

Abstract
Microarray analysis revealed genes of the posterior HOXD locus normally involved in bone formation to be over-expressed in primary Ewing sarcoma (ES). The expression of posterior HOXD genes was not influenced via ES pathognomonic EWS/ETS translocations. However, knock down of the dickkopf WNT signaling pathway inhibitor 2 (DKK2) resulted in a significant suppression of HOXD10, HOXD11 and HOXD13 while over-expression of DKK2 and stimulation with factors of the WNT signaling pathway such as WNT3a, WNT5a or WNT11 increased their expression. RNA interference demonstrated that individual HOXD genes promoted chondrogenic differentiation potential, and enhanced expression of the bone-associated gene RUNX2. Furthermore, HOXD genes increased the level of the osteoblast- and osteoclast-specific genes, osteocalcin (BGLAP) and platelet-derived growth factor beta polypeptide (PDGFB), and may further regulate endochondral bone development via induction of parathyroid hormone-like hormone (PTHLH). Additionally, HOXD11 and HOXD13 promoted contact independent growth of ES, while in vitro invasiveness of ES lines was enhanced by all 3 HOXD genes investigated and seemed mediated via matrix metallopeptidase 1 (MMP1). Consequently, knock down of HOXD11 or HOXD13 significantly suppressed lung metastasis in a xeno-transplant model in immune deficient mice, providing overall evidence that posterior HOXD genes promote clonogenicity and metastatic potential of ES.
Keywords: HOXD, metastasis, Ewing sarcoma, WNT signaling, endochondral development
INTRODUCTION
Ewing sarcomas (ES) are bone or soft tissue tumors with a prominent stemness phenotype, mostly occurring in children and adolescents. These highly malignant sarcomas frequently arise in diaphysal bones possibly descending from a neuroectodermal or mesenchymal stem cell in transition from an undifferentiated state to a more differentiated phenotype of the chondro-osseous lineage [1–6]. Genetically, ES are defined by EWS/ETS translocations [1, 7, 8].
In the clinical setting, prognosis for patients with metastatic ES at diagnosis is clearly worse than for those without metastases [9]. Especially the development of metastases in bones is a catastrophic event in the clinical course of ES patients [10, 11].
Recently, we demonstrated the pro-metastatic gene dickkopf WNT signaling pathway inhibitor 2 (DKK2) to be critical for malignant growth of ES [5]. It can act as either an agonist or antagonist of WNT/β-catenin signaling, depending on the cellular context and the presence of the co-factor Kremen 2 [12–14]. In ES Kremen 2 is absent and DKK2 stimulates canonical β-catenin signaling. Further, DKK2 promotes bone infiltration and osteolysis in vivo and subsequent analyses defined DKK2 as a key factor in osteotropic malignancy [5].
Our subsequent analysis revealed several genes of the HOXD cluster to be over-expressed in ES. Class I homeobox genes (HOX) are transcription factors known to be involved in embryonic development and body segmentation [15]. In mammals they are organized in 4 different chromosomal loci (HOXA at 7p15.3, HOXB at 17q21.3, HOXC at 12q13.3 and HOXD at 2q31) comprising 39 genes that can be aligned with each other into 13 antero-posterior paralogous groups [16]. HOX genes are also expressed in adult human organs [17] where they appear to regulate cell identity [18], cell differentiation [19, 20], including metabolic processes [21]. In addition, posterior HOXD genes such as HOXD11, HOXD12 and HOXD13 were shown to not only regulate patterning but also to directly influence bone formation and the ossification pattern of bones. In part this effect is mediated via runt-related transcription factor 2 (RUNX2) [22]. HOX genes are further implicated in neoplastic transformation resulting in leukemia [23] as well as solid cancers derived from various organs [24, 25].
Here we demonstrate posterior HOXD genes such as HOXD10, HOXD11 and HOXD13 are significantly up-regulated in ES. We show that inhibition of DKK2 expression significantly suppresses the expression of HOXD10, HOXD11 and HOXD13 while over-expression of DKK2 and factors stimulating the WNT signaling pathway such as WNT3a, WNT5a or WNT11 further increased their expression. RNA interference of genes of the posterior HOXD locus revealed individual HOXD genes to be important for chondrogenic differentiation potential. In contrast, osteogenic differentiation was not impacted by HOXD gene loss of function but the expression of bone-associated genes such as RUNX2 was enhanced. Finally, HOXD11 and HOXD13 further promote metastatic growth and invasiveness that seemed to be mediated via matrix metallopeptidase 1 (MMP1).
RESULTS
Posterior HOXD genes are over-expressed in Ewing sarcoma (ES)
Microarray analysis disclosed genes of the posterior HOXD locus to be clearly over-expressed in primary ES. Interestingly, only HOXD10, HOXD11 and HOXD13 of the homeobox loci, normally involved in bone formation and ossification pattern of bones [22, 26], were significantly up-regulated in ES in comparison to neuroblastoma, normal and fetal tissue (Figure 1A). They were within the 50 most up-regulated genes with the strongest over-expression in ES compared to normal tissue (see Supplementary Material, Table S1). Extended analysis of other sarcoma and carcinoma on publically available expression data revealed that only fibrosarcoma demonstrated a similar up-regulation of genes of the posterior HOXD locus (see Supplementary Material, Figure S1A, S1B). Furthermore, qRT-PCR confirmed a significantly lower expression of HOXD10, HOXD11 and HOXD13 in neuroblastoma and osteosarcoma (see Supplementary Material, Figure S2A).
Figure 1. HOXD gene expression and regulation in ES.
