

ABSTRACT
Intravenous drug use is one of the major risk factors for HIV-infection in HIV-related pulmonary arterial hypertension patients. We previously demonstrated exaggerated pulmonary vascular remodeling with enhanced apoptosis followed by increased proliferation of pulmonary endothelial cells on simultaneous exposure to both opioids and HIV protein(s). Here we hypothesize that the exacerbation of autophagy may be involved in the switching of endothelial cells from an early apoptotic state to later hyper-proliferative state. Treatment of human pulmonary microvascular endothelial cells (HPMECs) with both the HIV-protein Tat and morphine resulted in an oxidative stress-dependent increase in the expression of various markers of autophagy and formation of autophagosomes when compared to either Tat or morphine monotreatments as demonstrated by western blot, transmission electron microscopy and immunofluorescence. Autophagy flux experiments suggested increased formation rather than decreased clearance of autolysosomes. Inhibition of autophagy resulted in a significant increase in apoptosis and reduction in proliferation of HPMECs with combined morphine and Tat (M+T) treatment compared to monotreatments whereas stimulation of autophagy resulted in opposite effects. Significant increases in the expression of autophagy markers as well as the number of autophagosomes and autolysosomes was observed in the lungs of SIV-infected macaques and HIV-infected humans exposed to opioids. Overall our findings indicate that morphine in combination with viral protein(s) results in the induction of autophagy in pulmonary endothelial cells that may lead to an increase in severity of angio-proliferative remodeling of the pulmonary vasculature on simian and human immunodeficiency virus infection in the presence of opioids.
KEYWORDS: apoptosis, autophagy, endothelial cells, HIV-Tat, morphine, proliferation
Introduction
The advent of antiretroviral therapy has clearly led to an improved survival among human immunodeficiency virus (HIV)-1 infected individuals. Nevertheless, this advancement has been accompanied by serious noninfectious cardiovascular complications including coronary heart disease and pulmonary arterial hypertension (PAH).1 PAH results from increased vascular resistance in the pulmonary arteries that most often results in right heart failure and death.2 HIV-1 infection is one of the major causes of pulmonary hypertension in the world.3 The survival rate of HIV-related PAH (HRPAH) patients is significantly reduced to one-half compared with HIV-infected individuals without PAH. However, not all HIV-infected patients develop HRPAH and may require a second hit from another risk factor to develop the disease. Intravenous drug use (IVDU) has been found to be the most common risk factor associated with HIV-infection in individuals diagnosed with HRPAH. A range of 53% to 70% of HRPAH cases were reported to be in individuals who were IVDUs including the recent report by Quezada et al. suggesting 10% prevalence of HRPAH in the HIV-infected cohort from Spain.4-7 Our previous findings consistently suggest augmentation of pulmonary arteriopathy and endothelial dysfunction in HIV-infected IVDUs (mainly opioids and/or cocaine abusers) compared to HIV-infected nondrug users or uninfected IVDUs.8-11
Our previous study on lung tissues from simian immunodeficiency virus (SIV)-infected macaques exposed to morphine, an opioid (VM group), demonstrates significant pulmonary vascular remodeling including the presence of early- and advanced-stage complex (plexiform) lesions when compared with either the SIV-infected (V group) or morphine-treated uninfected (M group) macaques.10 Furthermore, the pulmonary endothelial cells (pECs) lining the vessels showing medial hypertrophy or initial stage intimal lesions in lung sections from VM macaques demonstrated an increase in positivity for both TUNEL (apoptosis marker) and MKI67(proliferation marker). However, the cells within the advanced stage of fully occluded plexiform or intimal fibrotic lesions stained negative for apoptosis but were highly positive for MKI67. This observation was supported by cell culture studies demonstrating enhanced apoptosis with the maximum increase at d 3 followed by enhanced proliferation of apoptosis-resistant endothelial cells that peaked at d 6, upon simultaneous treatment with HIV-transactivator of transcription (Tat) protein and morphine compared to either treatment alone.10 Apoptosis, followed by enhanced proliferation of endothelial cells, has been proposed to be a critical step in the development of plexiform lesions associated with the pulmonary hypertension.12 However, what leads to the transition of endothelial cells from initial apoptosis to apoptosis-resistant hyper-proliferation remains unknown.
Accumulating evidence suggests autophagy to be a major cytoprotective pathway. Although autophagy has been reported to cause cellular senescence and growth arrest in tumor cells,13-15 it also plays a fundamental role in causing chemotherapy resistance by favoring cell survival and uncontrolled proliferation,16,17 making it a plausible therapeutic target.18,19 Autophagy is triggered immediately in response to stress leading to apoptosis or is activated as a defense mechanism upon persistent stress exposure in favor of cell survival and uncontrolled proliferation.16,17 Elevated autophagy markers in lung tissue from patients20 with pulmonary hypertension and in the pulmonary hypertensive murine models20,21 were noted recently. However, the evidence on the role of autophagy in PAH is not yet conclusive. On one hand, increased autophagy is protective in hypoxia model of PAH20 whereas, on the other hand, it is believed to be detrimental in a monocrotaline model of PAH.22 Moreover, the autophagy response is cell dependent and may increase in one cell type and decrease in another in the same disease process. Therefore, further research is needed to provide better insight in understanding the role of autophagy in the pathogenesis of pulmonary vascular remodeling.
Based on literature16,23,24 and our earlier report10 we now hypothesize that the chronic increase in autophagy in pulmonary endothelial cells on combined exposure to HIV protein(s) and morphine results in a hyperproliferative state of endothelial cells leading to irreversible occlusion of pulmonary arterioles. Here we demonstrate the cumulative effect of morphine on HIV-Tat-mediated autophagy in primary human pulmonary microvascular endothelial cells (HPMECs) by analyzing in detail various autophagy markers. Inhibition of autophagy resulted in attenuation whereas stimulation of autophagy resulted in exacerbation of M+T-mediated enhanced proliferation of endothelial cells. These cell-culture findings are later confirmed by analyzing autophagy in the lungs from SIV-infected, morphine-exposed macaques with angio-obliterated pulmonary vessels10 and in the lungs from HIV+ IVDUs that demonstrate enhanced pulmonary vascular remodeling.9 To our knowledge, this study is the first endeavor into exploring the potential role of autophagy in the enhancement of proliferation of apoptosis-resistant endothelial cells mediated by HIV-protein and drug abuse.
Results
Increased expression of autophagy markers with simultaneous exposure of endothelial cells to morphine and HIV-Tat
To determine the role of autophagy in M+T-mediated endothelial dysfunction, we first assessed the levels of autophagic markers involved in the initiation and assembly of the initial phagophore membrane formation,25 ULK1 and BECN1, and the molecules of the conjugation system involved in autophagosome formation, ATG5 and ATG7.17 As represented by the densitometry graphs in Fig. 1A to D there was a significant increase in the expression of ULK1, BECN1, ATG5 and ATG7 within 3 h in HPMECs with combined treatment of morphine and HIV-Tat compared to untreated control cells. Maximum significant increase in the expression of all these autophagy-associated proteins was observed at 6 h of M+T combined treatment compared to all other time points tested, and expression at this time point is shown in the representative western blots. This increase in the expression of autophagy proteins on combined treatment was also higher than that observed in cells treated with either Tat or morphine alone with statistically significant increase in the ULK1 expression at both 3 h and 6 h post treatment. In addition, a significant increase in the expression of ULK1 and BECN1 was also noted in the case of morphine or Tat treatment alone compared to control, at 6 h time point. ULK1 expression in response to combined treatment followed a downward trend at later time points but was still higher when compared with monotreatments. Nevertheless, the expression of BECN1 on combined treatment continued to be significantly higher compared to untreated cells until 48 h whereas ATG7 expression slowly dropped after 6 h but was significantly higher compared to untreated and morphine-treated cells for 12 to 48 h. This increase in the expression of autophagy markers continued for 3 d of combined treatment compared to untreated cells, beyond which the autophagy activity dropped closer to the level of untreated cells. Further, we confirmed that the increase in autophagy with morphine and/or Tat treatment was mainly through specific binding of M+T to OPRM1/µ-opioid and VEGF (vascular endothelial growth factor) receptors, respectively. As seen in Fig. 1E, pretreatment of cells with naloxone, an opioid receptor blocker26 and/or Su5416, the inhibitor of VEGF receptor27 resulted in attenuation of morphine and/or Tat-mediated increase in the expression of ULK1 and BECN1 at 6 h post-treatment when compared with morphine and/or Tat-treated cells in absence of these inhibitors.
Figure 1.
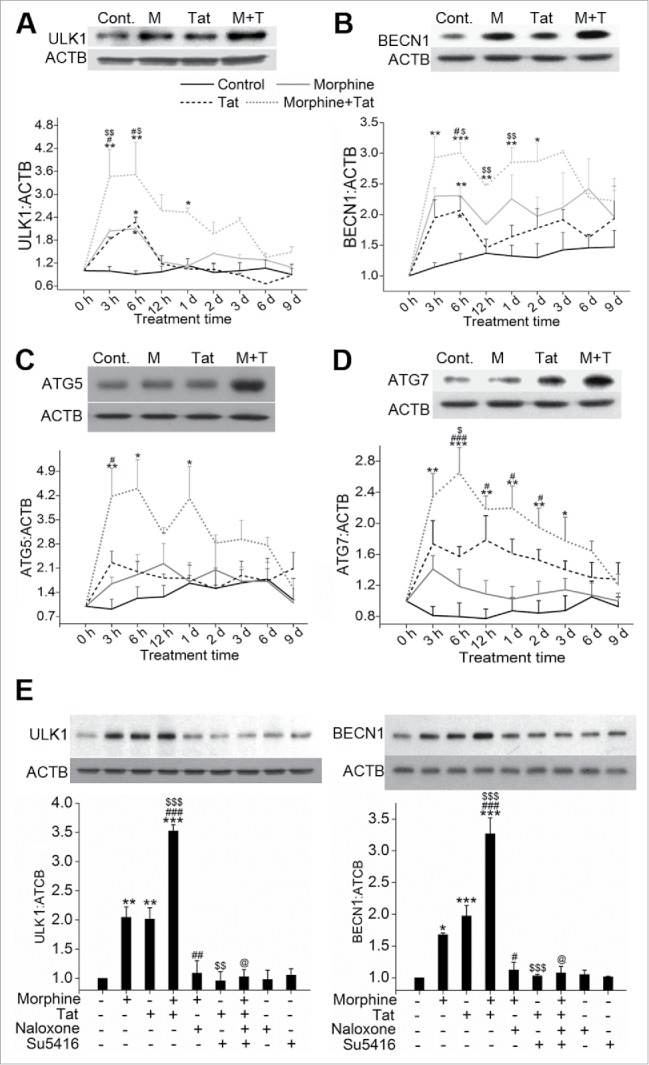
Enhanced expression of autophagy markers in pulmonary endothelial cells on combined treatment with morphine and Tat compared to monotreatments. Confluent Human pulmonary microvascular endothelial cells (HPMEC) were treated with 1 µM morphine (M) and/or 25 ng/ml Tat (T) in 0.5% serum-containing medium from 3 h to 9 d followed by western blot for (A) ULK1, (B) BECN1, (C) ATG5 and (D) ATG7. The graphs are the densitometry analysis of protein expression at various time points from at least 3 independent experiments. The western blots above each graph represent analysis of the 6-h treatment. The blot probed for BECN1 was stripped and reprobed for ATG5 and ATG7. (E) Cells were pretreated with naloxone (1 µM) and/or Su5416 (0.5 µM) 20 min before 6 h morphine and/or Tat treatment followed by western blot of ULK1 and BECN1 expression. The representative blots are shown with densitometry graphs representing mean +SEM from 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. morphine, $P < 0.05, $$P < 0.01, $$$P < 0.001 vs. Tat, @P < 0.001 vs. M+T.
