchanged in most lung tumors [6, 7].
Garrett M.F. et al. demonstrated that NIH-3T3 cells stably expressed mutant MET showed anchorage-independent growth. This constitutive activity was sensitive to MET inhibitors in NIH-3T3 cells harboring MET exon 14 alterations [23]. It is interesting that five independent studies almost simultaneously reported the efficacy of MET inhibitors (crizotinib and capmatinib) for treating patients harboring MET exon 14 skipping [15–18, 23]. Of the five studies, 11 patients are evaluable for responses to the MET inhibitor. The response rate was 83.3% (nine of 12 lesions) and the median progression free survival was five months (range of two to 13 months). The antitumor activity shown in early phase clinical trials strongly supports MET exon 14 skipping as a bona fide target of the MET inhibitor. However, the responses to the MET inhibitor are quite heterogeneous. There still remains a substantial amount of tumor cells that are intrinsically resistant to the MET tyrosine kinase inhibitor. The molecular basis of the response heterogeneity is largely unknown, and no biomarker is available for predicting a response.
Remarkably, almost four-fifths of the patients with MET exon 14 skipping in our cohort had EGFR and/or HER2 gene copy number gains. Whether this observed consequence was a coincidence or an intrinsic connection remained unclear. One patient harboring MET exon 14 skipping simultaneously with EGFR, HER2 gene copy number gains received Iressa and maintained his disease for 12 months. It might be difficult to Figure out which alternation of genes played a more important role. Specific MET inhibitors should be an excellent choice according to the reported data; EGFR-TKIs, however may come to rescue as a replacement, especially when patients have had EGFR gene alternations. Since these three genes are essential in cell signaling, affecting various kinds of activities in cells such as survival, proliferation and invasion, completely understanding of the interaction of these genes is a necessity, and further experiments are urgently demanded. Future possible therapeutic strategy may hide in this network by double or triple targeting these molecules.
In summary, MET exon 14 skipping defined a new molecular subset of NSCLC with identifiable clinical characteristics. These patients are typically older, female, and non-smokers who might benefit from MET targeted therapy. Most of the lung tumors with MET exon 14 skipping had EGFR and/or HER2 gene copy number gains. Additionally, the therapeutic EGFR inhibitors might be an alternative treatment for patients with MET mutant NSCLC.
MATERIALS AND METHODS
Patients and tissues
From October 2007 to June 2013, a total of 1770 frozen surgically resected NSCLC tumor tissues, including 1305 lung adenocarcinomas, 48 adenosquamous carcinoma and 417 squamous cell carcinomas, were prospectively collected in the Department of Thoracic Surgery of Fudan University Shanghai Cancer Center. This study was approved by the Institutional Review Board of the Fudan University Shanghai Cancer Center. All patients provided written informed consent. Two pathologists (Y.L. and X.X.S.) confirmed the diagnosis by H&E staining and verified by immunohistochemistry as TTF1, P63, Nepsin A and CK5/6. DNA and RNA were collected as previously reported [19, 20].
Mutational analyses
As previously reported [5, 19–21], mutational status of EGFR, KRAS, HER2, BRAF, CTNNB1, PIK3CA, ALK, RET, ROS1, NRG1, FGFR1, FGFR2, and FGFR3 was detected. MET (exon 13 to 21) was PCR amplified using cDNA for direct sequencing and exon 14 deleted cases were verified by sequencing of the PCR product of MET exon 13 to 15. Primers were shown in Supplementary Table S1. Quantitative Real-Time PCR was used to detect MET gene copy number gain as previously reported [22].
Immunohistochemistry (IHC)
Standard 5-μm formalin-fixed paraffin-embedded sections were subjected to IHC analysis using anti-MET (Cell Signaling Technology), P40 (ΔP63, Fuzhou Maxim Biotech, Ltd. China), and TTF-1 (clone: SPT24, Ready- to-use antibody, Fuzhou Maxim Biotech, Ltd. China) antibodies.
Clinicopathological characteristics
Clinical and pathologic data were prospectively collected for analyses, including age at diagnosis, sex, smoking history, histologic type, pathologic TNM stage, tumor size, and tumor differentiation. Patients were observed in clinic or by telephone for disease recurrence and survival from the date of diagnosis. Of the 1770 NSCLC patients, 56 patients who were incidentally found to have pleural metastasis were excluded from survival analysis. Seventy-nine patients of loss to follow-up were also excluded. A total of 1635 NSCLC patients were included for analysis with a median follow-up time of 32.83 months.
Statistical analysis
Associations between mutations and clinical and pathologic characteristics were analyzed by the χ2 test or Fisher's exact test. Survival curves were estimated using the Kaplan-Meier method, with differences in survival assessed by the log-rank test. Data were analyzed using Statistical Package for the Social Sciences Version 19.0 Software (SPSS Inc.). The two-sided significance level was set at p < 0.05.
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
We thank Dr. Matthew Meyerson and Dr. William Pao for valuable advice regarding this study.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FINANCIAL SUPPORT
The authors have reported to Oncotarget that no potential conflicts of interest exist with any companies/organizations whose products or services may be discussed in this article.