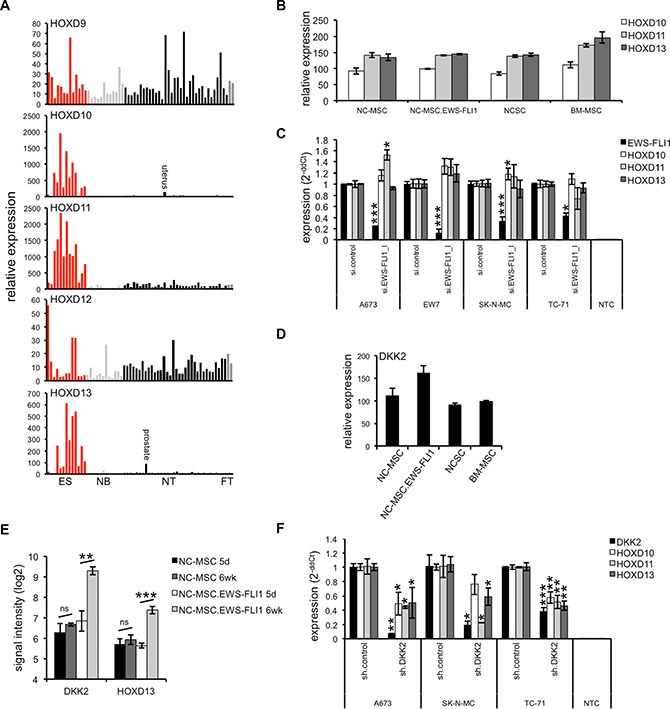
(A) Expression profile of HOXD9 – HOXD13 in ES (red) in comparison to neuroblastoma (NB; light gray), normal (NT; black) and fetal tissue (FT; dark gray). ES and NB RNA were hybridized onto HG U133A arrays (Affymetrix; GSE1825, GSE15757; [50]) and compared to a published microarray study of normal tissue (GSE2361). Each bar represents the expression signal of an individual array. (B) HOXD genes seem not induced after over-expression of EWS-FLI1 in mesenchymal and neural crest stem cells. NC-MSC: control vector transduced neural crest-derived MSC after 5 days in self-renewal media, NC-MSC.EWS-FLI1: EWS-FLI1 transduced NC-MSC after 5 days in self-renewal media, NCSC: undifferentiated, freshly isolated neural crest stem cells, BM-MSC: undifferentiated adult bone marrow derived MSC. (C) Expression of HOXD genes is not affected after suppression of EWS-FLI1 in four different ES lines using RNA interference as measured by qRT-PCR. Data are mean ± SEM; t-test. (D) Expression of DKK2 in undifferentiated stem cell populations (NC-MSC, NC-MSC.EWS-FLI1, NCSC and BM-MSC; from GEO dataset GEO21511, CEL files were RMA normalized by use of expression console software, Affymetrix). (E) Expression of DKK2 and HOXD13 in NC-MSC cells following exposure to differentiation conditions. NC-MSC transduced with GFP-only (NC-MSC) or EWS-FLI1-GFP (NC-MSC.EWS-FLI1) lentiviral vectors were passaged for 5 days in self-renewal media (5 d) and then transferred to differentiation media for 6 weeks (6 wk). Gene expression profiling studies of triplicate samples reveals that exposure to differentiation conditions resulted in up-regulation of DKK2 and HOXD13 in EWS-FLI1+ cells. (F) Suppression of DKK2 by specific shRNA [5] in different ES lines results in a significantly down-regulation of HOXD10, HOXD11 and HOXD13 as measured by qRT-PCR. Data are mean ± SEM; t-test.
To investigate the putative origin of this increased HOXD gene expression in ES we analyzed public array data of neuroectodermal or mesenchymal stem cells [27] presumed originating for ES. First we recognized a low level of expression of HOXD10, HOXD11 and HOXD13 in neural crest-derived mesenchymal stem cells (NC-MSC) as well as in undifferentiated, freshly isolated neural crest stem cells (NCSC) or adult bone marrow derived MSC (BM-MSC). Interestingly, HOXD genes seem not to be further increased after transduction of NC-MSC with EWS-FLI1 (Figure 1B). Neither over-expression of EWS-FLI1 in MSC lines (see Supplementary Material, Figure S2B) nor its knock down in ES lines by specific siRNA (Figure 1C, see Supplementary Material, Figure S2C) [28] did significantly influence posterior HOXD gene expression. Furthermore, enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) containing PRC2 complex, often involved regulating broad regions of the HOX locus during development and in adult tissue [29, 30], does not influence the expression of HOXD10, HOXD11 or HOXD13 in ES (see Supplementary Material, Figure S2D). These observations complement previous results demonstrating absence of H3K27me3 across the HOXD cluster in ES [31].
HOXD genes are stimulated via DKK2
Increasing evidence indicates that developmental canonical WNT/β-catenin signaling and HOX gene expression may interact: e.g. there are results demonstrating WNT-dependent regulation of HOXB8 in zebrafish during lateral line cell migration [32] or a HOXD13 dependent expression of WNT5a for the regulation of cell polarity in the cartilage growth plate [33]. Recently, we demonstrated DKK2 to be an agonist of the WNT/β-catenin pathway in ES [5]. Here we analyzed whether DKK2 may impact on the expression of posterior HOXD genes in ES.
Surprisingly, we first observed an increased DKK2 expression after acute up-regulation of EWS-FLI1 in NC-MSC (Figure 1D). Subsequent expression profiling studies of control and EWS-FLI1+ NC-MSC demonstrated that exposure to differentiation conditions for 6 weeks resulted in an EWS-FLI1 dependent up-regulation of DKK2 and HOXD13 in differentiated NC-MSC (Figure 1E, see Supplementary Material, Figure S2E). Consistent with these results, suppression of DKK2 by specific shRNA [5] in different ES lines resulted in a significant down-regulation of HOXD10, HOXD11 and HOXD13 (Figure 1F). Further, down-regulation of TCF4, a transcription factor in the canonical WNT/β-catenin pathway, resulted in a partial inhibition of posterior HOXD gene expression (see Supplementary Material, Figure S3A). Thus, the WNT signaling pathway seems to be generally active in ES: (i) over-expression of DKK2 in ES cell lines as well as in the NB cell line SH-SY5Y (Figure 2A) increased posterior HOXD gene expression; (ii) treatment of different ES lines with WNT3a, WNT5a or WNT11 or combined resulted in a further induction of HOXD10, HOXD11 or HOXD13 as well as of the WNT/β-catenin target gene LEF1 (Figure 2B). This induction was independent of the level of endogenous WNT expression in ES cell lines (Figure 2C and [14]).
Figure 2. DKK2 increases HOXD gene expression through WNT signaling pathway.
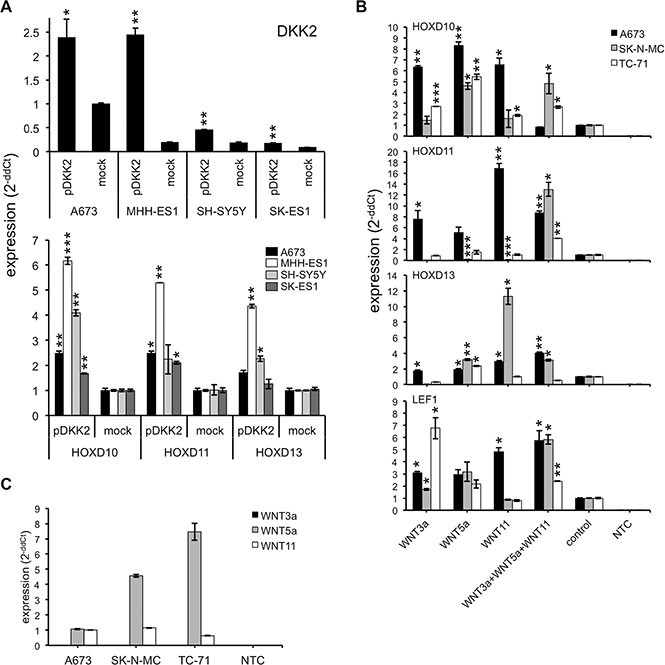
(A) Increased HOXD10, HOXD11 or HOXD13 expression (bottom) after transfection with cDNA encoding human DKK2 (top) in ES lines A673, MHH-ES1, SK-ES1 or NB line SH-SY5Y, respectively. Results of qRT-PCRs are shown. Data are mean ± SEM; t-test. (B) Analysis of HOXD gene expression in A673, SK-N-MC and TC-71 cells after incubation with recombinant human WNT3a, WNT5a or WNT11 and the combination of all three ligands. RNA was isolated after 1, 3, 6 and 12 h and the time point with the highest increase was shown. Data are mean ± SEM; t-test. (C) Expression of endogenous WNT3a, WNT5a and WNT11 mRNA in three ES cell lines (A673, SK-N-MC and TC-71) analyzed by qRT-PCR. Data are mean ± SEM; t-test.
