

ABSTRACT
Chemotactic migration is a fundamental behavior of cells and its regulation is particularly relevant in physiological processes such as organogenesis and angiogenesis, as well as in pathological processes such as tumor metastasis. The majority of chemotactic stimuli activate cell surface receptors that belong to the G protein-coupled receptor (GPCR) superfamily. Although the autophagy machinery has been shown to play a role in cell migration, its mode of regulation by chemotactic GPCRs remains largely unexplored. We found that ligand-induced activation of 2 chemotactic GPCRs, the chemokine receptor CXCR4 and the urotensin 2 receptor UTS2R, triggers a marked reduction in the biogenesis of autophagosomes, in both HEK-293 and U87 glioblastoma cells. Chemotactic GPCRs exert their anti-autophagic effects through the activation of CAPNs, which prevent the formation of pre-autophagosomal vesicles from the plasma membrane. We further demonstrated that CXCR4- or UTS2R-induced inhibition of autophagy favors the formation of adhesion complexes to the extracellular matrix and is required for chemotactic migration. Altogether, our data reveal a new link between GPCR signaling and the autophagy machinery, and may help to envisage therapeutic strategies in pathological processes such as cancer cell invasion.
KEYWORDS: autophagosome biogenesis, cell adhesion, chemotactic migration, CXCR4, GPCR, urotensin II
Introduction
Cell migration is a key aspect of many physiological processes, including angiogenesis, immune cell trafficking, and organogenesis, as well as pathological processes such as tumor progression and metastasis. In response to chemotactic stimuli, actin polymerization at the front of the cell drives the formation of membrane protrusions, referred to as lamellipodia. Subsequently, lamellipodia are stabilized by attachment to the extracellular matrix through integrin-mediated adhesion complexes. Once coupled to adhesion complexes, the actin cytoskeleton can then generate the forces necessary to translocate the cell body forward.1
The majority of chemotactic stimuli, including the chemotactic cytokines (chemokines), bind to cell surface receptors that belong to the G protein-coupled receptor (GPCR) superfamily. As a prototypical chemokine receptor, the GPCR named CXCR4 (C-X-C motif chemokine receptor 4) has received much attention. Activation of CXCR4 by its ligand, CXCL12 (C-X-C motif chemokine ligand 12), has been shown to play a crucial role in the chemotaxis of many cell types including leucocytes, endothelial cells, neurons2,3 and a wide range of cancer cells.4,5 Although not fully elucidated, the signaling mechanisms by which CXCR4 promotes cell migration appear to involve the coordinated activation of the small GTPases RAC and RHOA, which participate in the formation of lamellipodia and contractile actin-myosin filaments.6 Besides “classical” chemokine receptors, several other GPCRs display chemotactic activity. This includes receptors for bioactive lipids, and for vasoactive peptides such as angiotensin II, EDNs/endothelins and UTS2 (urotensin 2). UTS2 is the most potent vasoactive peptide identified so far, and binding of UTS2 to its cognate GPCR, UTS2R (urotensin 2 receptor), stimulates the migration of glioma cells,7 fibroblasts,8 endothelial progenitor cells,9 monocytes,10 LNCaP prostatic11 and colon cancer cells.12 Importantly, UTS2-induced migration of glioma cells, endothelial progenitors and LNCaP requires the activation of RHOA,7,9,11 pointing to the idea that CXCR4 and UTS2R engage similar signaling mechanisms during chemotaxis.