Morphine augments the formation of autophagosomes and autolysosomes in HIV-Tat-treated endothelial cells
To determine the autophagosome and autolysosome formation in response to Tat and /or morphine treatment, cells were immunostained for MAP1LC3B and treated with LysoTracker Red dye to track lysosomes. As represented in Fig. 2A, there was a remarkable increase in the green-colored MAP1LC3B puncta on combined treatment of HPMEC with M+T for 24 h compared to untreated cells or cells exposed to monotreatments. We also found a greater detection of yellow-orange stained autolysosomes as an indication of fusion between MAP1LC3B-positive autophagosomes and lysosomes in cells exposed to combined treatment (Fig. 2A). Quantification of green and yellow-orange fluorescence puncta demonstrated not only a significant increase in autophagosomes but also autolysosomes on simultaneous treatment with M+T when compared with monotreatments, indicating higher flux of LC3 toward lysosomes (Fig. S1A and B). We further confirmed the induction of autophagy in M+T-treated HPMECs by transfecting cells with pBABE-puro mCherry-EGFP-LC3B plasmid. The mCherry-EGFP-LC3B vector helps to distinguish the autophagosomes (EGFP positive, green) from autolysosomes (mCherry positive, red) as EGFP signal is lost or decreased in an acidic environment. As represented in Fig. 2B, combined treatment of morphine and HIV-Tat resulted in an increase in the number of autophagosomes as well as autolysosomes compared to monotreatments. However, we found a low number of red-only positive autolysosomes and this could be because we fixed the cells before viewing under a confocal microscope. Fixation restores the signal of GFP28 and this could have resulted in underestimation of mCherry-only positive signal in M+T-treated cells considering our observation in Fig. 2A. When we added the autophagy stimulator rapamycin to M+T-treated cells both GFP and mCherry signals increased remarkably. However, red-positive and green-positive puncta in cells treated with rapamycin alone were found to be far fewer compared to M+T treatment in the absence or presence of rapamycin (Fig. S1C and Fig. 2B). Furthermore, significant increase in the protein expression of LAMP1 on combined treatment with M+T compared to monotreatments is shown in Figure S1D.
Figure 2.

Increased number of autophagosomes or autolysosomes in HPMEC on combined treatment with morphine and Tat. (A) Confluent HPMEC were treated with morphine (1 μM) and/or HIV-Tat (25 ng/ml) for 24 h. Live cells were stained with LysoTracker Red dye for 30 min followed by immunofluorescence staining for LC3B (green). Magnification 60X. Scale bar: 50 μm. (B) HPMECs were transfected with 300 ng of pBABE-puro mCherry-EGFP-LC3B plasmid followed by morphine and/or Tat treatment at 48 h post-transfection. After 24 h of treatment, cells were fixed with 4% paraformaldehyde and viewed using a Nikon Eclipse E2000-U inverted confocal microscope. Green puncta (GFP) represent autophagosomes and red puncta (mCherry) represent autolysosomes. Magnification 100X. Scale bar: 50 μm. (C) Transmission electron microscopy (TEM) analysis of HPMEC treated with morphine and/or Tat. Cells treated for 24 h were trypsinized followed by fixation in glutaraldehyde and processed for TEM (3000X magnification). The column on the right shows magnified images of the insets. Scale bars: 500 nm.
We next confirmed these findings with TEM, which is one of the most widely used techniques to detect the presence of autophagic vesicles.29 We looked for the presence of double-membrane autophagosomes and for autolysosomes by TEM in HPMEC treated with morphine and/or Tat for 24 h. As represented in Fig. 2C and Figure S1E, quantification of autophagosomes and autolysosomes present within cells demonstrated maximum presence of these autophagic bodies in cells exposed to combined treatment of M+T and this increase was statistically significant compared to untreated or only morphine or tat treated cells.
Increased autophagic flux on combined treatment with morphine and HIV-Tat
To determine if the increase in autophagic bodies is due to the increase in the rate of autophagosome formation or a decrease in their degradation, cells were treated with bafilomycin A1 (BAF)30 followed by morphine and/or Tat treatment. As observed by immunofluorescence staining shown in Fig. 3A pretreatment of HPMECs with BAF resulted in a further increase in morphine and Tat-mediated green fluorescent MAP1LC3B puncta formation (lower panel) when compared with cells treated with only morphine or Tat (upper panel). The quantification of MAP1LC3B puncta in the morphine and/or Tat-treated cells in the presence or absence of BAF is represented in Figure S2A. The augmentation in autophagosome flux was further demonstrated by an increase in the number of green and yellow-positive puncta and the absence of red-only mCherry-positive puncta after treatment of GFP-mCherry-LC3B-transfected cells (Fig. 3B) with M+T in the presence of BAF. Western blot analysis also demonstrated an increase in the levels of MAP1LC3B-II on BAF pretreatment compared to the respective non-BAF M+T-treated cells as shown in Fig. 3C. Pretreatment of Tat-treated cells with BAF also resulted in significant increase in the MAP1LC3B-II:ACTB ratio compared to Tat treatment in the absence of BAF. However, treatment of cells with only morphine for 24 h did not demonstrate an increase in the MAP1LC3B puncta formation or in the MAP1LC3B-II expression when compared with untreated cells (Fig. 3A and C). Also as expected, an increase in MAP1LC3B puncta (Fig. 3A) and MAP1LC3B-II expression (Fig. 3C) was observed in only BAF-treated cells compared to untreated control. Figure S2C shows increase in the expression of MAP1LC3B-II in M+T-treated cells at various time intervals that further increased in the presence of BAF.
Figure 3.
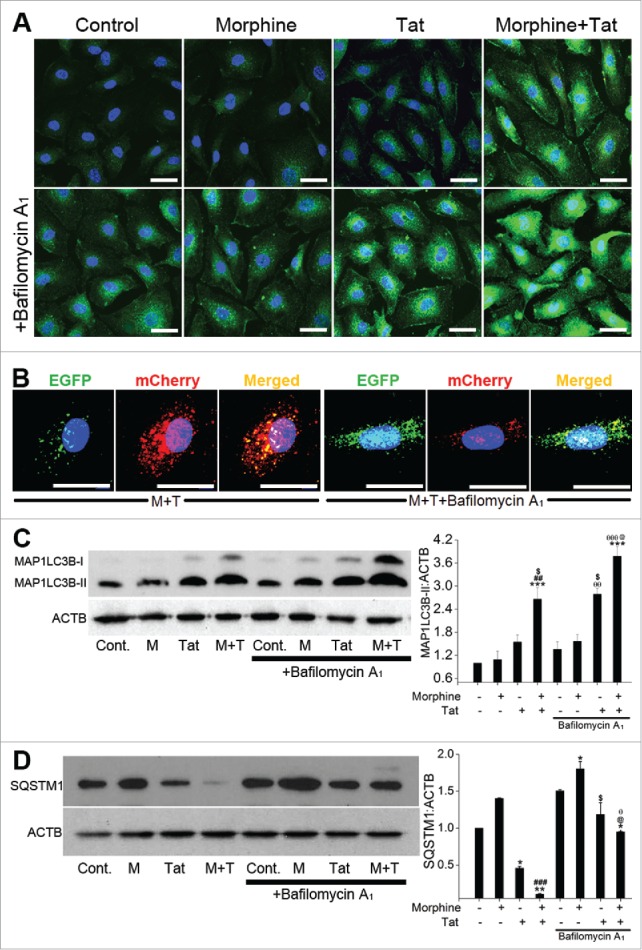
Enhanced autophagic flux on combined treatment with morphine and Tat. HPMECs untransfected (A, C, D) or transfected with pBABE-puro mCherry-EGFP-LC3B plasmid (B) were treated with morphine and/or HIV-Tat in the absence or presence of BAF. ((A)and B) For MAP1LC3B immunostaining (A) and visualization of GFP or mCherry LC3B puncta (B) cells were fixed with 4% paraformaldehyde at 24 h post-treatment and viewed using a confocal microscope. ((C)and D) Western blot analysis of MAP1LC3B-II (C) and SQSTM1 (D) at 24 h and 48 h post treatment, respectively. The graphs represent the densitometry analysis of western blots from 2 or 3 independent experiments. Mean +SEM, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control, ##P < 0.01, ###P < 0.001 vs. morphine, $P < 0.05 vs. Tat, @P < 0.05 vs. combined morphine and Tat (M+T), θP < 0.05, θθP < 0.01, θθθP < 0.001 vs BAF. Note: In order to visualize autophagosomes (yellow puncta) more clearly in M+T+BAF, we lowered the green and increased the red fluorescence intensities compared to the images captured for Figure 2.
Alteration in the autophagy flux was further confirmed by western blot analysis of SQSTM1/p62 (sequestosome 1) expression.31 As expected, combined treatment showed a significant reduction in the SQSTM1 level compared to untreated cell or monotreated cells (Fig. 3D). Pretreatment with BAF, resulted in an increase in the SQSTM1 expression compared to the corresponding non-BAF M+T-treated cells at 24 h (Fig. S2B) and 48 h post-treatment (Fig. 3D). Interestingly, as shown in Fig. 3D, 48 h of morphine treatment alone caused an increase in SQSTM1 expression, which further increased upon BAF treatment. Time-course studies found a remarkable reduction in SQSTM1 expression as early as 3 h to 6 d of combined treatment compared to untreated cells. The corresponding BAF-pretreated cells showed an increase in SQSTM1 expression as illustrated in Figure S2C. Overall, the results show that combined treatment of M+T leads to remarkable induction of autophagy in pulmonary microvascular endothelial cells.
Morphine and Tat-mediated increase in the levels of autophagy proteins was independent of changes in the mRNA levels
Since we saw an increase in protein expression of key autophagy proteins as early as 3 h post-treatment we next investigated if this corresponded to the increase in mRNA levels. Interestingly, as seen in Fig. 4A, we observed no significant alterations in the mRNA expression of ULK1, BECN1, ATG5 or ATG7 in morphine- and/or Tat-treated cells at any of the early time points tested; from 1 h to 6 h post-treatment. To confirm transcription-independent regulation of ATG protein levels by M+T we examined the expression of ATG proteins in the presence of the RNA synthesis inhibitor actinomycin D as well as in the presence of the protein synthesis inhibitor cycloheximide. We did not observe any significant changes in the levels of ULK1, BECN1, ATG5 and ATG7 in response to M+T treatment in the presence of actinomycin D when compared with cells treated with only M+T (Fig. 4B). However, we observed significant abrogation in the M+T induced expression of all these autophagy proteins in the cells pretreated with cycloheximide (Fig. 4C).
Figure 4.