Posterior HOXD genes contribute to chondrogenic as well as bone associated gene expression in ES
ES are bone or soft tissue neoplasms with a prominent immature stemness phenotype maintained by epigenetic repressors BMI1 and EZH2 [27, 28]. Further, posterior HOXD genes regulated by EZH2 during development are known to influence the ossification pattern of bones [22], so it seemed relevant to investigate whether the expression of posterior HOXD genes in ES may influence their differentiation capacity.
First, we investigated whether posterior HOXD genes may affect chondrogenic differentiation associated genes by incubating ES lines stably infected with HOXD10, HOXD11 or HOXD13 shRNA containing retroviruses (see Supplementary Material, Figure S3B) with specific differentiation media. The differentiation potential was analyzed using established marker genes, COL10A1 (collagen, type X, alpha-1), IHH (indian hedgehog) and SOX9 (SRY-box 9) for chondrogenic differentiation [34]. As shown in Figure 3A, early chondrogenic differentiation potential was impaired in SK-N-MC cells after HOXD10 and HOXD11 knock down as demonstrated by the decrease of IHH and SOX9 induction. In contrast, late chondrogenic differentiation as evaluated by COL10A1 seemed unaffected. Results for HOXD10 were reproducibly observed after knock down in A673 cells (see Supplementary Material, Figure S4A), while HOXD13 suppression seemed to support SOX9 expression during chondrogenic differentiation of SK-N-MC cells (Figure 3A), but these results were not reproducible in A673 cells (see Supplementary Material, Figure S4A). However, posterior HOXD genes seemed not to affect osteogenic differentiation, since no significant differences in Alizarin Red S staining (see Supplementary Material, Figure S4B) or osteogenic marker gene expression were observed (see Supplementary Material, Figure S4C).
Figure 3. Posterior HOXD genes promote chondrogenic differentiation and expression of bone associated genes.
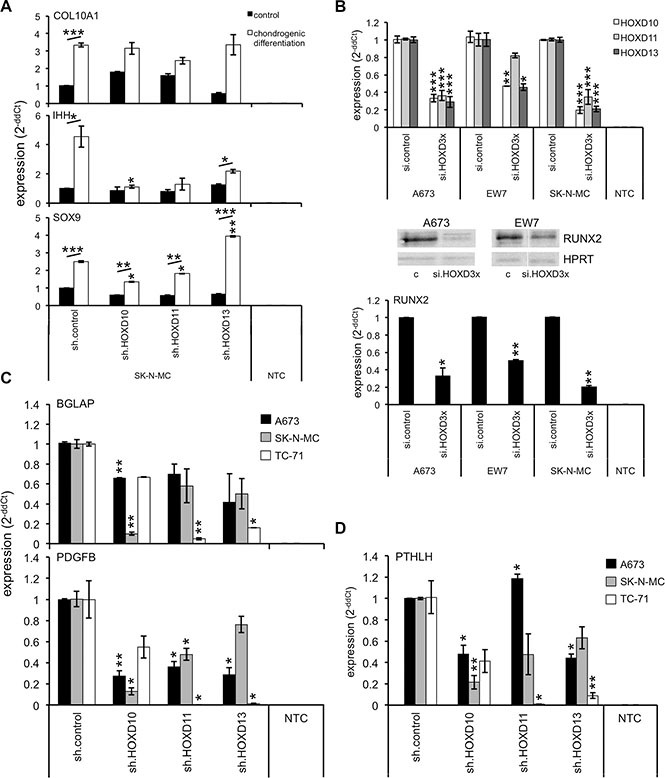
(A) Chondrogenic differentiation potential of ES lines with specific shRNA constructs was shown by the expression of specific chondrogenic marker genes COL10A1, IHH and SOX9 using qRT-PCR. Data are mean ± SEM; t-test. (B) Analysis of RUNX2 expression in A673, EW7 and SK-N-MC cells after transient combined HOXD knock down with specific siRNAs against HOXD10, HOXD11 and HOXD13 using qRT-PCR. Data are mean ± SEM; t-test. Middle panel western-blot of RUNX2 expression after triple HOXD knock down compared to respective controls (si.control: c). (C) mRNA analysis of BGLAP and PDGFB expression in 3 different ES lines after inhibition of HOXD10, HOXD11 or HOXD13 expression with specific shRNA. Data are mean ± SEM; t-test. (D) Expression analysis of PTHLH in ES after knock down of individual HOXD genes by qRT-PCR. Data are mean ± SEM; t-test.
Posterior HOXD genes are known to affect RUNX2 expression during ossification [22] and may do so via direct interaction of individual posterior HOXD genes [35]. In fact, when we analyzed individual ES lines after a triple knock down of HOXD10, HOXD11 and HOXD13 (Figure 3B, top) we reproducibly observed a clear inhibition of RUNX2 expression under normal culture conditions (Figure 3B, bottom). Interestingly, RUNX2 expression was not consistently affected after single knock down of each HOXD gene (data not shown) even after incubation in osteogenic differentiation media (see Supplementary Material, Figure S4C). But, posterior HOXD genes clearly promoted the expression of additional genes important for ossification or endochondral bone development in ES such as osteoblast-specific gene osteocalcin (bone gamma-carboxyglutamate (gla) protein, BGLAP), the pre-/osteoclast specific factor platelet-derived growth factor beta polypeptide (PDGFB) [36] (Figure 3C) or induction of osteolytic ES growth typical PTHLH [5] (Figure 3D), respectively.
HOXD genes enhance TRAP+ osteoclasts in osteotropic tumor growth
DKK2 was previously identified to be a critical mediator of osteolytic tumor growth in ES [5]. Here, we asked whether the effect of DKK2 is mediated via the activity of posterior HOXD genes and if these genes may influence bone invasion and osteolysis in vivo, too. We injected A673 cells stable infected with HOXD10, HOXD11 and HOXD13 shRNA into the tibiae of immunodeficient Rag2−/−γC−/− mice and analyzed bone infiltration and destruction by X-ray radiography and histology. In contrast to DKK2, stable knock down of HOXD genes did not significantly influence bone invasiveness, although suppression of HOXD10, HOXD11 and HOXD13 seemed to slightly reduce the invasive growth potential of tumor cells in the bone marrow (Figure 4A and 4B). Especially after injection of constitutive A673 sh.HOXD10 and sh.HOXD11 transfectants, the amount of tumor cells in the bone marrow was clearly reduced (Figure 4A). Furthermore, we observed no considerable differences in osteolytic bone destruction after suppression of several posterior HOXD genes, as measured by the quantification of TRAP+-stained osteoclasts in the bone. Interestingly, HOXD10, HOXD11 and HOXD13 knock down significantly decreased the number of TRAP+ cells within tumor tissue while their number in bones was not affected (Figure 4C and 4D).
Figure 4. HOXD genes enhance the amount of TRAP+ osteoclasts in the tumor.