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved lysosomal pathway involved in the degradation of long-lived proteins and cytoplasmic organelles. This process, which is essential for normal turnover of cellular compartments, is upregulated in response to nutrient starvation. One of the first events in autophagy is the formation of the phagophore. The edges of these phagophore membranes elongate and thereby engulf portions of cytoplasm. After the scission of the membrane edges, the structure becomes a completed autophagosome, which later fuses with lysosomes, resulting in the degradation of its lumenal content. Several highly conserved autophagy-related (ATG) proteins that function at key steps in the autophagy process have been identified.13 Initiation of the phagophore requires the BECN1/Beclin 1-containing class III phosphatidylinositol 3-kinase (PtdIns3K) complex, generation of phosphatidylinositol 3-phosphate (PtdIns3P), and recruitment of the PtdIns3P-binding protein called WIPI1.14 WIPI1 in turn recruits the ATG12–ATG5-ATG16L1 ternary complex, along with phosphatidylethanolamine-conjugated MAP1LC3B/LC3B (LC3B-II), which are essential for elongation of the phagophore membrane. Whereas the ATG12–ATG5-ATG16L1 complex decorates the phagophore and dissociates after completion of autophagosome formation,15 part of LC3B-II remains associated with fully formed autophagosomes.16,17
Numerous membrane sources are involved in the formation of autophagosomes, including the endoplasmic reticulum,18,19 mitochondria,20 and plasma membrane,21-23 suggesting that autophagy closely intersects with and depends on various membrane trafficking events inside the cell. Regarding the plasma membrane, it has been demonstrated that clathrin-mediated endocytosis induces the formation of ATG5-ATG16L1-positive vesicles, which traffic through recycling endosomes and ultimately fuel the phagophore.21,22
Autophagic activity has been shown to play a role in cell migration.24-26 Nevertheless, the functional connection between these 2 fundamental processes is still elusive, and the impact of chemotactic GPCRs on the autophagy machinery remains largely unexplored. Several lines of data demonstrated the autophagic degradation of key proteins involved in the initiation and maturation of adhesion complexes, such as integrins,24 RHOA,27 PXN (paxillin),28 the SRC kinase,29,30 VCL (vinculin) and ZYX (zyxin).31 We found here that ligand-induced activation of 2 chemotactic GPCRs, CXCR4 or UTS2R, triggers a marked reduction in the biogenesis of autophagosomes. Chemotactic GPCRs do not affect the early intracellular events of autophagy, such as class III PtdIns3K activity, but strongly reduce the targeting of ATG16L1 to pre-autophagosomal vesicles forming from the plasma membrane. We further demonstrated that the anti-autophagic activity of CXCR4 and UTS2R depends on the activation of CAPNs/calpains, favors the formation of adhesion complexes at lamellipodia and is required for chemotactic migration. Collectively, our data, obtained with 2 GPCRs and in 2 cell lines (HEK-293 and U87 glioma cells), lead us to propose that inhibition of autophagy represents a hallmark of chemotaxis and constitutes a necessary event for the maintenance of adequate pools of phospholipids and adhesion-complex proteins at the front of migrating cells.
Results
Activation of chemotactic GPCRs inhibits autophagosome biogenesis
We first evaluated autophagic activity by the use of the Cyto-ID autophagy dye, which has been shown to specifically bind to autophagosomal membranes.32 Cell fluorescence intensity, measured by flow cytometry, therefore correlates with the amount of autophagosomes.32 HEK-293 cells were transiently transfected with cDNAs encoding human CXCR4 or UTS2R, and treated with CXCL12 or UTS2, respectively. As shown in Figure 1A, activation of chemotactic GPCRs with their respective ligands significantly reduced Cyto-ID labeling, indicating a reduction in the total number of autophagosomes. This effect could be due to either a decrease in the flux of autophagosome biogenesis, or an increase in the turnover of autophagosomes due to activation of the lysosomal pathway. To distinguish between these 2 possibilities, we compared the effects of each ligand alone, or in combination with chloroquine (CQ), a lysosomotropic agent that neutralizes the acidic pH of lysosomes, thereby preventing autophagosome turnover.17 As expected, CQ treatment, by inducing accumulation of autophagosomes, evoked an increase in Cyto-ID labeling (Fig. 1B). CXCL12 or UTS2 treatment reduced Cyto-ID labeling to a similar extent in the absence or presence of CQ, indicating that the effects of the ligands likely correspond to inhibition of autophagosome biogenesis.
Figure 1.

