

ABSTRACT
Automatic classification of LTR retrotransposons is a big challenge in the area of massive genomics. Many tools were developed to detect them but automatic classification is somehow challenging. Here we propose a simple approach, LTRclassifier, based on HMM recognition followed by BLAST analyses (i) to classify plant LTR retrotransposons in their respective superfamily, and (ii) to provide automatically a basic functional annotation of these elements. The method was tested on various TE databases, and shown to be robust and fast. This tool is available as a web service implemented at IRD bioinformatics facility, http://LTRclassifier.ird.fr/.
KEYWORDS: classification, LTR retrotransposons, superfamily
Introduction
Because of the current availability of complete genome sequences, a lot of efficient tools have been developed to identify LTR (Long-terminal Repeat) retrotransposons.1-3 Those tools are generally based on structure recognition of those elements: terminal direct repeats (LTR), flanked by target site duplication, and with a reverse transcriptase (RT) motif between the 2 LTRs. However, only a very few of them is able to provide informations about the non-RT motifs, or to provide a classification in superfamily (copia or gypsy, Fig. 1; see Wicker et al.4).
Figure 1.
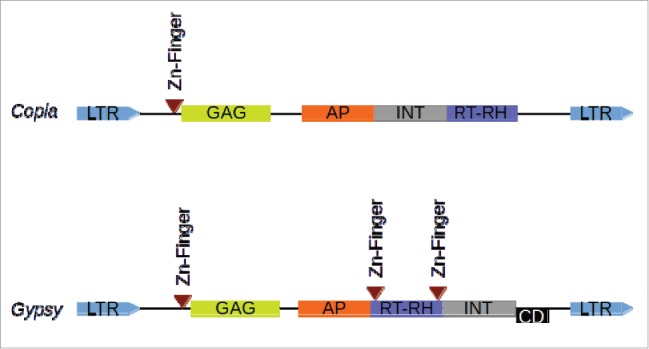
Structure of LTR retrotransposons. The order of RT-RH/INT determines the corresponding superfamily. For Ggypsy elements, the CD (Chromo Domain) is present for a subset of elements only (Athila related). GAG: Group-specific AntiGen; AP: Aspartic Protease; INT: INTegrase; RT-RH: Reverse Transcriptase - RnaseH; LTR: Long Terminal Repeat.
Some recent heavy implementations have been published for local analyses and classification,5-7 but neither on line tool nor light implementation was proposed yet. Here we described LTRclassifier, a web server based on a suite of perl scripts to classify a set of LTR retrotransposons in their superfamily. LTRclassifier was tested on different datasets of LTR retrotransposons, from a single sequence to thousands, and shown a very efficient recovery score in a short amount of time.
Results and discussion
Annotation and classification results
In terms of efficiency and step (Table 1), we can assign to a superfamily from 26% to 99% of elements, depending on the considered tested public TE database used as target for the analysis. Thus, with the exception of RepBase (see below the discussion on the limits), LTRclassifier was able to automatically classify about 75% of the total elements, with a sensibility of 80% to 98%, and a specificity higher than 95% (see Materials & Methods). Its precision is higher than 97%, as it wrongly annotated as LTR retrotransposon only 2.2% of the complete DNA elements from TREP (data not shown).
Table 1.
Benchmarking upon various nucleic databases of LTR retrotransposons (number of LTR are shown in bracket). N/A means no informations of classification was available from the database at the superfamily level.
| Database (number of LTR RT) | TIGR grasses (711) | TIGR Oryza (4033) | Retroryza (241) | RepBase (2356) | TREP (98) |
|---|---|---|---|---|---|
| Pfam classified, % | 38.5 | 45.9 | 90 | 0.2 | 55.1 |
| BLASTX classified, % | 29.7 | 35.7 | 7.9 | 25.8 | 20.3 |
| BLASTN classified, % | 1.3 | 5.1 | 0.8 | 0.2 | 0.3 |
| Total classified, % | 69.5 | 86.7 | 98.7 | 26.2 | 75.7 |
| Analysis time, 4 cores | 23′ 57″ | 102′ 44″ | 10′ 42″ | 53′ 13″ | 5′ 8″ |
| Sensitivity | 80 % | 95 % | N/A | 30 % | 92,5 % |
| Specificity | 95 % | 97,5 % | N/A | 97,5 % | 97,5 % |
LTRclassifier can annotate quickly (less than 2h for 4,000 LTR retrotransposon sequences), and with a high confidence (95 to 97.5% of specificity), the frames for any LTR retrotransposon sequence, complete element or not. It can detect the normal HMM motifs, protein motifs and nucleic similarity if needed, and provides position/annotation information for proteins and motifs in tabular text files. Moreover, it will provide informations about 2 types of abnormal structures to improve annotation: (i) detection of opposite strands as coding (e. g. Frame +1 for gag and -1 for RVE), (ii) detection of motifs from copia and gypsy.