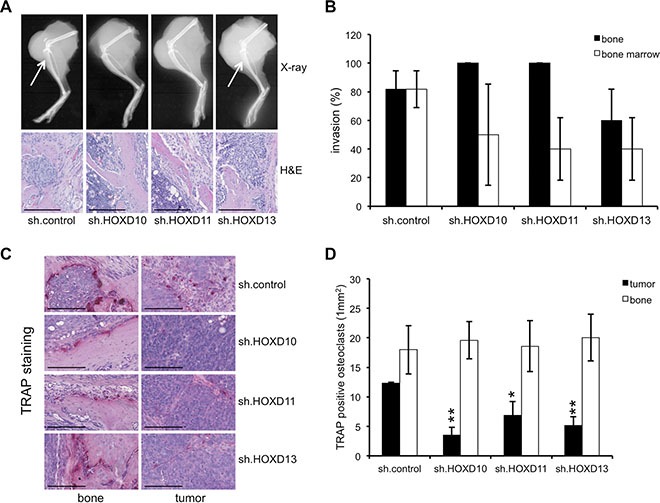
Analysis of bone invasiveness and osteolysis of constitutive A673 HOXD10, HOXD11 and HOXD13 shRNA infectants and negative controls in an orthotopic bone xeno-transplantation model (5–11 mice/group). Affected bones were assessed by X-ray radiography and histology. (A) Representative pictures of X-ray radiography and H&E staining showing no clear differences in bone invasiveness between A673 cells with constitutive HOXD10, HOXD11 or HOXD13 knock down and respective controls (X-ray, H&E, scale bar 0.25 mm). (B) Percentage of mice exhibiting infiltration of cortical bone or bone marrow infiltration after intra-tibial injection. (C) TRAP staining of osteoclasts for better visualization and to quantify osteolysis in bone and tumor tissues (scale bar 0.15 mm). (D) Average number of TRAP+ osteoclasts attached to the bone or in the tumor (1 mm2) was determined in at least three tumor samples/group (In each sample not less than 20 segments were analyzed).
Subsequent expression analysis of key players associated with preparing the pre-metastatic niche, homing and invasion to bone as well as the osteolytic growth potential [37] demonstrated in addition to PTHLH (Figure 3D) only reduced gene expression of interleukin 6 (IL6) after HOXD10 knock down (see Supplementary Material, Figure S5A). In contrast, hypoxia-inducible factor 1, alpha subunit (HIF1α) was reproducibly up-regulated after suppression of HOXD13 in different ES lines (see Supplementary Material, Figure S5A).
HOXD genes promote ES growth and invasiveness
To further elucidate the possible contribution of posterior HOXD genes to phenotype and tumor pathology of ES we analyzed contact dependent growth of HOXD10, HOXD11 and HOXD13 shRNA infectants with an impedance-based system. As shown in Figure 5A, we observed a significant inhibition of proliferation after suppression of HOXD13 expression in A673 and SK-N-MC cells, whereas no significant, reproducible contribution of HOXD10 and HOXD11 in this assay was detected. In colony forming assay in methylcellulose a strong reduction of colony formation after HOXD13 knock down, but also a significant contribution of HOXD11 to contact independent growth was observed (Figure 5B).
Figure 5. HOXD10, HOXD11 and HOXD13 promote growth and invasiveness of ES.
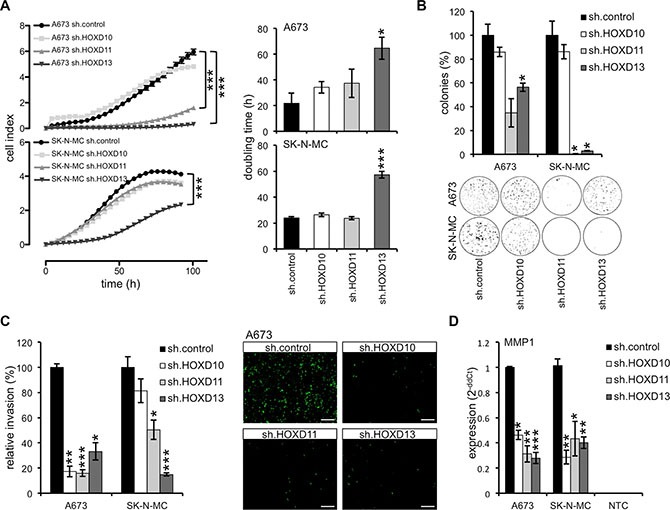
(A) Left, analysis of proliferation of constitutively infected ES lines with xCELLigence (hexaplicates/group). Cellular impedance was measured every 4 hours (relative cell index). Right, doubling time of constitutive A673 and SK-N-MC HOXD10, HOXD11 and HOXD13 shRNA infectants and respective controls (sh.control). Data are mean ± SEM of two independent experiments/cell line (hexaplicates/group). (B) Anchorage-independent colony formation in methylcellulose of ES lines with stable HOXD10, HOXD11 and HOXD13 knock down. Upper panel, data are mean ± SEM of two independent experiments (duplicates/group). Lower panel, macrographs show two representative experiments with A673 and SK-N-MC. (C) Analysis of invasiveness of ES lines through Matrigel after transfection with specific HOXD10, HOXD11 or HOXD13 shRNA constructs. Left, data are mean ± SEM of three independent experiments. Right, invasive A673 cells are shown after 48 hours incubation (scale bar 500 μm). (D) Expression of MMP1 mRNA after HOXD knock down (sh.HOXD10, sh.HOXD11 and sh.HOXD13) using qRT-PCR. Data are mean ± SEM; t-test.
To assess phenotypic markers associated with malignancy, we asked whether posterior HOXD genes may influence in vitro invasiveness, too. To do this, ES lines were analyzed in vitro on BioCoat invasion plates and cells that invaded into the Matrigel and migrated to the other side of the membrane were monitored 48 hours later. As shown in Figure 5C, A673 and SK-N-MC HOXD10, HOXD11 and HOXD13 shRNA infectants revealed a significant contribution of at least HOXD11 and HOXD13 to in vitro invasiveness. Recent results of our laboratory already indicated a strong contribution of MMP1 to invasion and metastasis of ES and its possible induction via several, presumably independent pathways [5, 38, 39]. Subsequent analysis of MMP1 expression in these HOXD shRNA infectants demonstrated a strong induction of MMP1 expression by all three HOXD genes (Figure 5D, see Supplementary Material, Figure S5B) signifying that the reduced invasive potential of HOXD11 or HOXD13 and probably HOXD10 silenced ES cells may be mediated at least in part via MMP1. In contrast to MMP1, mRNA expression of other matrix metallopeptidases (namely MMP7 or MMP9) were not affected after constitutive knock down of different HOXD genes (see Supplementary Material, Figure S5C).