CXCR4 and UTS2R inhibit autophagosome biogenesis. (A) HEK-293 cells expressing CXCR4 (left panels) or UTS2R (right panels) were treated (24 h) with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M). After treatment, cells were incubated with the Cyto-ID autophagy dye and fluorescence intensity was measured by flow cytometry. Data shown are cytometric profiles of a representative sample of each experimental group (10,000 cells per sample). a.u., arbitrary units; MFI, median fluorescence intensity; unlabeled, background signal without Cyto-ID incubation. Histograms show the average MFI ± SEM (n = 4; 10,000 cells per sample). (B) HEK-293 cells expressing CXCR4 (left panel) or UTS2R (right panel) were treated (24 h) with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M), with or without chloroquine (CQ; 5 × 10−5 M), as indicated. After treatment, cells were incubated with the Cyto-ID autophagy dye and fluorescence intensity was measured by flow cytometry. Data are expressed as median fluorescence intensity ± SEM (n = 4; 10,000 cells per sample). (C) HEK-293 cells expressing CXCR4 and the fluorescent protein EGFP-LC3B were treated (6 h) with or without CXCL12 (10−8 M) and chloroquine (5 × 10−5 M), as indicated. Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified on confocal images. Data represent means ± SEM, from at least 100 cells per group. Scale bars: 10 µm. (D) HEK-293 cells expressing UTS2R and the fluorescent protein EGFP-LC3B were treated (6 h) with or without UTS2 (10−9 M) and chloroquine (5 × 10−5 M), as indicated. Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified as in (C). Scale bars: 10 µm. (E) Left 2 panels: HEK-293 cells expressing CXCR4 and the fluorescent protein EGFP-LC3B were pretreated (1 h) with chloroquine (5 × 10−5 M), then placed in either rich (DMEM, 10% serum) or nutrient-deprived (HBSS) media containing chloroquine, and treated with or without CXCL12 (10−8 M) for the indicated times. After incubations, cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified as in (C). Right 2 panels: HEK-293 cells expressing UTS2R and the fluorescent protein EGFP-LC3B were pretreated (1 h) with chloroquine (5 × 10−5 M), then placed in either rich (DMEM, 10% serum) or nutrient-deprived (HBSS) media containing chloroquine, and treated with or without UTS2 (10−9 M) for the indicated times. After incubations, cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified as in (C). (F) HEK-293 cells expressing CXCR4 were treated (6 h) with or without CXCL12 (10−8 M). Cells were fixed and labeled with an anti-SQSTM1/p62 antibody (green). For each photographic field, SQSTM1 immunoreactivity was quantified and normalized to the number of nuclei (DAPI stained, blue). Data represent means ± SEM from 10 photographic fields per group. Scale bars: 20 µm. (G) HEK-293 cells expressing UTS2R were treated (6 h) with or without UTS2 (10−9 M). Cells were fixed and SQSTM1 immunoreactivity was measured as in (F). Scale bars: 20 µm. Statistical significance was evaluated using a Mann and Whitney test (A, B, F, G) or an unpaired t test (C, D, E). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not statistically different.
We next assessed autophagic activity by the use of enhanced green fluorescent protein (EGFP)-LC3B. LC3B protein is a well-established effector of autophagy and a bona fide marker for autophagosomes.33,34 Punctate EGFP-LC3B staining provides a measure of ongoing autophagy because it marks the successful processing of a cytosolic form, EGFP-LC3B-I, to EGFP-LC3B-II, a phospholipid-conjugated form that is targeted to phagophore membranes. We found that, in control conditions, the low number of EGFP-LC3B dots per cell precluded the accurate determination of UTS2 or CXCL12's inhibitory effects. Nevertheless, prevention of autophagosome degradation by the use of CQ evoked, as expected, a marked increase in the number of EGFP-LC3B puncta (Fig. 1C and 1D). Treatment with CXCL12 (Fig. 1C) or UTS2 (Fig. 1D) partially prevented the accumulation of the EGFP-LC3B-labeled autophagosomes in the presence of CQ, confirming that chemotactic receptors engage an intracellular signaling pathway leading to inhibition of autophagosome biogenesis. We next evaluated autophagosome accumulation in the presence of CQ, at steady state (Dulbecco's modified Eagle's medium [DMEM] with 10% serum), or upon serum starvation (Hank's balanced salt solution [HBSS] medium), a condition that stimulates autophagic flux. Activation of CXCR4 or UTS2R with their respective ligands markedly reduced the formation of EGFP-LC3B puncta in cells maintained in both complete or starvation medium (Fig. 1E), indicating that chemotactic GPCRs are able to inhibit autophagosome biogenesis under basal or stimulated conditions.