Limits of the approach on consensus sequences
As shown on Table 1, the results on RepBase are low in terms of classification (only 26,2% of LTR retrotransposons vs almost 70% for TIGR grasses database). Most sequences in RepBase are consensus sequences from large families and subfamilies of repeats, and are used with tools such as RepeatMasker,8 Censor 9 or BLASTN, to mask and annotate repetitive DNA in genomes. These consensus nucleic sequences do not represent true sequences and have induced frameshifts modifying the translation, and then not allowing the PfamScan/HMM analysis, as well as BLASTX one on a limited scale, to provide good results under our conditions (data not shown). The structure of RepBase is thus very efficient in terms of massive genome annotation (or transcriptome), but a consensus sequence cannot be used to annotate the element itself for its functional components.
Conclusion
A lot of currently available tools can detect genomic structure of LTR retrotransposons (LTRharvest,2 LTR_finder,10 LTR_Struct,1 REPET,11). However, only few provide informations about the functional annotation of those elements in term of proteins and motifs, and they are generally either heavy to install (REPCLASS,7 LTRdigest,12 TEclassifier from REPET, PASTEC13 eg.) and not available on-line (Table 2).
Table 2.
Comparison of existing classification tools.
| Software | URL | TE type | Interface | Remark |
|---|---|---|---|---|
| TEclass | http://www.compgen.uni-muenster.de/teclass | DNA transposons, LTRs, LINEs, SINEs | Command line | The input should not contain more than a few thousand sequences. The entered data must not exceed 1MB in size. |
| LTRSift | http://www.zbh.uni-hamburg.de/?id=378 | LTRs | Graphical/Desktop | gff3 file (annotation data) is required, it could be generated by LTRHarvest and LTRdigest. |
| RepClass | https://sourceforge.net/projects/repclass | DNA transposons, LTRs, non-LTRs, Hélitrons | Command line | Dependencies: wu-blast. No documentation available about installation or usage. |
| PASTEC | https://urgi.versailles.inra.fr/Tools/PASTEClassifier | DNA transposons, LTRs, non-LTRs, Hélitrons | Command line | Pastec is the REPET package Transposable Elements classifier used in the TEdenovo pipeline, and the corresponding standalone tool is PASTEClassifier.py. |
Here we present a fast on line tool able to annotate functionally a large set of LTR retrotransposons. The provided data are strong enough to make of LTRclassifier a efficient companion tool for any genome annotation improvement and LTR retrotransposon evolution study.
Materials and methods
Pipeline of annotation
The whole pipeline sequence is shown in Fig. 2. Firstly, the element sequences are translated in the 6 frames (positive and negative; for nucleic data only) using transeq tool from the EMBOSS package 14 and the translated sequences submitted to a PfamScan.pl v1.3 analysis,15 using the version 26.0 of Pfam database, to identify their putative functional motifs. Specific motifs were manually selected (Table 3) after large analyses using well defined LTR retrotransposons. The retain criteria were to be associated with normal LTR retrotransposon motifs AND only with them.
Figure 2.