HOXD11 and HOXD13 promote metastatic spread in vivo
Since HOXD11 and HOXD13 contributed to ES in vitro invasiveness we finally asked whether posterior HOXD genes may promote metastatic potential of ES in vivo. Thus, we injected stable HOXD10, HOXD11 and HOXD13 shRNA infected A673 cells and appropriate controls into the tail vein of Rag2−/−γC−/− mice. As shown in Figure 6A left, A673 control infectants grew to numerous confluent and necrotic tumor nodules within the lung, while A673 sh.HOXD11 and A673 sh.HOXD13 infectants revealed a significantly reduced metastatic phenotype that was statistically significant (Figure 6A, right). HOXD10 shRNA infectants formed lung tumors indistinguishable from control infectants with increased appearance of tumor nodules within the liver (Figure 6A, right). After serial sectioning, we observed that A673 sh.HOXD10 and sh.control tumor nodules in lungs were bigger with a higher amount of necrosis than the tumor nodules after injection of A673 cells with stable HOXD11 and HOXD13 knock down. These results were confirmed with SK-N-MC cells (Figure 6B): shRNA-mediated knock down of HOXD10, HOXD11 or HOXD13, respectively, resulted in a strong inhibition of lung metastasis (Figure 6B), while there was some unspecific increase of liver metastasis in this mouse model. This overall indicates that especially HOXD11 and HOXD13 are important for lung metastatic potential in ES.
Figure 6. HOXD11 and HOXD13 promote lung metastasis in ES.
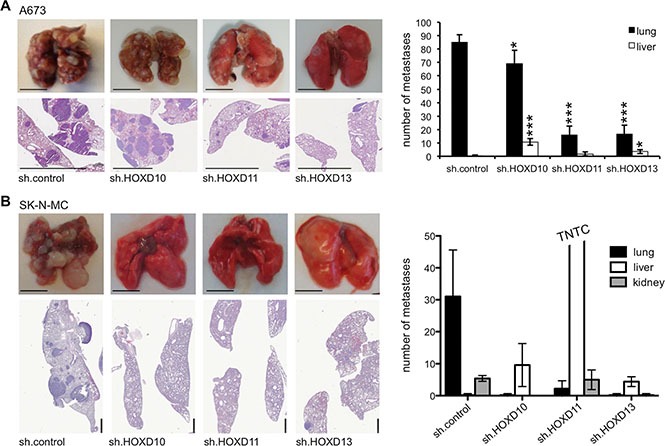
(A) In vivo analysis of the metastatic potential of A673 cells constitutively transfected with sh.control, sh.HOXD10, sh.HOXD11 and sh.HOXD13 in Rag2−/−γC−/− mice (5 mice/group). Affected organs were photographed and analyzed by histology. Left, representative pictures of whole organs and H&E staining sections are shown (scale bar 5 mm). Right, average number of apparent metastases in lung and liver tissues is plotted. (B) Similar experiments were carried out with SK-N-MC cells constitutively transfected with sh.control, sh.HOXD10, sh.HOXD11 and sh.HOXD13 in immune deficient Rag2−/−γC−/− mice (5 mice/group). Here knock down of HOXD10 also inhibited lung metastasis (scale bar 5 or 1mm).
DISCUSSION
Ewing sarcoma (ES) a bone and soft tissue malignancy is characterized by early metastasis to lung and bone [1]. Here we observed genes such as HOXD10, HOXD11 and HOXD13 normally involved in bone formation and ossification pattern of bones [22], to be significantly over-expressed in ES. Other genes of the posterior HOXD locus including HOXD9 and HOXD12 where not uniquely up-regulated in primary ES compared to other tumors.
In search for factors presumably involved in their increased expression in ES we investigated putative ES originating cells such as neural crest or mesenchymal stem cells [1–3]. Though, genes of the HOXD locus were already expressed in mesenchymal or neural crest derived cells (Figure 1B), they were not increased early after up-regulation of EWS-FLI1 in such stem cells nor did EWS-FLI1 knock down in ES lines influence HOXD10, HOXD11 or HOXD13 expression in vitro.
Remarkably, we identified HOXD10, HOXD11 and HOXD13 to be potential downstream targets of DKK2 using RNA interference analysis (Figure 1F). We previously demonstrated DKK2 to be an agonist of the canonical WNT/β-catenin pathway and critical mediator of osteolytic tumor growth in ES [5]. Here, analysis of mesenchymal and neuroectodermal stem cells demonstrated an up-regulation of DKK2 in EWS-FLI1 expressing neuroectodermal stem cells [27]. Long-term exposure in differentiation media resulted in a significant further up-regulation of DKK2 and HOXD13 in EWS-FLI1+ stem cells. DKK2 dependency of posterior HOXD gene expression persisted in ES lines and was further increased after transfection of DKK2 encoding cDNA into ES. In addition, treatment of ES lines with ligands of the canonical WNT/β-catenin signaling pathway resulted in an additional up-regulation of posterior HOXD genes as well as of the WNT/β-catenin target gene LEF1. This overall indicates a biologically active WNT signaling pathway in ES involved in posterior HOXD gene induction and expression.
Previous results already demonstrated agonists of WNT/β-catenin signaling to support skeletogenesis [40] or to play a role in terminal osteoblast differentiation into mineralized bone matrices [41]. HOXD11, HOXD12 and HOXD13 were shown to not only regulate patterning but also to directly influence bone formation and the ossification pattern of bones [22], with HOXD13 presumably being a master regulator of autopod skeletal morphogenesis [26]. In line with these results we observed early chondrogenic differentiation potential to be significantly impaired after HOXD10 and HOXD11 knock down as demonstrated by the decrease of IHH and SOX9 induction. Furthermore, posterior HOXD gene expression similarly stimulated RUNX2 expression, and enhanced the expression of osteoblast-specific osteocalcin (BGLAP), pre-/osteoclast specific PDGFB and PTHLH. The transcription factor RUNX2 binds to the oncogenic fusion protein EWS-FLI1 [42] and may promote osteogenic differentiation and development of perichondrial cells that differentiate directly into osteoblasts in diaphysal bones [43]. RUNX2 further affects cancer cell invasion and osteolysis [37, 44], which seems also true for ES as demonstrated by us in the context of DKK2 [5]. However, after suppression of posterior HOXD genes only a slight reduction of bone marrow invasiveness, but an obvious reduction of TRAP+ osteoclasts within tumor tissue was observed. This supports a critical role for posterior HOXD gene mediated regulation of RUNX2 and osteolytic capacity of ES itself, although the underlying, exact mechanisms are not understood [37]. PDGFB, although osteoclast-specific, has been shown to contribute to bone development [36]. Characteristic PTHLH expression was observed in osteochondrogenic progenitor cells highly susceptible for EWS-FLI1 mediated transformation [6]. These results overall suggest that posterior HOXD genes in ES mimic an immature endochondral developmental transcriptional program presumably important for bone associated tumor growth and metastasis.
The posterior HOXD gene-mediated expression program in ES not only affected the immature differential but similarly the invasive, metastatic phenotype of ES. Several HOX genes were previously implicated in neoplastic transformation resulting in leukemia [23] as well as solid cancers derived from various organs [24, 25]. Furthermore, the involvement of particular HOX genes such as HOXC13, HOXD3, HOXA1 in metastasis and invasiveness was recently demonstrated for melanoma, breast and prostate cancer, respectively [45–47].