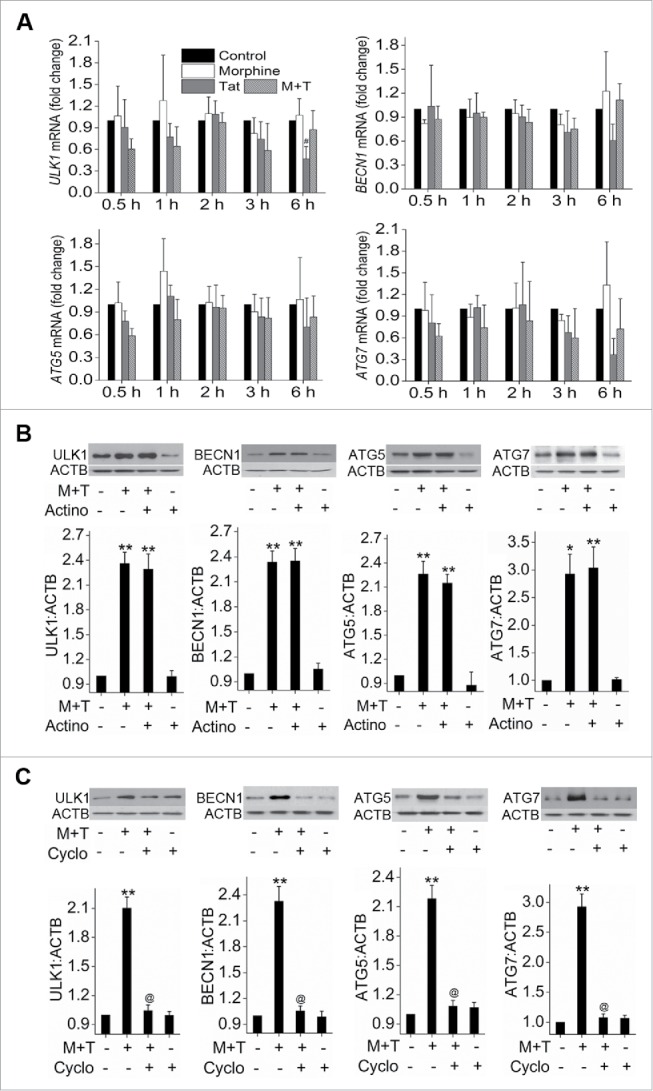
Transcription-independent increase in the expression of autophagy proteins on morphine and Tat treatment. (A) Confluent HPMEC were treated with morphine and/or Tat in 0.5% FBS containing endothelial cell medium from 30 min to 6 h followed by RNA extraction, cDNA preparation and qPCR for ULK1, BECN1, ATG5 and ATG7. (B and C) HPMEC at 80% confluence were treated with either (B) actinomycin D (3 μg/ml) or (C) cycloheximide (10 μg/ml)15 min before morphine and Tat treatment for 6 h followed by protein extraction for western blot analyses of ULK1, BECN1, ATG5 and ATG7. The graphs represent the densitometry of 4 independent experiments. Mean +SEM, *P < 0.01, **P < 0.001 vs. control, @P < 0.001 vs. combined morphine and Tat (M+T).
Negative modulation of morphine and Tat-mediated apoptosis by autophagy
Previously we have demonstrated that combined treatment with M+T initially results in an oxidative stress-mediated enhanced apoptosis of HPMECs with maximum increase observed at 3 d post-treatment, followed by enhanced proliferation that peaked at 6 d post-treatment.10 To investigate if autophagy is contributing to the M+T-mediated enhanced proliferation of apoptotic resistant endothelial cells, we first tested whether autophagy protects HPMECs against morphine-Tat induced apoptotic stress. We inhibited autophagy by a chemical method using 3-methyladenine (3-MA) or by a gene knockdown approach using siRNA against ULK1. Results represented in Figure S3 confirmed the efficiency of siRNA transfection. As observed in Fig. 5A and 5B, inhibition of autophagy by 3-MA or by knocking down ULK1 expression with the use of siRNA in cells treated with morphine and/or Tat for 3 d resulted in significant increase in cell apoptosis compared to corresponding morphine and/or Tat-treated cells in the absence of 3-MA treatment or ULK1 knockdown As previously reported by us,10 the combined M+T exposure for 3 d significantly increased endothelial apoptosis compared to monotreatments. Interestingly, autophagy inhibition lead to further significant enhancement of this M+T -mediated apoptosis indicating that autophagy protects the cells from undergoing severe apoptosis. In corroboration of these findings, stimulation of autophagy using pharmacological stimulators, rapamycin (Fig. 5C) or temozolomide (Fig. S4A) and overexpression of ULK1 protein (Fig. 5D) in M+T-treated HPMEC resulted in significant reduction of M+T-mediated apoptosis thus confirming the protective role of autophagy against pulmonary endothelial cell apoptosis. The overexpression of ULK1 in HPMEC transiently transfected with ULK1 expression plasmid was confirmed by western blot analysis as represented in Figure S5. Morphine and /or Tat treatment of cells transfected with scrambled siRNA or empty plasmid showed similar alterations in apoptosis as in case of untransfected morphine and /or Tat-treated cells.
Figure 5.
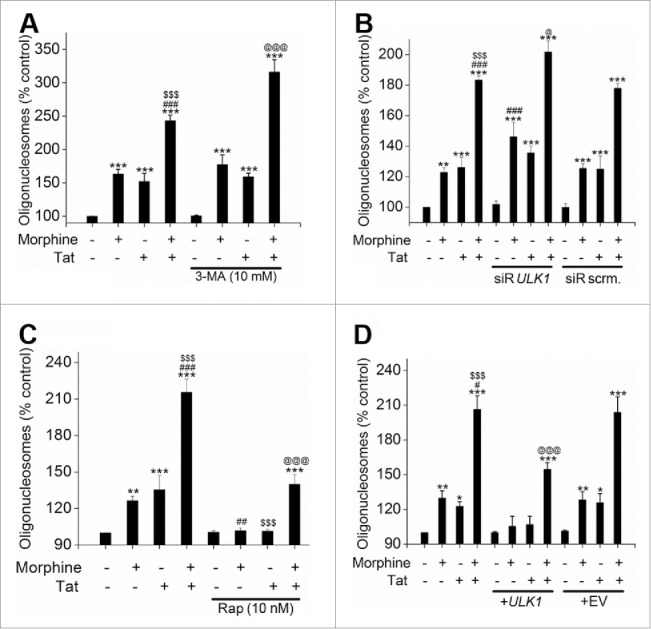
Autophagy protects HPMEC from morphine and Tat-mediated apoptosis. HPMEC (1.25 × 104 cells / well) were plated on 96-well plates followed by pretreatment with either 3-methyladinine (3-MA) (A) or rapamycin (C) before morphine and/or Tat treatment for 3 d followed by Cell Death Detection ELISA. Alternatively, cells were transfected with ULK1 siRNA or scrambled (scrm) siRNA (B) or with ULK1 expression plasmid or empty vector (EV) (D) using HiPerfect or GeneJuice reagent respectively as per the manufacturer's instructions before treatment with morphine and/or Tat. The data are average of 3 independent experiments performed in triplicates. Mean + SD, *P < 0.05, **P < 0.01, ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. morphine, $$$P < 0.001 vs. Tat, @P < 0.05, @@@P < 0.001 vs. morphine+Tat treatment.
Autophagy-dependent increased proliferation of morphine-Tat-treated HPMEC
Finally we analyzed whether autophagy, which was responsible for rescuing the cells from M+T-mediated apoptosis, could actually lead to hyperproliferation. As indicated in Fig. 6A and B, autophagy inhibition using either 3-MA or ULK1 siRNA resulted in significant reduction in morphine-Tat induced HPMEC proliferation after 6 d of exposure. On the other hand, stimulation of autophagy using rapamycin (Fig. 6C) or temozolomide (Fig. S4B) and overexpression of ULK1 using an expression plasmid (Fig. 6D) further increased the M+T-mediated HPMEC proliferation significantly. As expected we also observed a corresponding significant increase in cell proliferation on autophagy stimulation of cells treated with morphine or Tat alone. These results clearly highlight the key role of autophagy in promoting the cells that survive the M+T triggered apoptosis to develop a proliferative phenotype.
Figure 6.
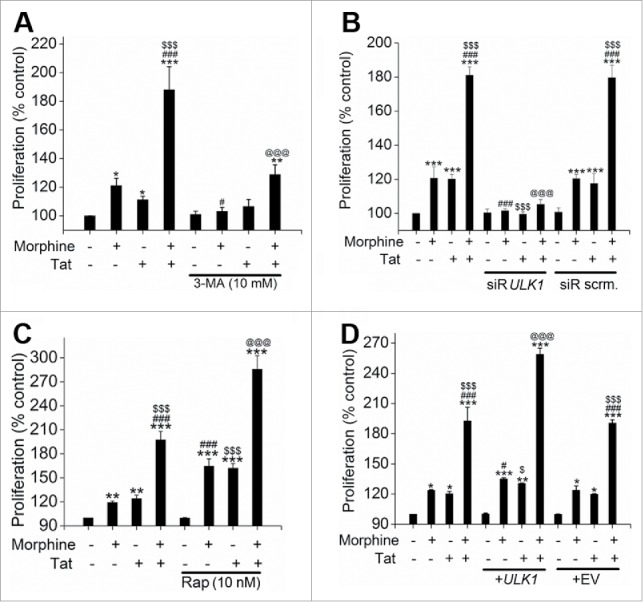
Autophagy promotes the morphine and Tat-treated HMPEC toward hyperproliferation. Autophagy was inhibited by treatment with 3-MA (A) or ULK1 knockdown (B) and was stimulated by rapamycin (C) or ULK1 overexpression (D) followed by morphine and/or Tat treatment. MTS Cell Proliferation Assay was later done at 6 d post-treatment. Cells (3 × 103 cells/well) were plated on 96-well plates followed by pretreatment with either 3-MA (A) or rapamycin (C) before morphine and/or Tat treatment. Cells were transfected with ULK1 siRNA or scrambled (scrm) siRNA using HiPerfect (B) or transfected with ULK1 expression plasmid or empty vector (EV) using GeneJuice reagent (D) as per the manufacturer's instructions followed by morphine and/or Tat treatment. The data shown are mean + SD of 3 independent experiments done in triplicates. *P < 0.05, ** P <0.01, ***P < 0.001 vs. control, #P < 0.05, ###P < 0.001 vs. morphine, $P < 0.05, $$$P < 0.001 vs. Tat, @@@P < 0.001 vs. morphine+Tat treatment.
Oxidative stress dependent increase in autophagy on morphine and Tat treatment of endothelial cells
Growing evidence suggests crosstalk between ROS and autophagy. While ROS is considered as one of the important inducers of autophagy,32 autophagy in turn is known to regulate oxidative stress by removal of ROS generating damaged organelles. In our earlier findings we reported significant increase in the generation of reactive oxygen species (ROS) on morphine-Tat treatment of HPMEC; however, we measured H2O2 and O2− generation only at 1 h post treatment.10 Now, to see if oxidative stress is involved in chronic induction of autophagy in response to chronic exposure of HPMEC to M+T, we did time-course studies. As shown in Fig. 7A, morphine or Tat alone could significantly increase the H2O2 generation from 1 h to 1 day post-treatment compared to control. In cells treated with both M+T a sharp peak in H2O2 generation was observed at early time points followed by chronic generation of H2O2 until 6 d post treatment. This increase in H2O2 on combined treatment was significantly more compared to monotreated cells at all time-points tested. Similarly, we observed significant increase in superoxide (O2−) radicals with a peak as early as 3 h post-treatment of M+T compared to monotreatments or untreated controls as shown in Fig. 7B. On blocking the interactions of M+T with cells using naloxone and Su5416, we were able to prevent M+T-mediated increase in total ROS generation (Fig. 7C).
Figure 7.