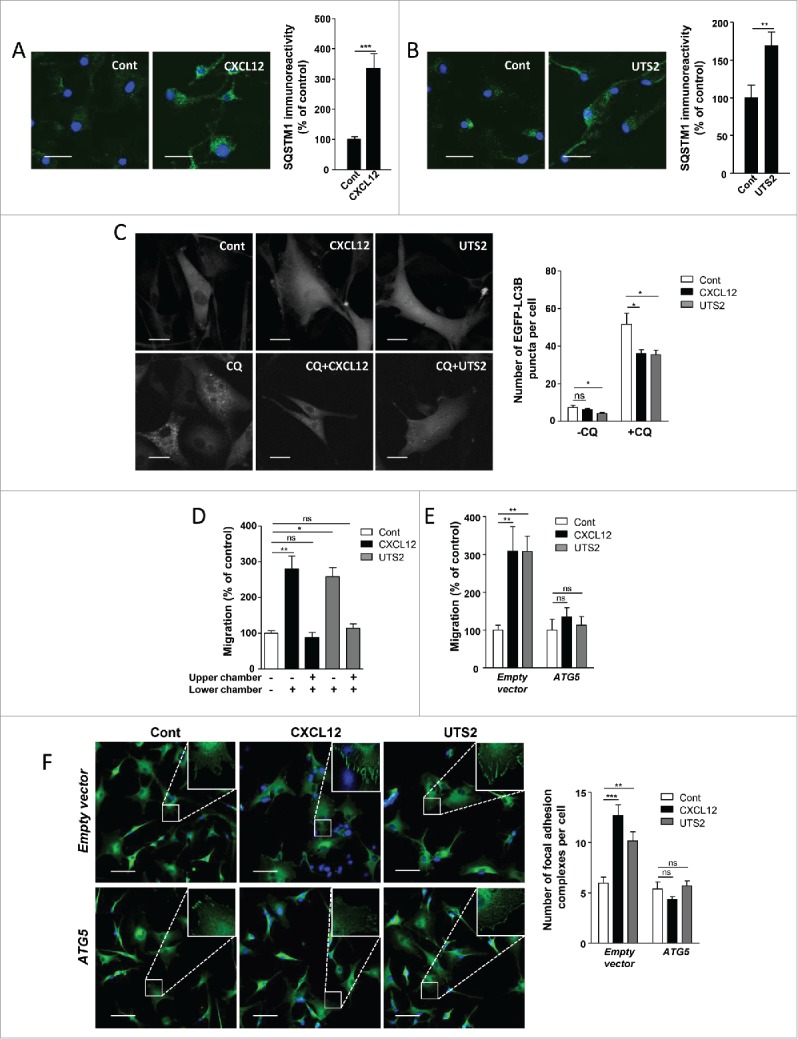
For further proof of CXCL12- and UTS2-evoked inhibition of autophagy, we performed an immunocytochemical analysis of endogenous SQSTM1/p62 levels. SQSTM1 is a ubiquitously expressed protein that can bind to ubiquitinated substrates and to LC3B on phagophores, and is itself degraded by autophagy.35,36 Therefore, impaired autophagy is accompanied by the accumulation of SQSTM1 in the cytosol, and formation of SQSTM1-ubiquitinated protein aggregates.37 As expected, a 6-h treatment with CXCL12 (Fig. 1F) or UTS2 (Fig. 1G) evoked a significant increase in SQSTM1 immunolabeling, which displayed a punctate pattern, reminiscent of cytosolic aggregates.