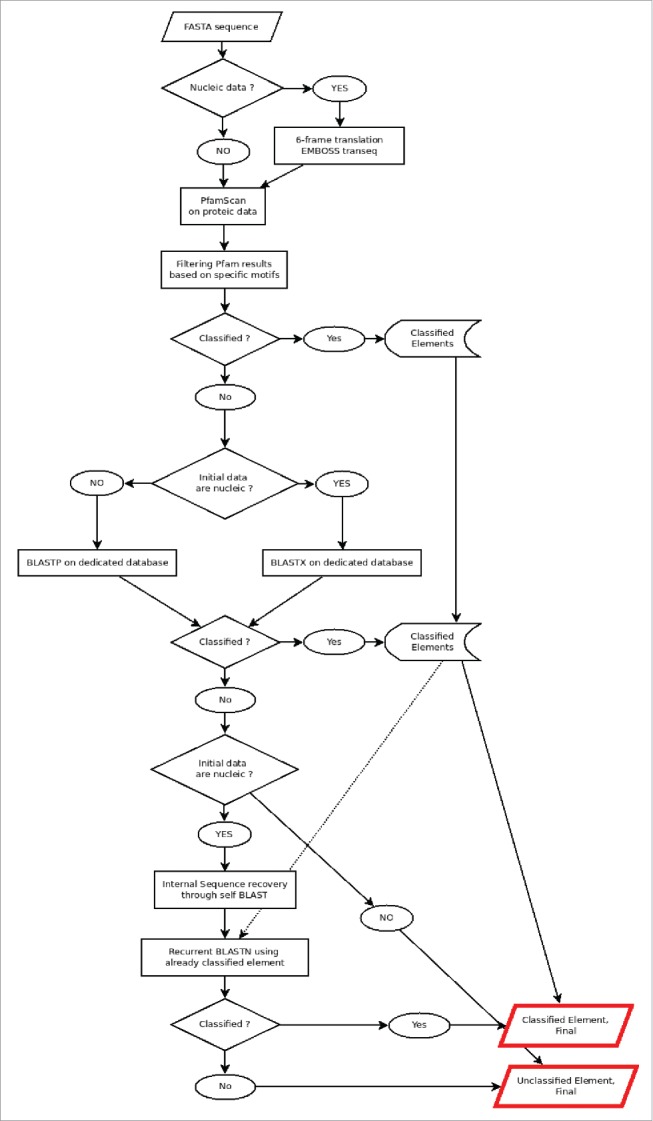
General scheme of the analysis. The 6-frames translation and the recurrent BLASTN steps are optional to nucleic data.
Table 3.
Pfam motifs retained for the classification.
| General Motifs | Copia Motifs | Gypsy Motifs | |
|---|---|---|---|
| DUF4283 | DUF4219 | Asp_protease | RVP_2 |
| RNase_H | gag_pre_integrase | Asp_protease_2 | RVT_1 |
| rve | RVT_2 | ATHILA | RVT_3 |
| rve_3 | UBN2 | Chromo | Transposase_28 |
| RVP | UBN2_2 | DUF390 | Zf-CCHC_4 |
| zf-CCHC | UBN2_3 | gag-asp_protease | Zf-RVT |
Note. #houba–>Copia UBN2_2-410..524-3 zf-CCHC-569..586-3 gag_pre-integrs-735..803-3 rve-817..932-3 RVT_2-1149..1392-3
The conserved results from PfamScan step are 85% significant (see PfamScan.pl documentation for more details), with a minimal score of 50. Other motifs are conserved but are not used for superfamily classification, but only to validate the sequence as a LTR retrotransposon. Based on the identified motifs, the elements are then classified as copia, gypsy, unclassified. The Pfam-classified elements are also provided with their functional annotation based on Pfam motifs location.
The not yet classified elements are then subject to a BLASTX16 analysis, using as reference database a home-made concatenation of the TREPprot17 and plant RepbaseProt18,19 (developed for REPET) databases. The retained criteria for classification here are 25% of identity, for a minimal length of 80 residues.
The final step for nucleic input is a recurrent BLASTN analysis, trying to classify still unclassified sequences using their homology with already classified ones. This BLASTN is performed on the internal sequence only, if determination of LTR position is possible. For that the system will use a BLASTN of the sequence vs itself and check if 2 highly similar sequences (the LTR) can be identified on each side of the target sequence; if yes, the internal sequence will be extracted and used in the next step. Else, the BLASTN analysis will be performed on the whole sequence. The limits are 80% of identity, on at least 80 bases, and a minimal length of 80 bases for the query (as described in Wicker et al.4). The here classified elements are then added to the database, and the analysis is re-iterated until no more element can be added to the classified ones.
The final output will be a set of files, as described in Table S1 and Fig. S1.
Databases description
The databases used for the annotation itself are Pfam (version 26.0), RepBaseProt (version 17.0 for REPET pipeline, 5,844 proteic sequences) and TREPprot (version 11.0, 193 proteic sequences). Pfam contains HMM motifs for functional annotation; TREPprot and RepBaseProt contain proteic sequences of already known TE from diverse organisms. Here we limited the set of proteic data to plant LTR retrotransposons sequences, but it can be theoretically extended to any kingdom to identify copia and gypsy elements.
Testing material (sequences and computer)
The tested sequences for implementation and benchmarking are RetrOryza,20 TIGR grasses repeats (one representative element per family), TIGR Oryza repeats (all annotated elements), RepBase (v. 17) and TREPtotal (v. 11). From those sequence libraries, we selected only the LTR retrotransposons sequences for our tests.
The benchmarks were performed using 4 threads per assay. The current version is using 2 threads for each analysis.
The sensibility was calculated as the percentage of element classified by LTRclassifier upon the total number of already classified elements in the reference tested set. The sensitivity was expressed as the percentage of correctly classified element by LTRclassifier compared to the reference tested data set (already annotated elements).