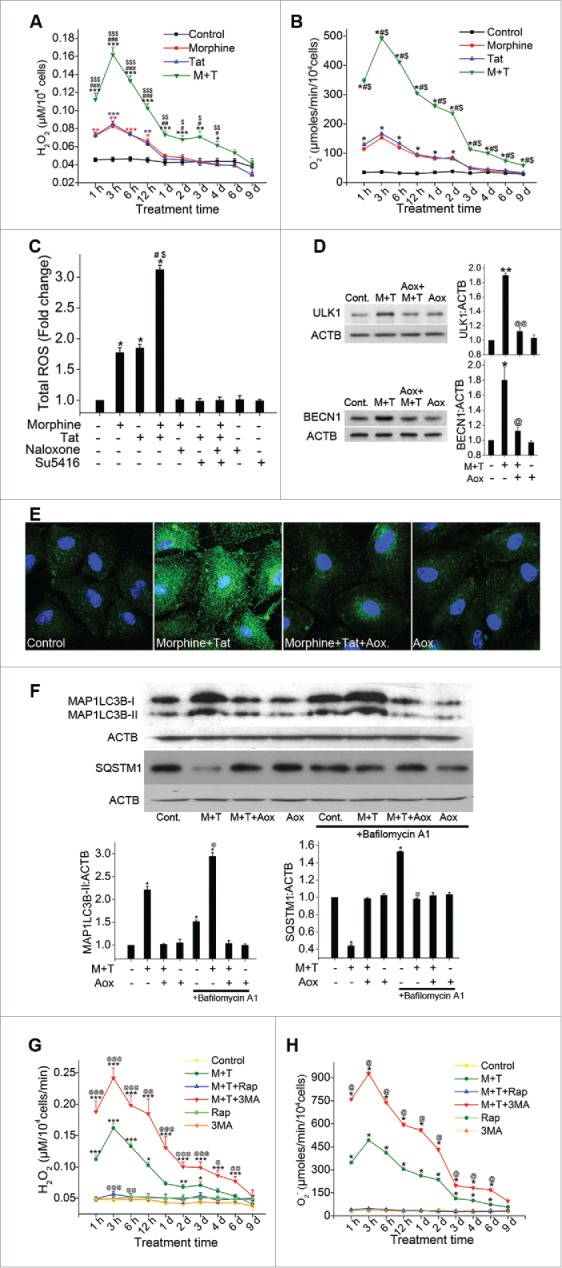
Morphine and Tat-mediated enhanced oxidative stress activates autophagy in endothelial cells. (A) Analysis of H2O2 or (B) superoxide levels in morphine (1 μM) and/or HIV-Tat (25 ng/ml)-treated HPMEC for the indicated time points by amplex red assay or SOD-inhibitable cytochrome C reductase assay, respectively. C) HPMEC (2 × 104 cells/well) plated on a 96-well plate were pretreated with naloxone (1 μM) and/or Su5416 (0.5 µM) followed by morphine and/or Tat for 3 h and measurement of total reactive oxygen species using the DCF assay kit. (D and E) Confluent HPMEC were pretreated with antioxidants for 30 min followed by 6 h Tat and morphine treatment for western blot analysis for ULK1 and BECN1 expression (D) and for MAP1LC3B immunofluorescence staining (100X magnification) (E). (F) HPMEC were pretreated with antioxidant cocktail (Aox) and/or BAF followed by combined morphine-Tat treatment for 24 h for western blot analyses of MAP1LC3B-II and SQSTM1 expression. The graphs represent densitometry from 3 independent experiments, mean+ SEM. (G and H) Modulation of morphine and Tat induced oxidative stress with autophagy inhibition and stimulation. Confluent HPMEC pretreated with either 3MA (10 mM) or rapamycin (10 nM) before morphine and Tat were assayed for (G) H2O2 and (H) O2− generation using amplex red and SOD-inhibitable cytochrome c reductase assays, respectively. Mean +SEM of at least 3 independent experiments. A, D, G) *P < 0.05, **P < 0.01, ***P < 0.001 vs. control, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. morphine, $P < 0.05, $$P < 0.01, $$$0.001 vs. Tat, @P < 0.05, @@P < 0.01, @@@P < 0.001 vs. combined morphine and Tat (M+T). (B, C, F, H) *P < 0.001 vs. control, #P < 0.001 vs. morphine, $0.001 vs. Tat, @P < 0.001 vs. M+T treatment. Scale bars: 50 μm .
Next we investigated if ROS is the initiator of the morphine-Tat-mediated increase in autophagy by pretreating the cells with antioxidant cocktail, NADPH oxidase inhibitor L-ascorbate, glutathione and α-tocopherol, for 20 min followed by 6 h of M+T treatment. As shown in Fig. 7D, there was a significant reduction in morphine-Tat-mediated increased ULK1 and BECN1 expression in presence of antioxidants. Immunofluorescence staining showed reduction in the LC3B puncta in morphine-Tat-treated HPMEC on pretreatment with antioxidants (Fig. 7E). We further confirmed the role of ROS in morphine-Tat-mediated autophagy induction by analysis of autophagy flux in the presence or absence of the antioxidant cocktail. As illustrated in Fig. 7F, cells treated with M+T in the presence of antioxidants did not show increase in the expression of MAP1LC3B-II expression both in the presence and absence of BAF when compared with only BAF-treated or untreated controls. However, as observed earlier in Fig. 3C, the morphine-Tat-treated cells in the absence of antioxidants showed significantly higher MAP1LC3B-II levels compared to untreated control and this increase further increased significantly in the presence of BAF (Fig. 7F).
Modulation of oxidative stress due to chronic induction of autophagy by Tat and morphine
While we investigated how ROS modulates M + T-mediated induction of autophagy we also examined how autophagy in turn modulates ROS. Effect of autophagy inhibition using 3-MA as well as autophagy stimulation using rapamycin on production of ROS was examined in HPMEC treated with morphine and/or Tat for 1, 3, 6, 12 h or 1 to 9 d. Inhibition of autophagy using 3-MA resulted in further increase in M+T-mediated induction of H2O2 and O2− radicals at all time intervals tested (Fig. 7G), whereas stimulation of autophagy using rapamycin could bring down the M+T-mediated increase in oxidative stress to the levels observed in control untreated cells (Fig. 7H). Overall, our findings suggest that excess generation of ROS in response to combined treatment with M+T upregulates autophagy which in turn helps in scavenging oxidative stress to a level that may impede cell death and promote cell proliferation.
Increased autophagy in the endothelium of the remodeled vessels of SIV-infected macaques exposed to morphine
Based on our earlier published data10 showing increased apoptosis and proliferation of endothelial cells within the early-stage vascular lesions and actively proliferating endothelial cells with absence of TUNEL positive cells within the advanced-stage lesions in SIV-infected macaques exposed to morphine (VM group), we speculated that autophagy may be more enhanced in these macaque lungs. Hence we first analyzed the expression of autophagic markers in the lungs of these macaques by western blot. As shown in Fig. 8A, there was a significant increase in the expression of BECN1, ATG5 and ATG7 in VM group compared to only virus-infected macaques (V group). The expression of ATG5 in VM lungs was also significantly higher compared to uninfected morphine exposed macaques (M group).
Figure 8.
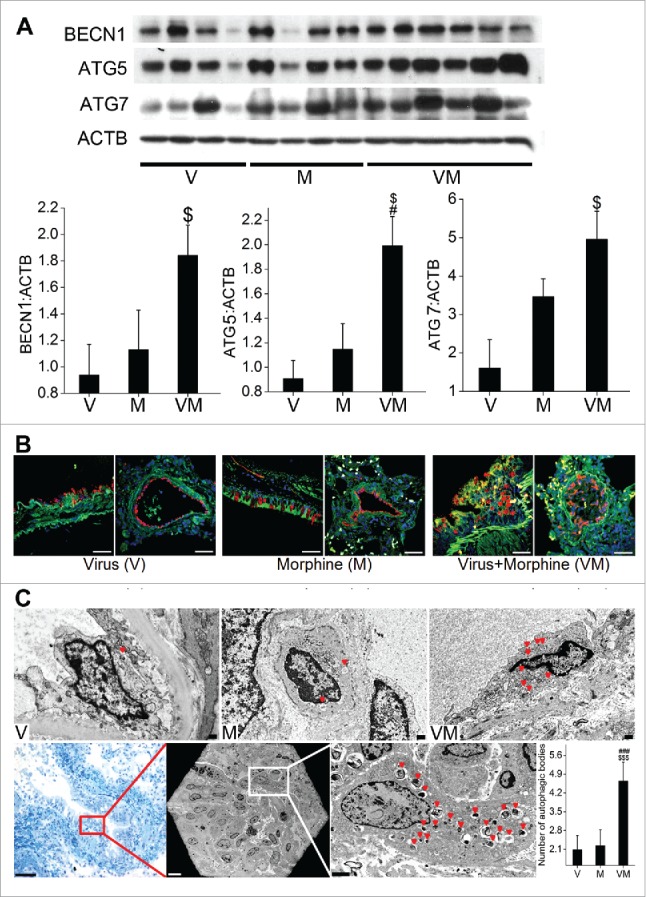
Enhanced autophagy in SIV-infected macaques exposed to morphine. (A) Total cellular extract from frozen macaque lung tissues from SIV (V group, n = 4), morphine (M group, n = 4) or SIV+morphine-injected macaques (VM group, n = 6) were probed or reprobed for the expression of autophagic markers BECN1, ATG5 and ATG7 by western blot. Graphs represent the average densitometry of the protein expression from each group. (B) The paraffin-embedded macaque lung sections immunostained for LC3B and endothelial marker (VWF). Magnification: 60X. Scale Bar 50 μm. (C) Representative TEM images of macaque lung sections. The upper panel represents 4000X magnification of endothelial cells lining the blood vessels from the macaques in the respective groups with red triangles pointing at the autophagic bodies (autophagosomes or autolysosomes). Lower panel images are of C088 macaque from the VM group. The first image (20X) is a crystal violet stained section from the cut block used for processing the tissue for TEM imaging, representing one of the areas with proliferating endothelial cells. The red box represents the approximate area of the TEM image shown in the middle, lower panel (250X). Further higher magnification image (2000X) of the inset shows a remarkably higher number of autophagic bodies in one of the cells. The graph is the average number of autophagic bodies in at least 30 endothelial cells per group (n = 3 for V or M group and n = 4 for VM group). #P < 0.05, ###P < 0.001 vs. M, $P < 0.05, $$$P < 0.001 vs V. Scale bars: 500 nm.
We also observed a notable increase in the expression of MAP1LC3B protein (stained green) in the VWF (von Willebrand factor)-positive (stained red) endothelial lining of pulmonary vessels in VM macaques by immunostaining the paraffin embedded lung sections (Fig. 8B). The V group showed a relatively weak and diffuse distribution of MAP1LC3B staining in the endothelial lining of vessels, whereas qualitatively more intense staining was observed in the M group as shown in the representative images. Maximum increase in staining intensity was observed in the VM group compared to the V or M groups as indicated by merged yellow-orange, intense staining well distributed throughout the endothelial cells lined as multiple layers or within the occluded vessels from VM macaques.
To further confirm these findings we examined the presence of autophagic bodies (autophagosomes or autolysosomes) by TEM in the pulmonary vessels of these macaques. As represented graphically in Fig. 8C, presence of maximum autophagic bodies was detected in the endothelial cells lining the vessel walls of VM lungs compared to V or M groups. The upper panel of Fig. 8C shows the representative endothelial cell from each group showing the presence of autophagosomes and autolysosomes. Lower panel shows the images from CO88 macaque from VM group that showed remarkably more vascular remodeling with advanced pulmonary vascular and obliterated plexiform lesions as reported earlier.10 The first image is from the cut block used for processing the tissue for TEM imaging, representing one of the areas with proliferating endothelial cells. The next is the TEM imaging from the same area under the grid followed by a closer look at these cells. As expected, we saw remarkably higher number of autophagic bodies in these endothelial cells with presence of autophagosomes.