CXCR4- and UTS2R-evoked inhibition of autophagy is not relayed by MTOR kinase and the class III PtdIns3K complex
As a first step to determine the signaling pathway relaying the anti-autophagic effect of CXCR4 and UTS2R, we next checked whether CXCL12 or UTS2 had an effect on PP242-induced autophagy. PP242 stimulates autophagy through inhibition of MTOR (mechanistic target of rapamycin [serine/threonine kinase]).38 In line with starvation-related results, EGFP-LC3B dot formation was markedly increased after incubation with PP242 (Fig. 2A and 2B). Activation of CXCR4 (Fig. 2A) or UTS2R (Fig. 2B) strongly reduced the effects of PP242 on EGFP-LC3B staining. These data were confirmed by the use of the Cyto-ID autophagy fluorescent probe. Treatment of cells with PP242 evoked an increase in Cyto-ID labeling, consistent with autophagy induction, and this effect was reversed by cotreatment with CXCL12 or UTS2 (Fig. S1). The fact that chemotactic GPCRs can still exert potent anti-autophagic activity in the presence of MTOR inhibitors suggests that they act downstream of this kinase.
Figure 2.
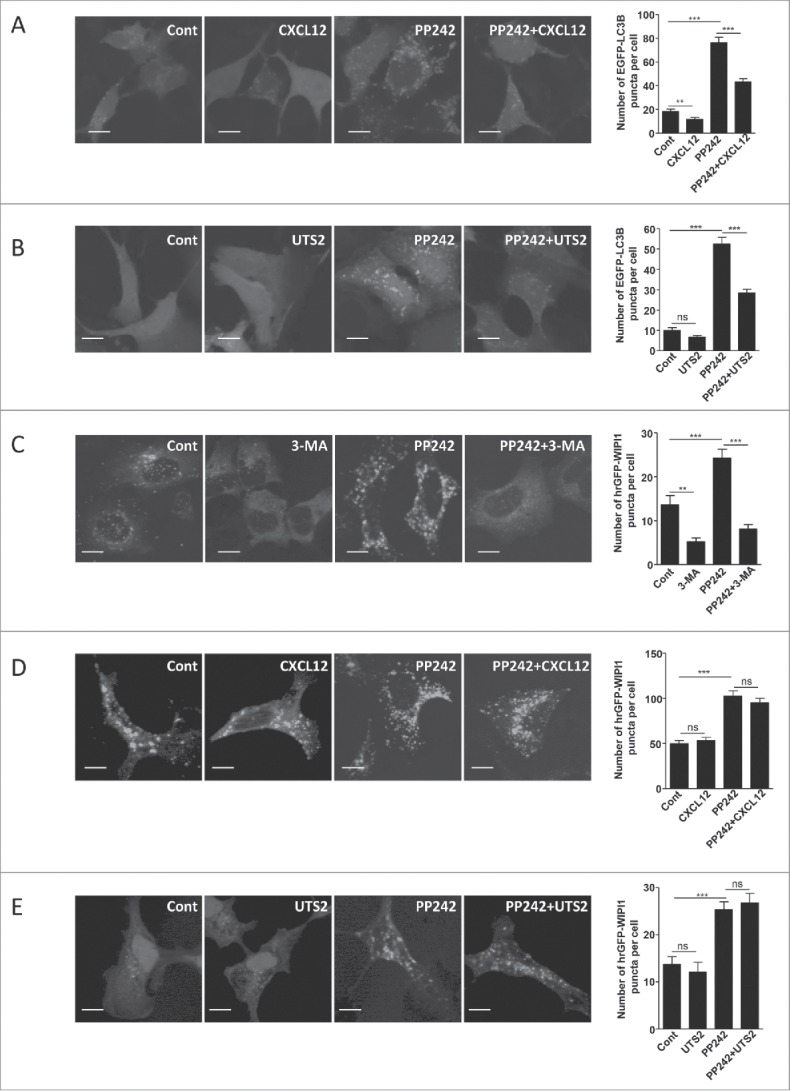
CXCR4- and UTS2R-evoked inhibition of autophagy does not depend on regulation of MTOR kinase or recruitment of WIPI1 to the phagophore. (A) HEK-293 cells expressing CXCR4 and the fluorescent protein EGFP-LC3B were treated (6 h) with or without CXCL12 (10−8 M), and the MTOR inhibitor PP242 (10−6 M), as indicated. Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified in confocal images. Data represent means ± SEM, from at least 100 cells per group. (B) HEK-293 cells expressing UTS2R and the fluorescent protein EGFP-LC3B were treated (6 h) with or without UTS2 (10−9 M), and the MTOR inhibitor PP242 (10−6 M), as indicated. Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified as in (A). (C) HEK-293 cells expressing the fluorescent protein hrGFP-WIPI1 were treated (4 h) with or without 3-MA (10−2 M), a class III PtdIns3K inhibitor, and PP242 (10−6 M), as indicated. Cells were fixed and the number of hrGFP-WIPI1 fluorescent dots per cell was quantified on confocal images. Data represent means ± SEM, from at least 100 cells per group. (D) HEK-293 cells expressing CXCR4 and the fluorescent protein hrGFP-WIPI1 were treated (6 h) with or without CXCL12 (10−8 M) and PP242 (10−6 M), as indicated. Cells were fixed and the number of hrGFP-WIPI1 fluorescent dots per cell was quantified as in (C). (E) HEK-293 cells expressing UTS2R and the fluorescent protein hrGFP-WIPI1 were treated (6 h) with or without UTS2 (10−9 M) and PP242 (10−6 M), as indicated. Cells were fixed and the number of hrGFP-WIPI1 fluorescent dots per cell was quantified as in (C). Statistical significance was evaluated in all experiments using an unpaired t test. **P < 0.01; ***P < 0.001; ns, not statistically different. Scale bars: 10 µm.