While in ES HOXD11 and especially HOXD13 promoted in vitro proliferation and contact independent growth, in vitro invasiveness of ES lines was dependent on HOXD10, HOXD11 as well as HOXD13. The enhanced invasive potential seemed mediated at least in part via MMP1, since down-regulation of individual HOXD genes likewise resulted in a repressed MMP1 expression profile. MMP1 is well described as an AP1 target gene [48] and additional results of our laboratory demonstrated a STAT1 dependency of MMP1 expression in ES [38]. Furthermore, there is also evidence that MMP1 is a direct target of EWS/ETS proteins, as a previous study has shown that EWS-ETV1 (ER81) and EWS-FLI1 fusion proteins can interact with the MMP1 promoter and collaborate with c-Jun and the cofactor p300 to activate MMP1 gene transcription in vitro [49]: But in our hands, EWS-FLI1 knock down suppressed MMP1 expression only in A673 cells but not in other ES lines. Previous results of our laboratory already indicated a strong contribution of MMP1 to metastasis of ES and its possible induction via several, presumably independent pathways that so far was not understood [5, 38, 39]. In line with these observations, knock down of HOXD11 or HOXD13 significantly suppressed metastasis in a xeno-transplant model in immune deficient Rag2−/−γC−/− mice. So expression of posterior HOXD genes in ES may generate an open transcriptional platform for MMP1 induction, that may be activated by multiple pathways, sensing e.g. oxidative stress via STAT1 [38] or other factors of the microenvironment [39]. However, details of these pathway interplays have to be further investigated.
In summary, posterior HOXD genes over-expressed and presumably deregulated via DKK2 and the canonical WNT/β-catenin pathway in ES seem critical mediators of an immature endochondral program of ES mediating a transcriptional profile important for ES bone malignancy and its metastatic potential.
MATERIALS AND METHODS
Cell lines
Cells were maintained in a humidified incubator at 37°C in 5–8% CO2 atmosphere in RPMI 1640 or DMEM medium (both Invitrogen) containing 10% heat-inactivated fetal bovine serum (FBS, Biochrom) and 100μg/ml gentamicin (Invitrogen). Cell lines were checked routinely for purity (e.g. EWS-FLI1 translocation product, surface antigen or HLA-phenotype) and Mycoplasma contamination.
Expression profiling
For comparative gene expression analysis of individual HOXD genes, RNA of 13 individual ES and 12 NB were hybridized onto HG U133A arrays (Affymetrix; GSE1825, GSE15757; [50]) and compared to a published microarray study of 36 normal tissues (NT) (GSE2361) of diverse origin including tissues of normal heart, thymus, spleen, ovary, kidney, skeletal muscle, pancreas, prostate, small intestine, colon, placenta, bladder, breast, uterus, thyroid, skin, salivary gland, trachea, cerebellum, brain, fetal brain, adrenal gland, bone marrow, amygdala, caudate nucleus, corpus, hippocampus, thalamus, pituitary gland, spinal cord, testis, liver, stomach, lung, fetal lung and fetal liver. All datasets were analyzed by using Microarray Suite 5.0, and scaled to the same target intensity of 500. Whole genome expression profiling of undifferentiated human embryonic stem cell (hESC)-derived neural crest cells, with and without EWS-FLI1, and adult bone marrow derived mesenchymal stem cells was performed as previously described [27], and data were downloaded from GEO (GSE 21511). To evaluate gene expression in neural crest stem cells that had been exposed to differentiation conditions, control and EWS-FLI1-transduced NC-MSC were transferred to serum containing differentiation media (Data are available at GEO GSE68898).
RNA interference
For transient RNA interference cells were transfected with small interfering RNA (siRNA) as previously described [28]. siRNA sequences are provided in the supplementary data.
Constructs and retroviral gene transfer
The production of stable infectants was done as described by Richter et al. [28].
Quantitative RT-PCR
Differential gene expression was analyzed by qRT-PCR using TaqManTM Universal PCR Master Mix and fluorescence detection with an AB7300 Real-Time PCR System (both Life Technologies) as previously described [28]. A list of used assays is provided in the supplementary data. NTC: non template control.
Constructs and transfection
Human cDNA ORF clone of DKK2 (NM_014421; OriGene) was transfected via electroporation into human cell lines. Stable transfectants were isolated after selection in 600μg/ml G418 (Sigma Aldrich).
WNT ligand stimulation
To analyze a possible involvement of WNT signaling triggered by WNT ligands on HOXD expression, we incubated A673, SK-N-MC and TC-71 with two different concentrations of recombinant human WNT3a (5036-WN, 0.1μg/ml or 0.3μg/ml), WNT5a (645-WN, 0.1μg/ml or 0.3μg/ml) or WNT11 (6179-WN, 0.6μg/ml or 1.2mg/ml; all R&D Systems). Additionally, we analyzed each ligand in combination. After 1, 3, 6 and 12h RNA was isolated and mRNA expression levels of HOXD10, HOXD11, HOXD13 and the WNT target gene LEF1 were analyzed by qRT-PCR.
Differentiation assay
For testing of chondrogenic cell differentiation, cells were cultured in specific differentiation media (STEMPRO Chondrogenesis Differentiation Kit, Invitrogen) according to the manufacturer's instructions. To validate differentiation efficacy, expression of the well-known chondrogenic marker genes COL10A1, IHH and SOX9 was monitored by qRT-PCR [34].
Proliferation assay
Cell proliferation was measured according to the manufacturer with an impedance-based instrument system (xCELLigence, Roche) enabling label-free real time cell analysis.
Colony forming assay
Cells were seeded in duplicate into a 35mm plate at a density of 5×103 cells per 1.5ml methylcellulose-based media (R&D Systems) according to the manufacturer's instruction and cultured for 14 days at 37°C/5% CO2 in a humidified atmosphere.
In vitro invasion assay
To study cell invasion BioCoat™ Angiogenesis System: Endothelial Cell invasion was used (BD Biosciences) according to the manufacturer's instructions.
Western blot
Procedures were described previously [28]. Following antibodies were used: anti-RUNX2 (mouse monoclonal, clone AS110, 05-1478 Millipore). Equal protein loading was controlled with rabbit polyclonal to HPRT antibodies (1:500; sc20975; Santa Cruz Biotechnology).
Mice
Immune deficient Rag2−/−γC−/− mice on a BALB/c background were obtained from the Central Institute for Experimental Animals (Kawasaki, Japan) and maintained in our animal facility under pathogen-free conditions in accordance with the institutional guidelines and approval by local authorities. Experiments were performed in 6-16 week old mice.
In vivo experiments
For the analysis of in vivo metastatic potential 2×106 ES cells were injected in a volume of 0.2ml into the tail vein of immunodeficient Rag2−/−γC−/− mice. Five weeks later mice were sacrificed and metastatic spread was monitored in individual organs. To examine bone invasiveness and osteolysis, mice were anesthetized with 500mg/ml Novaminsulfon (Ratiopharm) and isoflurane (Abbott) and injection was done as previously described [5]. In all experiments, tumors and affected tissues were recovered and processed for histological analyses. Intra-tibial tumor formation was monitored by X-ray radiography.
Histology
Visceral organs were fixed in phosphate buffered 4% formaldehyde and paraffin embedded. 3-5μm thick sections from all tissues were stained with hematoxylin and eosin (H&E). Hind limb bones were decalcified and paraffin embedded, the histological analysis with H&E was complemented by quantification of tartrate-resistant acid phosphatase (TRAP+) stained osteoclasts. All sections were reviewed and interpreted by two pathologists (J.C-W. and F.N.).