Increase in the expression of autophagy proteins and in number of autophagic bodies in human lungs from HIV-infected intravenous drug users
We next examined the expression of autophagy proteins in the frozen lungs from HIV-infected IVDUs (HIV+ IVDU) that showed more enhanced pulmonary arteriopathy than HIV-infected nondrug users (HIV) or uninfected IVDUs (IVDU) alone as reported in our previous findings.9 HIV-infected IVDUs showed significantly higher expression of BECN1 when compared with uninfected, non-IVDUs (normal) or with the HIV group (Fig. 9A). However, ATG5 and ATG7 expression was significantly higher in the HIV+ IVDU group compared to all other groups.
Figure 9.
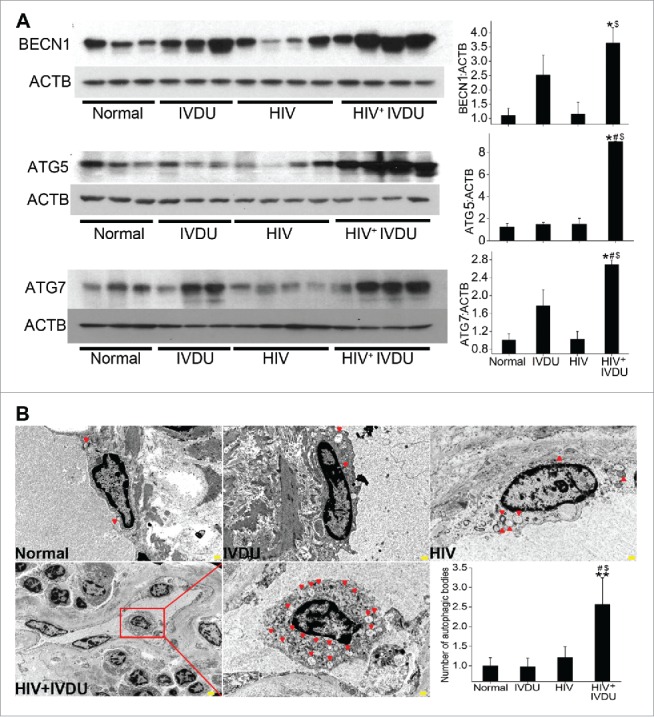
Significant increase in the expression of autophagy related proteins and number of autophagosomes or autolysosomes in the human lungs from HIV-infected IVDUs. (A) Western blot analysis of total lung extracts from normal, HIV-infected and /or IVDUs for BECN1, ATG5 and ATG7 expression. Graphs represent the densitometry analysis of the blots. (B) TEM analysis showing autophagic bodies (autophagosomes or autolysosomes) in the endothelial lining of the pulmonary blood vessels (3000X magnification). Lower panel shows the 800X magnification image with an area (red box) of the blood vessel from where representative 3000X image of HIV+ IVDU group was captured. The graph represents the average number of autophagic bodies counted in at least 24 cells per group (n = 2 per group). *P < 0.05, **P < 0.01 vs. normal, #P < 0.05 vs. IVDU, $P < 0.05 vs HIV. Scale bars: 500 nm.
Additionally, we performed TEM analysis on newly procured human lung tissues from all 4 mentioned groups. In concordance with our previous findings, these lungs also showed significant vascular remodeling in HIV+/− IVDUs including presence of medial hypertrophy, intimal lesions, blebbing and proliferation of endothelial cells as indicated by ACTA2 and VWF staining seen by brown coloration (Fig. S6). Correlating with these findings, TEM analysis represented in Fig. 9B showed remarkably higher number of autophagic bodies in the endothelial cells lining the pulmonary vessels of HIV+ IVDU compared to HIV or IVDU groups. The graph represents the overall significantly elevated counts of autophagic bodies in HIV+ IVDU humans compared to normal, HIV or IVDU groups.
Discussion
Endothelial injury has been proposed to be a critical step in the initiation and progression of vascular remodeling associated with the pulmonary hypertension.33 Researchers have observed that endothelial alterations precede the development of muscularization of pulmonary arteries in the animal model of PAH.34 The initial apoptotic injury of pECs followed by hyperproliferation of apoptosis-resistant cells33 is believed to be the cause of severe pulmonary hypertension characterized by complex pulmonary vascular lesions. Previously we reported that the treatment of pECs with morphine and HIV-protein Tat resulted in enhanced apoptosis followed by enhanced proliferation compared to either treatment alone.10 Accumulating evidence suggests autophagy to be a major cytoprotective pathway that is activated as a defense mechanism upon persistent stress exposure. Here we report remarkable induction of autophagy in HPMEC in response to exposure to the combined stress of HIV-Tat and morphine as early as 3 h of treatment. Maximum increase in the expression of autophagy-associated proteins was observed after 6 h in M+T-treated cells compared to control with continuation of higher expression of these proteins until 3 to 6 d. This chronic increase in autophagy with combined treatment was significantly more than that observed with either Tat or morphine treatment alone.
Recently Bernard et al. have reported transcriptional regulation of autophagy genes with induction in the expression of autophagy proteins as early as 1 h of serum starvation.35 However, our current findings indicate that the observed modulation of autophagy proteins in response to M+T is not dependent on transcriptional regulation as we did not observe changes in the mRNA expression of autophagy related proteins including LC3B (data not shown). Furthermore, no changes in the levels of these proteins in the presence of actinomycin D but significant inhibition in the presence of cycloheximide suggests involvement of translational or post-translation modifications such as phosphorylation, deubiquitination or acetylation that may provide stability to protein.36 The findings are critically justified given that autophagy-related proteins are constitutively expressed in unperturbed cells, and their post-translation modification and/or interaction with other regulatory autophagy molecules seems to be critical for their function rather than regulation of their gene expression.37 In a recent study USP19 (ubiquitin specific peptidase 19) was identified as a positive regulator of autophagy, which stabilizes BECN1 by protecting its degradation through the ubiquitin-proteasome pathway.38 In addition, Rodriguez et al. demonstrate modulation of total MAP1LC3B protein by BAG3-dependent regulation of MAP1LC3B mRNA translation in HeLa and HEK293 cells.39 Redox-dependent modification of ATG proteins has also been proved as regulatory sensors for autophagy proteins.40 Hence the regulation of autophagy-related proteins is highly complex that varies with cell type and biophysical environment. It would be interesting to expand our work on understanding the mechanism(s) involved in M+T-mediated increase in ATG proteins observed as early as 3 h post-treatment.
Mounting evidence suggests modulation of autophagy in response to HIV-141,42 or morphine.24 Depending on cell type, the effect of HIV-1 on autophagy varies with the course of productive infection or bystander effect of viral proteins. Although autophagy proteins are required for virus replication in CD4+T lymphocytes,43 the autophagy process is reported to restrict HIV infection in these cells. Recently Sagnier et al. report inhibition of virus replication in CD4+T cells by autophagy-mediated degradation of Tat.44 However the process of autophagy-mediated virus restriction is counteracted by blockage of autophagy on productive infection of CD4+T cells contributing to viral spread and persistence.44 Recently, HIV-Vif has been demonstrated to restrict autophagy by interacting with MAP1LC3B in productively infected CD4+T cells.45 Furthermore, Sagnier et al. report induction of autophagy-dependent apoptosis of uninfected bystander CD4+T cells by envelope (Env) glycoprotein.44 In contrast, Env protein does not induce autophagy in uninfected macrophages and autophagy is necessary for virus replication in these cells. While the initial stages of autophagy are preserved or induced upon HIV-infection46 of macrophages, the maturation stages of autophagy are inhibited on binding of HIV-Nef to BECN1,47 therefore preventing degradation and favoring higher virus yields.
Nevertheless, HIV-proteins play different role in case of noninfectable cells, as demonstrated in our current report and shown by others. HIV-Tat monotreatment increases autophagy in U87MG and A-172 glioblastoma cells.48 Similarly, morphine exposure induces autophagy in murine hippocampi and also in primary microglial cells as early as one d post-treatment.49 Zhao et al.24 have also reported induction of autophagy in response to morphine in a neuroblastoma cell line. Likewise we also observed increase in BECN1 and ATG7 expression on treatment with morphine alone as early as 3 h. Interestingly, this was accompanied by an increase in the SQSTM1 expression and no significant increase in MAP1LC3B-II with morphine treatment suggesting inhibition of autophagosome maturation and autolysosomal formation as recently reported by Wan et al. in bone marrow-derived macrophages treated with morphine and LPS.50 However, the time-course experiments on the combined effect of HIV-Tat and morphine revealed induction in ATG protein expression and autophagy flux until 24 h post-treatment with maximum induction observed at 6 h post-treatment. Higher levels of ATG proteins on combined treatment were observed at all time points tested. Contrary to this, El-Hage et al. previously have demonstrated decrease in autophagy on combined morphine-Tat treatment in human microglia.51 Although Dever et al.52 also report lower autophagy activity in neurons on M+T coexposure initially at 8 h post-treatment, increased autophagy was observed at 24 h of combined treatment, again establishing cell-type-dependent differences in the autophagy response.
The relationship between autophagy, cell death and cell survival is complex. Many scenarios demonstrate where autophagy inhibits apoptosis and is offered as a way to adapt to stress and cell survival. In other cases augmented autophagy leads to type II cell death.16,53 Tanaka et al. have earlier reported inhibition of apoptosis through interactions of MAP1LC3B with Fas-dependent apoptotic pathways.54 Additionally, studies have shown that activation of MAP1LC3B-I to MAP1LC3B-II results in promoting endothelial cell survival and inhibiting apoptotic properties.55 This is in line with our findings demonstrating further increase in the M+T-mediated apoptosis with concomitant reduction in the proliferation of cells on inhibition of autophagy in M+T-treated HPMEC. Alternatively, significant reduction in M+T-mediated early apoptosis and further exacerbation in the proliferation was observed in the stimulation of autophagy. Furthermore, reports suggest that autophagy can modulate unwarranted cell death by degrading the proapoptotic proteins. Also, studies suggest autophagy induction in response to apoptosis as a feedback mechanism and this may lead to cell survival.55
HIV-protein Tat, the transactivating factor of HIV-1 is actively secreted by infected cells and is known to promote growth and migration of endothelial cells through production of various growth factors as well as the induction of apoptosis of microvascular endothelial cells via caspase activation.56-59 HIV-Tat can elicit these responses by either entry into the target cells or interacting with cell-surface receptors such as KDR or integrins.60 Similar to Tat, morphine also stimulates both proapoptotic and prosurvival signals61-64 in various cell types via binding to its OPRM1 receptors.65 We have also found that the Tat and morphine-mediated effect on autophagy involves binding to KDR and OPRM1 receptor, respectively. Morphine has been shown to transactivate VEGF receptors via binding to its OPRM1 receptors.65 We have reported earlier attenuation in the phosphorylation of KDR at 24 h but augmentation in its phosphorylation and total expression at 6 d treatment with HIV-Tat and morphine. These changes in the activation of KDR correlated with the initial apoptosis and later hyperproliferation of pECs on chronic exposure to both HIV-proteins and morphine.10 Interestingly, studies have shown that BECN166 or ATG567 knockdown impairs VEGF-stimulated angiogenesis. In light of these reports and considering our previous and current findings, it may be possible that M+T-mediated chronic stimulation of autophagy lead to VEGFR stimulation and associated angiogenic activation of the pulmonary endothelium.