Following MTOR inhibition, formation of the phagophore requires the recruitment of the BECN1-containing class III PtdIns3K complex, generation of PtdIns3P and recruitment of the PtdIns3P-binding protein called WIPI1.14 Quantification of WIPI1 puncta can then be used to monitor autophagy initiation, and constitutes an indicator of class III PtdIns3K activity.14 As a validation step, we first monitored the number of humanized recombinant (hr)GFP-WIPI1 puncta upon treatment of HEK-293 cells with a class III PtdIns3K inhibitor, 3-methyladenine (3-MA), the autophagy inducer PP242, or a combination of both drugs. As expected, treatment with 3-MA significantly decreased, whereas PP242 significantly increased, the number of hrGFP-WIPI1 puncta (Fig. 2C). Cotreatment of PP242 with 3-MA abolished the effect of PP242 (Fig. 2C), which is in agreement with the sequential involvement of MTOR and the class III PtdIns3K in autophagosome biogenesis. Treatment of HEK-293 cells with CXCL12 or UTS2 had no effect on the number of hrGFP-WIPI1 fluorescent dots, under basal conditions or following autophagy induction by PP242 (Fig. 2D and 2E). These data strongly suggest that chemotactic GPCRs exert their effects independently of the class III PtdIns3K and recruitment of WIPI1 to the phagophore.
Chemotactic GPCRs inhibit the recruitment of ATG16L1 to pre-autophagosomal endocytic vesicles
Recruitment of WIPI1 to the phagophore allows in turn the recruitment of additional proteins of the autophagy machinery, i.e. the ATG12–ATG5-ATG16L1 ternary complex and LC3B. Previous data indicated that endocytic formation of ATG16L1-ATG5-positive vesicles from the plasma membrane plays a key role during autophagy induction, by providing a source of phospholipids to the forming autophagosome membrane.21-23 We therefore hypothesized that activation of chemotactic GPCRs, at the plasma membrane, may locally affect the formation of these pre-autophagosomal vesicles.
We first tested in our model the importance of endocytosis in the formation of autophagosomes by blocking the scission of endocytic vesicles with Dynasore, an inhibitor of DNM/dynamin.39 Treatment of HEK-293 cells with Dynasore prevented the CQ-dependent accumulation of EGFP-LC3B puncta (Fig. 3A), indicating that, in agreement with previous reports,21 endocytic activity was required for the biogenesis of autophagosomes. ATG16L1 has been previously shown to form a complex, via the AP2/adaptin complex, with CLTC (clathrin heavy chain) and to accumulate at the plasma membrane following Dynasore treatment.21 Accordingly, we observed, in the presence of Dynasore, an increase in the number of ATG16L1-positive structures, located at the plasma membrane (Fig. 3B and 3C; Fig. S2). Activation of CXCR4 or UTS2R with their respective ligands inhibited the Dynasore-induced accumulation of ATG16L1-positive structures (Fig. 3D), suggesting that chemotactic GPCRs exert their anti-autophagic action by reducing the targeting of ATG16L1 to these pre-autophagosomal vesicles.