RESEARCH SUPPORT
This work was supported by the National Natural Science Foundation of China (81330056, 81401886, 81401891, 81422029, 81472173 and 81372525), the Key Project of Science and Technology Commission of Shanghai Municipality (JGGG1302), Wu Jieping Medical Foundation (320.6750.14317) and Shen-Kang Center Project (SKMB1201), Shanghai rising star program (16QA1403500).
Role of the sponsors
The sponsors had no role in the design of the study, the collection and analysis of the data, or the preparation of the manuscript.
REFERENCES
- 1.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–80. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O'Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 4.Li H, Pan Y, Li Y, Li C, Wang R, Hu H, Zhang Y, Ye T, Wang L, Shen L, Sun Y, Chen H. Frequency of well-identified oncogenic driver mutations in lung adenocarcinoma of smokers varies with histological subtypes and graduated smoking dose. Lung Cancer. 2013;79:8–13. doi: 10.1016/j.lungcan.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 5.Pan Y, Wang R, Ye T, Li C, Hu H, Yu Y, Zhang Y, Wang L, Luo X, Li H, Li Y, Shen L, Sun Y, et al. Comprehensive Analysis of Oncogenic Mutations in Lung Squamous Cell Carcinoma with Minor Glandular Component. Chest. 2014;145:473–9. doi: 10.1378/chest.12-2679. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, Yu SB, Kim J, Lee ER, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–19. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M, Toschi L, Finocchiaro G, Destro A, et al. Increased MET Gene Copy Number Negatively Affects Survival of Surgically Resected Non–Small-Cell Lung Cancer Patients. J Clin Oncol. 2009;27:1667–74. doi: 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 10.Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, Gibbs JB, Pan BS. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007;67:2081–8. doi: 10.1158/0008-5472.CAN-06-3495. [DOI] [PubMed] [Google Scholar]
- 11.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 12.Camidge DR, Ou SH, Shapiro G, Otterson GA, Villaruz LC, Villalona-Calero MA, Iafrate J, Varella-Garcia M, Dacic S, Cardarella S, Zhao W, Tye L, Stephenson P, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC) J Clin Oncol. 2014;32:5s. [Google Scholar]
- 13.Spigel DR, Edelman MJ, O'Byrne k, Paz-Ares L, Shames DS, Yu W, Paton VE, Mok T. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial. J Clin Oncol. 2014;32:5s. doi: 10.1200/JCO.2016.69.2160. [DOI] [PubMed] [Google Scholar]
- 14.Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, Holcomb T, Pujara K, Stinson J, Fu L, Severin C, Rangell L, Schwall R, et al. Somatic Mutations Lead to an Oncogenic Deletion of Met in Lung Cancer. Cancer Res. 2006;66:283–289. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 15.Jenkins RW, Oxnard GR, Elkin S, Sullivan EK, Carter JL, Barbie DA. Response to Crizotinib in a Patient With Lung Adenocarcinoma Harboring a MET Splice Site Mutation. Clin Lung Cancer. 2015;16:e101–4. doi: 10.1016/j.cllc.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waqar SN1, Morgensztern D, Sehn J. MET Mutation Associated with Responsiveness to Crizotinib. J Thorac Oncol. 2015;10:e29–31. doi: 10.1097/JTO.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendenhall MA, Goldman JW. MET-Mutated NSCLC with Major Response to Crizotinib. J Thorac Oncol. 2015;10:e33–4. doi: 10.1097/JTO.0000000000000491. [DOI] [PubMed] [Google Scholar]
- 18.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, Borsu L, Schultz N, Berger MF, Rudin CM, Ladanyi M. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015 doi: 10.1158/2159-8290.CD-14-1467. pii: CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y, Ren Y, Fang Z, Li C, Fang R, Gao B, Han X, Tian W, Pao W, Chen H, Ji H. Lung Adenocarcinoma From East Asian Never-Smokers Is a Disease Largely Defined by Targetable Oncogenic Mutant Kinases. J Clin Oncol. 2010;28:4616–20. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R, Wang L, Li Y, Hu H, Shen L, Shen X, Pan Y, Ye T, Zhang Y, Luo X, Zhang Y, Pan B, Li B, et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer. Clin Cancer Res. 2014;20:4107–14. doi: 10.1158/1078-0432.CCR-14-0284. [DOI] [PubMed] [Google Scholar]
- 21.Wang R, Pan Y, Li C, Zhang H, Garfield D, Li Y, Ye T, Hu H, Luo X, Li H, Zhang Y, Zhang J, Zhou X, et al. Analysis of major known driver mutations and prognosis in resected adenosquamous lung carcinomas. J Thorac Oncol. 2014;9:760–8. doi: 10.1097/JTO.0b013e3182a406d1. [DOI] [PubMed] [Google Scholar]
- 22.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill JA, Lee C, Jentz D, Hoover R, Ou SH, Salgia R, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:842–9. doi: 10.1158/2159-8290.CD-15-0285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.