A growing number of reports suggests reactive oxygen species (ROS) as one of the important regulators of autophagy.68 Chen et al. have shown that ROS induces autophagy independent of apoptosis in transformed and cancer cells.69 In our previous publication we report a significant increase in the generation of H2O2 and O2− in HPMECs on the combined treatment with Tat and morphine when compared to either treatment alone.10 Furthermore, pretreatment of cells with antioxidants prevents the M+T-mediated increase in initial apoptosis followed by enhanced proliferation of HPMECs.10 In our current findings we demonstrate the attenuation of M+T-mediated enhanced autophagy of endothelial cells in the presence of antioxidants. It is likely that initial burst in ROS generation during early treatments of Tat and morphine induces a simultaneous increase in both autophagy and apoptosis leading to early cell death. However, the continuous activation of controlled autophagy in response to chronic exposure to Tat and morphine may prevent the accumulation of cytotoxic levels of ROS by removal of damaged organelles, thereby aiding the cells to adapt to chronic stress leading to cell survival. Future studies will be focused on determining if chronic activation of autophagy mitigates the cytotoxic levels of ROS by removal of ROS generating damaged mitochondria.
Our ex vivo data regarding significantly increased autophagy protein expression in VM group of macaques compared to V or M groups alone is noteworthy because in the same VM macaque group we have earlier10 reported the presence of significant severe pulmonary arteriopathy with angio-obliteration due to enhanced proliferation of pECs. Our current TEM data on the lung sections from these macaques show significantly higher number of autophagic bodies in the endothelial cells lining the vessels with medial hypertrophy and in the cells of fully occluding vessels. In addition our current findings of significantly higher expression of autophagy proteins in HIV-infected lung tissues from IV opioids and/or cocaine abusers that we previously have reported to have enhanced pulmonary vascular remodeling compared to lungs from normal, HIV or IVDU alone9 further highlights the association of autophagy with pulmonary endothelial dysfunction and vascular remodeling. As the vascular remodeling in these SIV-infected macaque and HIV-infected human lungs exposed to illicit drugs also involved smooth muscle hyperplasia, we also looked at the status of autophagy in morphine-Tat-exposed hyperproliferative human pulmonary arterial smooth muscle cells (HPASMCs).10 Surprisingly, we did not observe any differences in the autophagosome formation in cells exposed to either combined or monotreatments when compared to untreated cells (Fig. S7) both in the presence or absence of BAF. This suggests that the involvement of dynamic process of autophagy in protecting cells from initial morphine-Tat-mediated apoptosis leading to cell survival is unique to endothelial cells.
Taken together, our current and previous findings suggest a potential link between autophagy and HIV and /or opioid-mediated enhancement of proliferation of apoptosis-resistant pulmonary endothelial cells. We speculate that initial exposure of pulmonary endothelial cells to M+T, results in independent activation of both autophagy and apoptosis, however, chronic increase in autophagy brings down oxidative stress and apoptosis that allows the cells to adapt to stress leading to cell survival and uncontrolled proliferation of pECs (Fig. 10). Autophagy and its regulatory proteins have recently gained considerable focus not only in the context of PAH but also in the pathogenesis of other chronic lung diseases in general,23 and understanding which agents could further enhance it or vice versa, is important as a preventive strategy. Further research is needed to understand exactly the mechanism(s) involved in HIV-protein(s) or opioid-induced autophagy and associated lung injury to develop novel therapeutic approaches in future.
Figure 10.
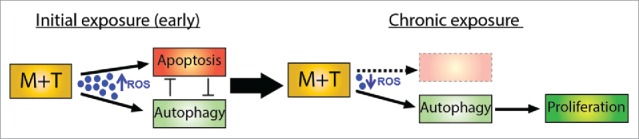
Schematic representation of autophagy-mediated hyperproliferation of pulmonary endothelial cells on chronic exposure to morphine and Tat.
Materials and methods
Cell culture and treatments
Human primary pulmonary microvascular endothelial cells (ScienCell Laboratories, 3000) were grown in endothelial cell basal medium containing 5% fetal bovine serum (FBS), endothelial cell growth supplements and penicillin-streptomycin (ScienCell Laboratories, 1001). At 80% confluency, the medium was replaced with endothelial cell medium containing 0.5% FBS. Cells were then treated with morphine (1 µM; Sigma-Aldrich, M8777) in the presence or absence of recombinant HIV-Tat (25 ng/ml; ProSpec, HIV-124) daily for various time intervals. The concentration of M+T was based on our previous published findings.10
Western blot analysis
HPMEC treated with morphine and/or Tat from 3 h to 9 d were lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (Santa Cruz Biotechnology, sc-24948) and then used for western blot analysis as described previously.70 For measurement of autophagic flux, cells were pretreated with 10 nM of BAF (Sigma-Aldrich, 11707) for 30 min before morphine and/or Tat treatment followed by protein extraction after 24 h and western blot analysis for MAP1LC3B (Cell Signaling Technology, 2775) and SQSTM1 (EMD Millipore, MABC32) expression. Total cellular extract from frozen macaque or human lung tissues was also probed for the expression of autophagic markers by western blot analysis. Blots were probed or reprobed with antibodies against ULK1, BECN1, ATG5, and MAP1LC3B (Cell Signaling Technology, 8054, 3738, 2630, 2775 respectively), ATG7 (EMD Millipore, 04-1055) or LAMP1 (Abcam, ab24170). The NIH ImageJ software was used for densitometric analysis of immunoblots.
MAP1LC3B detection by immunofluoroscence
HPMEC, pretreated with/or without BAF (10 nM) followed by morphine and/or Tat for 24 h, were stained live with LysoTracker® Red dye (50 nM; Invitrogen, L7528), formaldehyde-fixed, followed by MAP1LC3B (1:400) staining. Fixed cells were then incubated in the appropriate Alexa Fluor 488-goat anti-rabbit (Invitrogen, A11008) secondary antibody and mounted with Prolong Gold antifade reagent with DAPI (Invitrogen, P36935). Alternatively, cells were preincubated with antioxidant cocktail 0.2 mM L-ascorbate (Sigma, 795437), 0.5 mM glutathione and 3.5 µM α-tocopherol (Santa Cruz Biotechnology, sc-29094, sc-294383 respectively), 30 min before Tat and morphine treatment for 6 h followed by immunostaining for MAP1LC3B. Labeled cells were imaged using inverted Nikon Eclipse E2000-U laser-scanning confocal microscope (Nikon Inc., USA) using the linear sequential scan mode function (laser lines: diode 408, argon 488 and helium-neon 543 lines, excitation/emission filters, 450/35 nm, 515/30 nm and 605/75 nm). Maximum intensity projections of a Z stack were generated using ECZ1 software (Nikon Corporation, Melville, NY).
Transfection
HPMEC were transfected with 300 ng of pBABE-puro mCherry-EGFP-LC3B plasmid (gift from Jayanta Debnath, Addgene plasmid #22418) using GeneJuice® reagent (Novagen, 70967) in serum free ECM. The medium was changed to complete ECM after 8 h followed by treatment after 2 d with morphine and/or tat in the presence or absence of either BAF (1 nM) or rapamycin (10 nM). At 24 h post-treatment, cells were fixed with 4% paraformaldehyde and viewed using confocal microscope as described above.
Transmission electron microscopy
Direct visualization and quantification of autophagosomes and autolysosomes (autophagic bodies) in HPMEC treated with or without morphine and/or Tat; and in the endothelial cells present in the pulmonary vessels of macaque and human lung tissues was performed using TEM. Briefly, M+T-treated cells, or frozen macaque or human lung were fixed in 2% glutaraldehyde in sodium cacodylate and post-fixed in 1% osmium tetroxide. Cells and tissue samples were dehydrated in graded series of ethanol; the samples were infiltrated and embedded in Embed 812 resin (EMS, 14120), sectioned at 80 nm on a Leica UC-7 Ultramicrotome (Leica Microsystem Inc., USA), stained in uranyl acetate and lead citrate and viewed with a J.E.O.L. JEM1400 transmission electron microscope (JEOL USA, INC.,) at 100 kV. Autophagic bodies were quantified in cells from each treatment type as described by others.71,72
RT-PCR
Confluent HPMEC were treated with morphine and/or Tat for 30 min, 1 h, 2 h, 3 h and 6 h followed by RNA extraction using Trizol reagent (Life Technologies 15596026). Quantitative RT-PCR was performed for checking the expression of ULK1, BECN1, ATG5, ATG7 and MAP1LC3B using SYBR green reagent as mentioned in our previous publications.8
Dichlorodihydrofluorescein (DCF) assay
HPMEC were incubated with DCF (Cell Biolabs, STA-342) for 30 min followed by morphine and/or Tat treatment for 3 h in the presence or absence of naloxone (1 µM) and/or Su5416 (0.5 µM) (Sigma, N7758, S8442 respectively). Cells were then lysed and intracellular ROS was measured by florescence quantification as described previously.11
Measurement of H2O2 and superoxide (O2−) radicals
After 80% confluency, HPMEC plated on 96-well plates were treated with morphine and or Tat in the presence or absence of either rapamycin (10 nM) or 3-methyladenine (10 mM) for various time intervals followed by the measurement of extracellular H2O2 using Amplex red assay kit (Invitrogen, A22188) and superoxide radicals by SOD/superoxide dismutase-inhibitable cytochrome C reductase assay as described by us previously in detail.10 The generated H2O2 or O2− was normalized with the respective cell count in each well using CyQUANT® Cell Proliferation Assay Kit (Invitrogen, C7026) as per manufacturer's instructions.
Apoptosis ELISA
HPMEC (1.25 × 104/well) were pretreated with either 3-MA or temozolomide (TMZ, 50 µM) (Sigma, M9281, T2577 respectively) 20 min before morphine and/or Tat treatment for 3 d followed by apoptosis ELISA (Roche Applied Science, 11544675001) as described previously.10 Alternatively, ELISA was conducted on cells transfected with siRNA against ULK1 or scrambled siRNA (Ambion, 4427037) or with plasmid EX-M0809-M02-ULK1 (Genecopoeia, M0809) using HiPerfect (Qiagen, 301704) or GeneJuice reagent (Novagen, 70967-3) respectively as per the manufacturers' instructions.
MTS cell proliferation assay
HPMEC (3 × 103/well) treated with 3-MA or TMZ or transfected with ULK1 siRNA or ULK1 plasmid before morphine and/or Tat exposure as mentioned above for 6 d were analyzed for the difference in cell proliferation using CellTiter 96® Aqueous One Solution Cell Proliferation
Assay (Promega, G3580). We initially used 3 different concentrations of TMZ (10, 50 and 100 µM and found that 100 µM had a toxic effect whereas the 10 µM concentration did not show a significant difference between TMZ-treated and untreated in case of combined M+T treatment.