Figure 3.
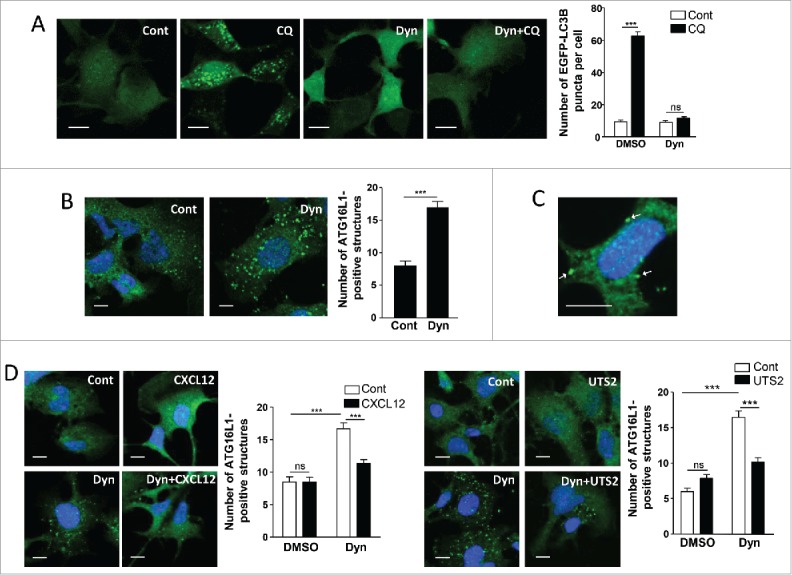
Chemotactic GPCRs inhibit the recruitment of ATG16L1 to pre-autophagosomal endocytic structures. (A) HEK-293 cells expressing the fluorescent protein EGFP-LC3B were treated (6 h) with or without chloroquine (CQ; 5 × 10−5 M) and Dynasore (Dyn; 10−4 M), as indicated. Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified in confocal images. Data represent means ± SEM, from at least 100 cells per group. (B) HEK-293 cells were starved in HBSS for 1 h with or without Dynasore (10−4 M). Cells were fixed and labeled with an anti-ATG16L1 antibody. For each photographic field, the number of ATG16L1-positive structures was quantified and normalized to the number of nuclei. Data represent means ± SEM from 15 photographic fields per group. (C) Magnified image of ATG16L1-positive structures located at the plasma membrane (arrows) of Dynasore-treated cells. (D) HEK-293 cells expressing CXCR4 (left panels) or UTS2R (right panels) were starved in HBSS for 1 h with the appropriate ligand (CXCL12, 10−8 M; UTS2, 10−9 M), in the absence or presence of Dynasore (10−4 M), as indicated. Cells were fixed and the number of ATG16L1-positive structures was quantified as in (B). Scale bars: 10 µm. Statistical significance was evaluated using a Mann and Whitney test (B, D) or an unpaired t-test (A). ***P < 0.001; ns, not statistically different.