Statistical analysis
Descriptive statistics is used to determine parameters like mean, standard deviation and standard error of the mean (SEM). Differences were analyzed by unpaired two-tailed student's t-test as indicated using Excel (Microsoft) or Prism 5 (GraphPad Software); p values < 0.05 were considered statistically significant (*p < 0.05; **p < 0.005; ***p < 0.0005).
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
We thank Andreas Kirschner and David Schirmer for their valuable assistance and their support.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest or financial interests.
Authors' contributions
Conception and design: K. von Heyking, G.H.S. Richter; Development of methodology: K. von Heyking, M. Ertl; Acquisition of data (provided animals, provided facilities, etc.): K. von Heyking, L. Roth, M. Ertl, J. Calzada-Wack, O. Schmidt, G.H.S. Richter; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): K. von Heyking, J. Calzada-Wack, F. Neff, E.R.Lawlor, S. Burdach, G.H.S. Richter; Writing, review, and/or revision of the manuscript: K. von Heyking, J. Calzada- Wack, E.R.Lawlor, S. Burdach, G.H.S. Richter; Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): E.R.Lawlor, G.H.S. Richter; Study supervision: K. von Heyking, S. Burdach, G.H.S. Richter
GRANT SUPPORT
This work was supported by grants from the Else-Kröner-Fresenius Stiftung (2013_A49), and the Wilhelm-Sander Stiftung (2009.901.2). It is part of the Translational Sarcoma Research Network (TransSaRNet; 01GM1104B), “Rare Diseases” and Prospective Validation of Biomarkers in Ewing Sarcoma for Personalized Translational Medicine (PROVABES; 01KT1311), Funding Programs of the Federal Ministry of Education and Research (BMBF), Germany.
REFERENCES
- 1.Mackintosh C, Madoz-Gurpide J, Ordonez JL, Osuna D, Herrero-Martin D. The molecular pathogenesis of Ewing's sarcoma. Cancer biology & therapy. 2010;9:655–667. doi: 10.4161/cbt.9.9.11511. [DOI] [PubMed] [Google Scholar]
- 2.Ewing J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J Clin. 1972;22:95–98. doi: 10.3322/canjclin.22.2.95. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt D, Harms D, Burdach S. Malignant peripheral neuroectodermal tumours of childhood and adolescence. Virchows Arch A Pathol Anat Histopathol. 1985;406:351–365. doi: 10.1007/BF00704304. [DOI] [PubMed] [Google Scholar]
- 4.Cavazzana AO, Miser JS, Jefferson J, Triche TJ. Experimental evidence for a neural origin of Ewing's sarcoma of bone. The American journal of pathology. 1987;127:507–518. [PMC free article] [PubMed] [Google Scholar]
- 5.Hauer K, Calzada-Wack J, Steiger K, Grunewald TG, Baumhoer D, Plehm S, Buch T, Prazeres da Costa O, Esposito I, Burdach S, Richter GH. DKK2 mediates osteolysis, invasiveness, and metastatic spread in Ewing sarcoma. Cancer Res. 2013;73:967–977. doi: 10.1158/0008-5472.CAN-12-1492. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka M, Yamazaki Y, Kanno Y, Igarashi K, Aisaki K, Kanno J, Nakamura T. Ewing's sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J Clin Invest. 2014;124:3061–3074. doi: 10.1172/JCI72399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annual review of pathology. 2012;7:145–159. doi: 10.1146/annurev-pathol-011110-130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dockhorn-Dworniczak B, Schafer KL, Dantcheva R, Blasius S, Winkelmann W, Strehl S, Burdach S, van Valen F, Jurgens H, Bocker W. Diagnostic value of the molecular genetic detection of the t(11;22) translocation in Ewing's tumours. Virchows Archiv. 1994;425:107–112. doi: 10.1007/BF00230345. [DOI] [PubMed] [Google Scholar]
- 9.Cotterill SJ, Ahrens S, Paulussen M, Jurgens HF, Voute PA, Gadner H, Craft AW. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. Journal of clinical oncology. 2000;18:3108–3114. doi: 10.1200/JCO.2000.18.17.3108. [DOI] [PubMed] [Google Scholar]
- 10.Burdach S, Jurgens H. High-dose chemoradiotherapy (HDC) in the Ewing family of tumors (EFT) Crit Rev Oncol Hematol. 2002;41:169–189. doi: 10.1016/s1040-8428(01)00154-8. [DOI] [PubMed] [Google Scholar]
- 11.Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12:6243s–6249s. doi: 10.1158/1078-0432.CCR-06-0931. [DOI] [PubMed] [Google Scholar]
- 12.Mao B, Niehrs C. Kremen2 modulates Dickkopf2 activity during Wnt/LRP6 signaling. Gene. 2003;302:179–183. doi: 10.1016/s0378-1119(02)01106-x. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Mao J, Sun L, Liu W, Wu D. Second cysteine-rich domain of Dickkopf-2 activates canonical Wnt signaling pathway via LRP-6 independently of dishevelled. The Journal of biological chemistry. 2002;277:5977–5981. doi: 10.1074/jbc.M111131200. [DOI] [PubMed] [Google Scholar]
- 14.Uren A, Wolf V, Sun YF, Azari A, Rubin JS, Toretsky JA. Wnt/Frizzled signaling in Ewing sarcoma. Pediatric blood & cancer. 2004;43:243–249. doi: 10.1002/pbc.20124. [DOI] [PubMed] [Google Scholar]
- 15.Gehring WJ, Hiromi Y. Homeotic genes and the homeobox. Annual review of genetics. 1986;20:147–173. doi: 10.1146/annurev.ge.20.120186.001051. [DOI] [PubMed] [Google Scholar]
- 16.Apiou F, Flagiello D, Cillo C, Malfoy B, Poupon MF, Dutrillaux B. Fine mapping of human HOX gene clusters. Cytogenetics and cell genetics. 1996;73:114–115. doi: 10.1159/000134320. [DOI] [PubMed] [Google Scholar]
- 17.Cillo C. HOX genes in human cancers. Invasion & metastasis. 1994;14:38–49. [PubMed] [Google Scholar]
- 18.Cillo C, Cantile M, Faiella A, Boncinelli E. Homeobox genes in normal and malignant cells. Journal of cellular physiology. 2001;188:161–169. doi: 10.1002/jcp.1115. [DOI] [PubMed] [Google Scholar]
- 19.Magli MC, Barba P, Celetti A, De Vita G, Cillo C, Boncinelli E. Coordinate regulation of HOX genes in human hematopoietic cells. Proc Natl Acad Sci U S A. 1991;88:6348–6352. doi: 10.1073/pnas.88.14.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cantile M, Procino A, D'Armiento M, Cindolo L, Cillo C. HOX gene network is involved in the transcriptional regulation of in vivo human adipogenesis. Journal of cellular physiology. 2003;194:225–236. doi: 10.1002/jcp.10210. [DOI] [PubMed] [Google Scholar]