Macaque lung tissues and sections
The snap-frozen archival lung tissues from SIV-infected rhesus macaques (virus+morphine [VM] group) or uninfected macaques (morphine [M] only group) exposed to morphine for up to 59 wk and SIV-infected macaques unexposed to morphine (virus only [V] group) were assessed for markers of autophagy by western blot. The details about virus infection, morphine treatment and comparison of pulmonary arteriopathy are mentioned in our previous publication.10 Frozen tissues were also sectioned for analysis of autophagosomes or autolysosomes by TEM. The paraffin-embedded lung sections were immunostained for LC3B (Cell Signaling Technology, 2775) and VWF (Dako, M0616) as described previously.10
Human lung tissues and sections
We previously reported enhanced pulmonary vascular remodeling in HIV-infected lung tissues from IV opioids and/or cocaine abusers.9 Total lung extract from the same human frozen lungs were used to analyze the expression of autophagy proteins by western blot. These human lung tissues as reported previously9 were obtained at the time of autopsy from HIV-infected individuals with (HIV+ IVDU group, n = 4) or without IVDU (HIV group, n = 4) and uninfected individuals with (IVDU group, n = 3) from the Manhattan HIV Brain Bank (R24MH59724; U01MH083501; New York, NY). Normal uninfected archival controls (normal group, n = 3) were from National Disease Research Interchange (NDRI, Philadelphia, PA). The clinical, demographic and pathological characteristics of the lung samples and more details of these human subjects are included in our previous report.9 Meanwhile additional frozen lung tissues from unidentified individuals within the normal, HIV, IVDU and HIV+ IVDU groups (n = 2 each) were procured from NDRI and were used for TEM analysis. These IVDUs were also heroin and/or cocaine abusers. Paraffin embedded lung sections were used for ACTA2 and VWF staining.9
Statistical analysis
Statistical analysis was performed using one-way analysis of variance with a post-hoc Bonferroni correction for multiple comparisons. Two-sided P values were calculated for analyzing all in vitro experiments using GraphPad Prism. The results were judged statistically significant when the Bonferroni corrected P values were less than 0.05. A nonparametric Wilcoxon Rank Sum test at a significance level of 0.05 was used for western blot and TEM quantification of autophagosomes from macaque and human lung tissues due to limited sample size.
Supplementary Material
Abbreviations
- ACTB
actin β
- ACTA2
α-smooth muscle actin
- ATG5
autophagy-related 5
- ATG7
autophagy-related 7
- BECN1
Beclin 1
- ECM
endothelial cell medium
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HIV-1
human immunodeficiency virus
- HPMECs
human pulmonary microvascular endothelial cells
- HRPAH
human immunodeficiency virus-related pulmonary arterial hypertension
- IVDU
intravenous drug use
- KDR
kinase insert domain receptor
- LAMP1
lysosomal associated membrane protein 1
- 3-MA
3-methyladenine
- M group
morphine group
- M+T
morphine and Tat
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- O2-
superoxide
- PAH
pulmonary arterial hypertension
- pECs
pulmonary endothelial cells
- RIPA
radioimmunoprecipitation assay
- ROS
reactive oxygen species
- SIV
simian immunodeficiency virus
- SQSTM1/p62
squestosome 1
- Su5416
Semaxanib
- Tat
HIV-transactivator of transcription
- TEM
transmission electron microscopy
- ULK1
unc-51 like autophagy activating kinase 1
- VM group
virus group
- VEGF
vascular endothelial growth factor
- VM group
virus + morphine
- VWF
von Willebrand factor
Disclosure of potential conflicts of interest
The authors have no conflicts of interest to be disclosed.
Acknowledgments
We wish to acknowledge the Electron Microscopy Research Lab (EMRL) facility and Barbara Fegley for assistance with the tissue preparation, sectioning, electron microscopy and counting of autophagic bodies.
Funding
This work was supported by NIH grants: R01DAO34542, R56HL129875 and R03DAO31589 and American Heart Association's Scientist Development grant 11SDG7500016 awarded to N.K.D. and by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (NIH) P20 GM103418. The EMRL is supported in part, by NIH COBRE grant 9P20GM104936. The JEOL JEM-1400 TEM used in the study was purchased with funds from NIH grant S10RR027564.
References
- [1].Monsuez JJ, Charniot JC, Escaut L, Teicher E, Wyplosz B, Couzigou C, Vignat N, Vittecoq D. HIV-associated vascular diseases: structural and functional changes, clinical implications. Int J Cardiol 2009; 133:293-306; PMID:19131130; http://dx.doi.org/ 10.1016/j.ijcard.2008.11.113 [DOI] [PubMed] [Google Scholar]
- [2].Cicalini S, Chinello P, Petrosillo N. HIV infection and pulmonary arterial hypertension. Expert Rev Respir Med 2011; 5:257-66; PMID:21510735; http://dx.doi.org/ 10.1586/ers.11.10 [DOI] [PubMed] [Google Scholar]
- [3].Barnett CF, Hsue PY, Machado RF. Pulmonary hypertension: an increasingly recognized complication of hereditary hemolytic anemias and HIV infection. Jama 2008; 299:324-31; PMID:18212317; http://dx.doi.org/ 10.1001/jama.299.3.324 [DOI] [PubMed] [Google Scholar]
- [4].Mehta NJ, Khan IA, Mehta RN, Sepkowitz DA. HIV-Related pulmonary hypertension: analytic review of 131 cases. Chest 2000; 118:1133-41; PMID:11035689; http://dx.doi.org/ 10.1378/chest.118.4.1133 [DOI] [PubMed] [Google Scholar]
- [5].Opravil M, Sereni D. Natural history of HIV-associated pulmonary arterial hypertension: trends in the HAART era. Aids 2008; 22 Suppl 3:S35-40; PMID:18845920; http://dx.doi.org/ 10.1097/01.aids.0000327514.60879.47 [DOI] [PubMed] [Google Scholar]
- [6].Nunes H, Humbert M, Sitbon O, Morse JH, Deng Z, Knowles JA, Le Gall C, Parent F, Garcia G, Herve P, et al.. Prognostic factors for survival in human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2003; 167:1433-9; PMID:12615632; http://dx.doi.org/ 10.1164/rccm.200204-330OC [DOI] [PubMed] [Google Scholar]
- [7].Quezada M, Martin-Carbonero L, Soriano V, Vispo E, Valencia E, Moreno V, de Isla LP, Lennie V, Almeria C, Zamorano JL. Prevalence and risk factors associated with pulmonary hypertension in HIV-infected patients on regular follow-up. AIDS 2012; 26:1387-92; PMID:22526521; http://dx.doi.org/ 10.1097/QAD.0b013e328354f5a1 [DOI] [PubMed] [Google Scholar]
- [8].Dalvi P, O'Brien-Ladner A, Dhillon N. Downregulation of bone morphogenetic protein receptor axis during HIV-1 and cocaine-mediated pulmonary smooth muscle hyperplasia: implications for HIV-related pulmonary arterial hypertension. Arteriosclerosis, Thrombosis, and Vascular Biology 2013; 33:2585-95; PMID:24008158; http://dx.doi.org/ 10.1161/ATVBAHA.113.302054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, Ladner AO. Effect of cocaine on human immunodeficiency virus-mediated pulmonary endothelial and smooth muscle dysfunction. Am J Respir Cell Mol Biol 2011; 45:40-52; PMID:20802087; http://dx.doi.org/ 10.1165/rcmb.2010-0097OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Spikes L, Dalvi P, Tawfik O, Gu H, Voelkel NF, Cheney P, O'Brien-Ladner A, Dhillon NK. Enhanced pulmonary arteriopathy in simian immunodeficiency virus-infected macaques exposed to morphine. Am J Respir Crit Care Med 2012; 185:1235-43; PMID:22447963; http://dx.doi.org/ 10.1164/rccm.201110-1909OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mermis J, Gu H, Xue B, Li F, Tawfik O, Buch S, Bartolome S, O'Brien-Ladner A, Dhillon NK. Hypoxia-inducible factor-1 alpha/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respir Res 2011; 12:103; PMID:21819559; http://dx.doi.org/ 10.1186/1465-9921-12-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2007; 293:L548-54; PMID:17526595; http://dx.doi.org/ 10.1152/ajplung.00428.2006 [DOI] [PubMed] [Google Scholar]
- [13].Cho YY, Kim DJ, Lee HS, Jeong CH, Cho EJ, Kim MO, Byun S, Lee KY, Yao K, Carper A, et al.. Autophagy and cellular senescence mediated by Sox2 suppress malignancy of cancer cells. PloS one 2013; 8:e57172; PMID:23451179; http://dx.doi.org/ 10.1371/journal.pone.0057172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mosieniak G, Adamowicz M, Alster O, Jaskowiak H, Szczepankiewicz AA, Wilczynski GM, Ciechomska IA, Sikora E. Curcumin induces permanent growth arrest of human colon cancer cells: link between senescence and autophagy. Mech Ageing Dev 2012; 133:444-55; PMID:22613224; http://dx.doi.org/ 10.1016/j.mad.2012.05.004 [DOI] [PubMed] [Google Scholar]
- [15].El Hasasna H, Athamneh K, Al Samri H, Karuvantevida N, Al Dhaheri Y, Hisaindee S, Ramadan G, Al Tamimi N, AbuQamar S, Eid A, et al.. Rhus coriaria induces senescence and autophagic cell death in breast cancer cells through a mechanism involving p38 and ERK1/2 activation. Scientific Rep 2015; 5:13013; PMID:26263881; http://dx.doi.org/ 10.1038/srep13013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol 2010; 22:241-5; PMID:19945838; http://dx.doi.org/ 10.1016/j.ceb.2009.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ryter SW, Lee SJ, Smith A, Choi AM. Autophagy in vascular disease. Proc Am Thoracic Soc 2010; 7:40-7; PMID:20160147; http://dx.doi.org/ 10.1513/pats.200909-100JS [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, et al.. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4:e838; PMID:24113172; http://dx.doi.org/ 10.1038/cddis.2013.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 2015; 4:184-92; PMID:25590798; http://dx.doi.org/ 10.1016/j.redox.2014.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, et al.. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 2011; 183:649-58; PMID:20889906; http://dx.doi.org/ 10.1164/rccm.201005-0746OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Qipshidze N, Tyagi N, Metreveli N, Lominadze D, Tyagi SC. Autophagy mechanism of right ventricular remodeling in murine model of pulmonary artery constriction. Am J Physiol Heart Circ Physiol 2012; 302:H688-96; PMID:22101525; http://dx.doi.org/ 10.1152/ajpheart.00777.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res 2013; 112:1159-70; PMID:23446737; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.300483 [DOI] [PubMed] [Google Scholar]
- [23].Ryter SW, Nakahira K, Haspel JA, Choi AM. Autophagy in pulmonary diseases. Annu Rev Physiol 2012; 74:377-401; PMID:22035347; http://dx.doi.org/ 10.1146/annurev-physiol-020911-153348 [DOI] [PubMed] [Google Scholar]
- [24].Zhao L, Zhu Y, Wang D, Chen M, Gao P, Xiao W, Rao G, Wang X, Jin H, Xu L, et al.. Morphine induces Beclin 1- and ATG5-dependent autophagy in human neuroblastoma SH-SY5Y cells and in the rat hippocampus. Autophagy 2010; 6:386-94; PMID:20190558; http://dx.doi.org/ 10.4161/auto.6.3.11289 [DOI] [PubMed] [Google Scholar]
- [25].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Ann Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/ 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Seidel M, Billert H, Kurpisz M. Regulation of eNOS expression in HCAEC cell line treated with opioids and proinflammatory cytokines. Kardiologia polska 2006; 64:153-8; discussion 9-60; PMID:16502366 [PubMed] [Google Scholar]