Availability and implementation
The perl scripts are available at http://LTRclassifier.ird.fr/LTRclassifierScripts.tar.gz, under GPLv3.
The version implemented on the IRD bioinformatics cluster is available at http://LTRclassifier.ird.fr/. The input is limited to 8 Mbytes of data, with a minimum of a single sequence.
Supplementary Material
Abbreviations
- BLAST
basic local alignment search tool
- HMM
Hidden Markov Models
- LTR
Long Terminal Repeats
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Authors want to thanks Benoit Piegu for the consensus problem identification, and the two anonymous reviewers for their comments.
ORCID
Francois Sabot http://orcid.org/0000-0002-8522-7583
References
- [1].McCarthy EM, McDonald JF. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 2003; 19:362-7; PMID:12584121; http://dx.doi.org/ 10.1093/bioinformatics/btf878 [DOI] [PubMed] [Google Scholar]
- [2].Ellinghaus D, Kurtz S, Willhoeft U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 2008; 9:18; PMID:18194517; http://dx.doi.org/ 10.1186/1471-2105-9-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Flutre T, Duprat E, Feuillet C, Quesneville H. Considering transposable element diversification in de novo annotation approaches. PLoS One 2011; 6:e16526; PMID:21304975; http://dx.doi.org/ 10.1371/journal.pone.0016526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al.. A unified classification system for eukaryotic transposable elements. Nat Rev Genet 2007; 8:973-82; PMID:17984973; http://dx.doi.org/ 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- [5].Abrusán G, Grundmann N, DeMester L, Makalowski W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009; 25:1329-30; http://dx.doi.org/ 10.1093/bioinformatics/btp084 [DOI] [PubMed] [Google Scholar]
- [6].Steinbiss S, Kastens S, Kurtz S. LTRsift: a graphical user interface for semi-automatic classification and postprocessing of de novo detected LTR retrotransposons. Mob DNA 2012; 3:18; PMID:23131050; http://dx.doi.org/ 10.1186/1759-8753-3-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feschotte C, Keswani U, Ranganathan N, Guibotsy ML, Levine D. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol Evol 2009; 1:205-20; PMID:20333191; http://dx.doi.org/ 10.1093/gbe/evp023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Smit, AFA, Hubley, R, Green, P. RepeatMasker Open-3.0. 1996-2010. http://www.repeatmasker.org [Google Scholar]
- [9].Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 2006; 7:474; PMID:17064419; http://dx.doi.org/ 10.1186/1471-2105-7-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 2007; 35:W265-8; PMID:17485477; http://dx.doi.org/ 10.1093/nar/gkm286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Flutre T, Duprat E, Feuillet C, Quesneville H. Considering transposable element diversification in de novo annotation approaches. PLoS One 2011; 6:e16526; PMID:21304975; http://dx.doi.org/ 10.1371/journal.pone.0016526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Steinbiss S, Willhoeft U, Gremme G, Kurtz S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res 2009; 37:7002-13; PMID:19786494; http://dx.doi.org/ 10.1093/nar/gkp759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hoede C, Arnoux S, Moisset M, Chaumier T, Inizan O, Jamilloux V, Quesneville H. PASTEC: an automatic transposable element classification tool. PLoS One 2014; 9:e91929; PMID:24786468; http://dx.doi.org/ 10.1371/journal.pone.0091929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rice P, Longden I, Bleasby A. EMBOSS: The European molecular biology open software suite. Trends Genet 2000; 16:276-7; PMID:10827456; http://dx.doi.org/ 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- [15].Mistry J, Bateman A, Finn RD. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 2007; 8:298; PMID:17688688; http://dx.doi.org/ 10.1186/1471-2105-8-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403-10; PMID:2231712; http://dx.doi.org/ 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- [17].Wicker T, Matthews DE, Keller B. TREP: a database for Triticeae repetitive elements. Trends Plant Sci 2002; 7:561-2; http://dx.doi.org/ 10.1016/S1360-1385(02)02372-5 [DOI] [Google Scholar]
- [18].Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462-7; PMID:16093699; http://dx.doi.org/ 10.1159/000084979 [DOI] [PubMed] [Google Scholar]
- [19].Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 2015; 6:11; PMID:26045719; http://dx.doi.org/ 10.1186/s13100-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chaparro C, Guyot R, Zuccolo A, Piégu B, Panaud O. RetrOryza: a database of the rice LTR-retrotransposons. Nucleic Acids Res 2007; 35:D66-70; PMID:17071960; http://dx.doi.org/ 10.1093/nar/gkl780 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.