- 21.Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, Barshack I, Seijffers R, Kopolovic J, Kaiser N, Karasik A. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nature medicine. 2000;6:568–572. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- 22.Villavicencio-Lorini P, Kuss P, Friedrich J, Haupt J, Farooq M, Turkmen S, Duboule D, Hecht J, Mundlos S. Homeobox genes d11-d13 and a13 control mouse autopod cortical bone and joint formation. J Clin Invest. 2010;120:1994–2004. doi: 10.1172/JCI41554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–6776. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- 24.Cillo C, Faiella A, Cantile M, Boncinelli E. Homeobox genes and cancer. Experimental cell research. 1999;248:1–9. doi: 10.1006/excr.1999.4451. [DOI] [PubMed] [Google Scholar]
- 25.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nature reviews Cancer. 2010;10:361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 26.Kuss P, Villavicencio-Lorini P, Witte F, Klose J, Albrecht AN, Seemann P, Hecht J, Mundlos S. Mutant Hoxd13 induces extra digits in a mouse model of synpolydactyly directly and by decreasing retinoic acid synthesis. J Clin Invest. 2009;119:146–156. doi: 10.1172/JCI36851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Levetzow C, Jiang X, Gwye Y, von Levetzow G, Hung L, Cooper A, Hsu JH, Lawlor ER. Modeling initiation of Ewing sarcoma in human neural crest cells. PloS one. 2011;6:e19305. doi: 10.1371/journal.pone.0019305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, Staege MS, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106:5324–5329. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 30.Soshnikova N, Duboule D. Epigenetic temporal control of mouse Hox genes in vivo. Science. 2009;324:1320–1323. doi: 10.1126/science.1171468. [DOI] [PubMed] [Google Scholar]
- 31.Svoboda LK, Harris A, Bailey NJ, Schwentner R, Tomazou E, von Levetzow C, Magnuson B, Ljungman M, Kovar H, Lawlor ER. Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics. 2014;9:1613–1625. doi: 10.4161/15592294.2014.988048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breau MA, Wilkinson DG, Xu Q. A Hox gene controls lateral line cell migration by regulating chemokine receptor expression downstream of Wnt signaling. Proc Natl Acad Sci U S A. 2013;110:16892–16897. doi: 10.1073/pnas.1306282110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuss P, Kraft K, Stumm J, Ibrahim D, Vallecillo-Garcia P, Mundlos S, Stricker S. Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Developmental biology. 2014;385:83–93. doi: 10.1016/j.ydbio.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 34.Vater C, Kasten P, Stiehler M. Culture media for the differentiation of mesenchymal stromal cells. Acta biomaterialia. 2011;7:463–477. doi: 10.1016/j.actbio.2010.07.037. [DOI] [PubMed] [Google Scholar]
- 35.Bruneau S, Johnson KR, Yamamoto M, Kuroiwa A, Duboule D. The mouse Hoxd13(spdh) mutation, a polyalanine expansion similar to human type II synpolydactyly (SPD), disrupts the function but not the expression of other Hoxd genes. Developmental biology. 2001;237:345–353. doi: 10.1006/dbio.2001.0382. [DOI] [PubMed] [Google Scholar]
- 36.Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, Li C, Xie L, Crane J, Wan M, Zhen G, Bian Q, Yu B, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nature medicine. 2014;20:1270–1278. doi: 10.1038/nm.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nature reviews Cancer. 2011;11:411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grunewald TG, Diebold I, Esposito I, Plehm S, Hauer K, Thiel U, da Silva-Buttkus P, Neff F, Unland R, Muller-Tidow C, Zobywalski C, Lohrig K, Lewandrowski U, et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Molecular cancer research. 2012;10:52–65. doi: 10.1158/1541-7786.MCR-11-0524. [DOI] [PubMed] [Google Scholar]
- 39.Richter GH, Fasan A, Hauer K, Grunewald TG, Berns C, Rossler S, Naumann I, Staege MS, Fulda S, Esposito I, Burdach S. G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. The Journal of pathology. 2013;230:70–81. doi: 10.1002/path.4170. [DOI] [PubMed] [Google Scholar]
- 40.Yamaguchi TP, Bradley A, McMahon AP, Jones S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development. 1999;126:1211–1223. doi: 10.1242/dev.126.6.1211. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Liu P, Liu W, Maye P, Zhang J, Zhang Y, Hurley M, Guo C, Boskey A, Sun L, Harris SE, Rowe DW, Ke HZ, et al. Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet. 2005;37:945–952. doi: 10.1038/ng1614. [DOI] [PubMed] [Google Scholar]
- 42.Li X, McGee-Lawrence ME, Decker M, Westendorf JJ. The Ewing's sarcoma fusion protein, EWS-FLI, binds Runx2 and blocks osteoblast differentiation. Journal of cellular biochemistry. 2010;111:933–943. doi: 10.1002/jcb.22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 44.Sugawara M, Kato N, Tsuchiya T, Motoyama T. RUNX2 expression in developing human bones and various bone tumors. Pathology international. 2011;61:565–571. doi: 10.1111/j.1440-1827.2011.02706.x. [DOI] [PubMed] [Google Scholar]
- 45.Cantile M, Scognamiglio G, Anniciello A, Farina M, Gentilcore G, Santonastaso C, Fulciniti F, Cillo C, Franco R, Ascierto PA, Botti G. Increased HOX C13 expression in metastatic melanoma progression. J Transl Med. 2012;10:91. doi: 10.1186/1479-5876-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shah N, Jin K, Cruz LA, Park S, Sadik H, Cho S, Goswami CP, Nakshatri H, Gupta R, Chang HY, Zhang Z, Cimino-Mathews A, Cope L, et al. HOXB13 mediates tamoxifen resistance and invasiveness in human breast cancer by suppressing ERalpha and inducing IL-6 expression. Cancer Res. 2013;73:5449–5458. doi: 10.1158/0008-5472.CAN-13-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Liu G, Shen D, Ye H, Huang J, Jiao L, Sun Y. HOXA1 enhances the cell proliferation, invasion and metastasis of prostate cancer cells. Oncol Rep. 2015;34:1203–1210. doi: 10.3892/or.2015.4085. [DOI] [PubMed] [Google Scholar]
- 48.Buttice G, Duterque-Coquillaud M, Basuyaux JP, Carrere S, Kurkinen M, Stehelin D. Erg, an Ets-family member, differentially regulates human collagenase1 (MMP1) and stromelysin1 (MMP3) gene expression by physically interacting with the Fos/Jun complex. Oncogene. 1996;13:2297–2306. [PubMed] [Google Scholar]
- 49.Fuchs B, Inwards CY, Janknecht R. Upregulation of the matrix metalloproteinase-1 gene by the Ewing's sarcoma associated EWS-ER81 and EWS-Fli-1 oncoproteins, c-Jun and p300. FEBS Lett. 2003;553:104–108. doi: 10.1016/s0014-5793(03)00984-0. [DOI] [PubMed] [Google Scholar]
- 50.Burdach S, Plehm S, Unland R, Dirksen U, Borkhardt A, Staege MS, Muller-Tidow C, Richter GH. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle. 2009;8:1991–1996. doi: 10.4161/cc.8.13.8929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.