- [27].Mendel DB, Laird AD, Smolich BD, Blake RA, Liang C, Hannah AL, Shaheen RM, Ellis LM, Weitman S, Shawver LK, et al.. Development of SU5416, a selective small molecule inhibitor of VEGF receptor tyrosine kinase activity, as an anti-angiogenesis agent. Anti-cancer drug design 2000; 15:29-41; PMID:10888034 [PubMed] [Google Scholar]
- [28].Brock R, Hamelers IH, Jovin TM. Comparison of fixation protocols for adherent cultured cells applied to a GFP fusion protein of the epidermal growth factor receptor. Cytometry 1999; 35:353-62; PMID:10213201; http://dx.doi.org/ 10.1002/(SICI)1097-0320(19990401)35:4%3c353::AID-CYTO8%3e3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- [29].Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol 2010; 221:117-24; PMID:20225337; http://dx.doi.org/ 10.1002/path.2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang B, Ling S, Lin WC. 14-3-3Tau regulates Beclin 1 and is required for autophagy. PloS one 2010; 5:e10409; PMID:20454448; http://dx.doi.org/ 10.1371/journal.pone.0010409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883; http://dx.doi.org/ 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- [32].Essick EE, Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid Med Cell Longev 2010; 3:168-77; PMID:20716941; http://dx.doi.org/ 10.4161/oxim.3.3.12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J 2005; 19:1178-80; PMID:15897232 [DOI] [PubMed] [Google Scholar]
- [34].Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol 1988; 255:H1484-91; PMID:3144186 [DOI] [PubMed] [Google Scholar]
- [35].Bernard A, Jin M, Gonzalez-Rodriguez P, Fullgrabe J, Delorme-Axford E, Backues SK, Joseph B, Klionsky DJ. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr Biol 2015; 25:546-55; PMID:25660547; http://dx.doi.org/ 10.1016/j.cub.2014.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jin S, Tian S, Chen Y, Zhang C, Xie W, Xia X, Cui J, Wang RF. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J 2016; 35:866-80; PMID:26988033; http://dx.doi.org/ 10.15252/embj.201593596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rodriguez AE, Lopez-Crisosto C, Pena-Oyarzun D, Salas D, Parra V, Quiroga C, Morawe T, Chiong M, Behl C, Lavandero S. BAG3 regulates total MAP1LC3B protein levels through a translational but not transcriptional mechanism. Autophagy 2016; 12:287-96; PMID:26654586; http://dx.doi.org/ 10.1080/15548627.2015.1124225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu Z, Lenardo MJ. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev Cell 2007; 12:484-5; PMID:17419989; http://dx.doi.org/ 10.1016/j.devcel.2007.03.016 [DOI] [PubMed] [Google Scholar]
- [41].Wang X, Gao Y, Tan J, Devadas K, Ragupathy V, Takeda K, Zhao J, Hewlett I. HIV-1 and HIV-2 infections induce autophagy in Jurkat and CD4+ T cells. Cell Signall 2012; 24:1414-9; PMID:22406083; http://dx.doi.org/ 10.1016/j.cellsig.2012.02.016 [DOI] [PubMed] [Google Scholar]
- [42].Faruqi M. Peptides: activating autophagy. Nat Rev Drug Discov 2013; 12:190; PMID:23449301; http://dx.doi.org/ 10.1038/nrd3970 [DOI] [PubMed] [Google Scholar]
- [43].Eekels JJ, Sagnier S, Geerts D, Jeeninga RE, Biard-Piechaczyk M, Berkhout B. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol J 2012; 9:69; PMID:22424437; http://dx.doi.org/ 10.1186/1743-422X-9-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sagnier S, Daussy CF, Borel S, Robert-Hebmann V, Faure M, Blanchet FP, Beaumelle B, Biard-Piechaczyk M, Espert L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol 2015; 89:615-25; PMID:25339774; http://dx.doi.org/ 10.1128/JVI.02174-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Borel S, Robert-Hebmann V, Alfaisal J, Jain A, Faure M, Espert L, Chaloin L, Paillart JC, Johansen T, Biard-Piechaczyk M. HIV-1 viral infectivity factor interacts with microtubule-associated protein light chain 3 and inhibits autophagy. AIDS 2015; 29:275-86; PMID:25490467; http://dx.doi.org/ 10.1097/QAD.0000000000000554 [DOI] [PubMed] [Google Scholar]
- [46].Espert L, Varbanov M, Robert-Hebmann V, Sagnier S, Robbins I, Sanchez F, Lafont V, Biard-Piechaczyk M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PloS one 2009; 4:e5787; PMID:19492063; http://dx.doi.org/ 10.1371/journal.pone.0005787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al.. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009; 186:255-68; PMID:19635843; http://dx.doi.org/ 10.1083/jcb.200903070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bruno AP, De Simone FI, Iorio V, De Marco M, Khalili K, Sariyer IK, Capunzo M, Nori SL, Rosati A. HIV-1 Tat protein induces glial cell autophagy through enhancement of BAG3 protein levels. Cell Cycle 2014; 13:3640-4; PMID:25483098; http://dx.doi.org/ 10.4161/15384101.2014.952959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pan J, He L, Li X, Li M, Zhang X, Venesky J, Li Y, Peng Y. Activating Autophagy in Hippocampal Cells Alleviates the Morphine-Induced Memory Impairment. Mol Neurobiol 2016; Epub ahead of print; PMID:26873855; http://dx.doi.org/25850855 10.1007/s12035-016-9735-3 [DOI] [PubMed] [Google Scholar]
- [50].Wan J, Ma J, Anand V, Ramakrishnan S, Roy S. Morphine potentiates LPS-induced autophagy initiation but inhibits autophagosomal maturation through distinct TLR4-dependent and independent pathways. Acta Physiol (Oxf) 2015; 214:189-99; PMID:25850855; http://dx.doi.org/ 10.1111/apha.12506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].El-Hage N, Rodriguez M, Dever SM, Masvekar RR, Gewirtz DA, Shacka JJ. HIV-1 and morphine regulation of autophagy in microglia: limited interactions in the context of HIV-1 infection and opioid abuse. J Virol 2015; 89:1024-35; PMID:25355898; http://dx.doi.org/ 10.1128/JVI.02022-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dever SM, Rodriguez M, Lapierre J, Costin BN, El-Hage N. Differing roles of autophagy in HIV-associated neurocognitive impairment and encephalitis with implications for morphine co-exposure. Front Microbiol 2015; 6:653; PMID:26217309; http://dx.doi.org/ 10.3389/fmicb.2015.00653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025-40; PMID:15657430; http://dx.doi.org/ 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol 2012; 46:507-14; PMID:22095627; http://dx.doi.org/ 10.1165/rcmb.2009-0415OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jin Y, Choi AM. Cross talk between autophagy and apoptosis in pulmonary hypertension. Pulmonary Circulation 2012; 2:407-14; PMID:23372925; http://dx.doi.org/ 10.4103/2045-8932.105029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chi D, Henry J, Kelley J, Thorpe R, Smith JK, Krishnaswamy G. The effects of HIV infection on endothelial function. Endothelium 2000; 7:223-42; PMID:11201521; http://dx.doi.org/ 10.3109/10623320009072210 [DOI] [PubMed] [Google Scholar]
- [57].Albini A, Soldi R, Giunciuglio D, Giraudo E, Benelli R, Primo L, Noonan D, Salio M, Camussi G, Rockl W, et al.. The angiogenesis induced by HIV-1 tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat Med 1996; 2:1371-5; PMID:8946838; http://dx.doi.org/ 10.1038/nm1296-1371 [DOI] [PubMed] [Google Scholar]
- [58].Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res 2003; 74:255-65; PMID:14515355; http://dx.doi.org/ 10.1002/jnr.10762 [DOI] [PubMed] [Google Scholar]
- [59].Avraham HK, Jiang S, Lee TH, Prakash O, Avraham S. HIV-1 Tat-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J Immunol 2004; 173:6228-33; PMID:15528360; http://dx.doi.org/ 10.4049/jimmunol.173.10.6228 [DOI] [PubMed] [Google Scholar]
- [60].Barillari G, Sgadari C, Fiorelli V, Samaniego F, Colombini S, Manzari V, Modesti A, Nair BC, Cafaro A, Sturzl M, et al.. The Tat protein of human immunodeficiency virus type-1 promotes vascular cell growth and locomotion by engaging the alpha5beta1 and alphavbeta3 integrins and by mobilizing sequestered basic fibroblast growth factor. Blood 1999; 94:663-72; PMID:10397733 [PubMed] [Google Scholar]
- [61].Hsiao PN, Chang MC, Cheng WF, Chen CA, Lin HW, Hsieh CY, Sun WZ. Morphine induces apoptosis of human endothelial cells through nitric oxide and reactive oxygen species pathways. Toxicology 2009; 256:83-91; PMID:19070643; http://dx.doi.org/ 10.1016/j.tox.2008.11.015 [DOI] [PubMed] [Google Scholar]
- [62].Liu HC, Anday JK, House SD, Chang SL. Dual effects of morphine on permeability and apoptosis of vascular endothelial cells: morphine potentiates lipopolysaccharide-induced permeability and apoptosis of vascular endothelial cells. J Neuroimmunol 2004; 146:13-21; PMID:14698842; http://dx.doi.org/ 10.1016/j.jneuroim.2003.09.016 [DOI] [PubMed] [Google Scholar]
- [63].Ding WG, Zhou HC, Cui XG, Li WZ, Guo YP, Zhang B, Liu W. Anti-apoptotic effect of morphine-induced delayed preconditioning on pulmonary artery endothelial cells with anoxia/reoxygenation injury. Chin Med J (Engl) 2008; 121:1313-8; PMID:18713554 [PubMed] [Google Scholar]
- [64].Gupta K, Kshirsagar S, Chang L, Schwartz R, Law PY, Yee D, Hebbel RP. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res 2002; 62:4491-8; PMID:12154060 [PubMed] [Google Scholar]
- [65].Chen C, Farooqui M, Gupta K. Morphine stimulates vascular endothelial growth factor-like signaling in mouse retinal endothelial cells. Curr Neurovasc Res 2006; 3:171-80; PMID:16918381; http://dx.doi.org/ 10.2174/156720206778018767 [DOI] [PubMed] [Google Scholar]
- [66].Ramakrishnan S, Nguyen TM, Subramanian IV, Kelekar A. Autophagy and angiogenesis inhibition. Autophagy 2007; 3:512-5; PMID:17643071; http://dx.doi.org/ 10.4161/auto.4734 [DOI] [PubMed] [Google Scholar]
- [67].Du J, Teng RJ, Guan T, Eis A, Kaul S, Konduri GG, Shi Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol Cell Physiol 2012; 302:C383-91; PMID:22031599; http://dx.doi.org/ 10.1152/ajpcell.00164.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gibson SB. Investigating the role of reactive oxygen species in regulating autophagy. Methods Enzymol 2013; 528:217-35; PMID:23849868; http://dx.doi.org/ 10.1016/B978-0-12-405881-1.00013-6 [DOI] [PubMed] [Google Scholar]
- [69].Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15:171-82; PMID:17917680; http://dx.doi.org/ 10.1038/sj.cdd.4402233 [DOI] [PubMed] [Google Scholar]
- [70].Dhillon NK, Li F, Xue B, Tawfik O, Morgello S, Buch S, O'Brien Ladner A. Effect of Cocaine on HIV-mediated Pulmonary Endothelial and Smooth Muscle Dysfunction. Am J Respir Cell Mol Biol 2011; 45(1):40-52; PMID:20802087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Barth JM, Szabad J, Hafen E, Kohler K. Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ 2011; 18:915-24; PMID:21151027; http://dx.doi.org/ 10.1038/cdd.2010.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011; 12:408-15; PMID:21478880; http://dx.doi.org/ 10.1038/ni.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.