The anti-autophagic effects of chemotactic GPCRs depend on ATG5
Previous studies have identified a membrane binding site on ATG515 and revealed that recruitment of ATG16L1 to membranes is highly dependent on ATG5.40 We therefore wondered whether chemotactic GPCR activation could inhibit the recruitment of ATG16L1 to endocytic vesicles by altering the function of ATG5. As a first step, we tested if siRNA knockdown of ATG5 could mimic the effects of chemotactic GPCR activation. In order to make sure that siRNA effects would not be due to off-target effects, we performed experiments with 4 independent siRNAs interacting with different regions of the ATG5 transcript. Effectiveness of each siRNA was verified by RT-qPCR (Fig. S3A) and western blot analyses (Fig. S3B). As expected and in agreement with previous reports,41 the knockdown of ATG5 reduced the biogenesis of autophagosomes, as evaluated by the accumulation of EGFP-LC3B puncta following CQ treatment (Fig. 4A), and reduced autophagic flux, as evaluated by analysis of SQSTM1 levels (Fig. S3C). We next tested the involvement of ATG5 in ATG16L1-targeting to endocytic vesicles. As was the case for chemotactic GPCR activation, siRNA knockdown of ATG5, using any of the individual siRNAs, markedly reduced the Dynasore-induced accumulation of ATG16L1-positive structures at the plasma membrane (Fig. 4B), indicating that ATG5 is required to initiate the formation of pre-autophagosomal vesicles from this compartment.
Figure 4.

The anti-autophagic effects of chemotactic GPCRs depend on ATG5. (A) HEK-293 cells were transfected with a vector encoding EGFP-LC3B, together with one of the 4 siRNAs directed against ATG5 (siATG5-1, siATG5-2, siATG5-3 and siATG5-4) or with nontargeting siRNA (siCont). Cells were placed in DMEM without serum for 6 h with or without chloroquine (CQ; 5 × 10−5 M), fixed, and the number of EGFP-LC3B fluorescent dots per cell was quantified in confocal images. Data represent means ± SEM, from at least 100 cells per group. (B) HEK-293 cells transfected with one of the 4 siRNAs directed against ATG5 (siATG5-1, siATG5-2, siATG5-3, siATG5-4) or with nontargeting siRNA (siCont) were starved in HBSS for 1 h with or without Dynasore (Dyn; 10−4 M), fixed and labeled with an anti-ATG16L1 antibody. For each photographic field, the number of ATG16L1-positive structures was quantified and normalized to the number of nuclei. Data represent means ± SEM from 15 photographic fields per group. (C) HEK-293 cells expressing MYC-ATG5 together with CXCR4 (left panels) or UTS2R (right panels) were placed in DMEM without serum for 1 h with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M). Cells were fixed and labeled with an anti-MYC antibody. For each photographic field, the fraction of MYC-ATG5 protein localized at the plasma membrane was evaluated. Data represent means ± SEM from 15 photographic fields per group. Scale bars: 10 µm. (D) HEK-293 cells expressing MYC-ATG5 together with CXCR4 (left panels) or UTS2R (right panels) were placed in DMEM without serum for 1 h with the appropriate ligand (CXCL12, 10−8 M; UTS2, 10−9 M). Cells were fixed and labeled with an anti-CDH2 antibody. For each photographic field, the fraction of CDH2 protein localized at the plasma membrane was evaluated. Data represent means ± SEM from 15 photographic fields per group. Scale bars: 10 µm. (E) HEK-293 cells were transfected with EGFP-LC3B, together with either ATG5, the ATG5K130R mutant or an empty vector, and either CXCR4 (left panel) or UTS2R (right panel). Cells were placed in DMEM without serum for 6 h with or without chloroquine (5 × 10−5 M) and CXCL12 (10−8 M, left panel) or UTS2 (10−9 M, right panel). Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified as in (A). (F) HEK-293 cells were transfected with ATG5 or an empty vector together with CXCR4 (left panel) or UTS2R (right panel). Cells were starved in HBSS for 1 h with or without Dynasore (10−4 M) and CXCL12 (10−8 M, left panel) or UTS2 (10−9 M, right panel). Cells were fixed and the number of ATG16L1-positive structures was quantified as in (B). Statistical significance was evaluated using a Mann and Whitney test (B, C, D, F) or an unpaired t test (A, E). *P < 0.05; ***P < 0.001; ns, not statistically different.
We next tested whether CXCL12 or UTS2 treatment could affect the subcellular localization of ATG5. Under control conditions, immunodetection of a MYC-tagged ATG5 showed that a significant fraction of this protein localized at the plasma membrane (Fig. 4C). Quantification of the “plasma membrane/whole cell” immunoreactive signal ratio revealed that CXCR4 and UTS2R activation reduced the pool of ATG5 protein localized at the plasma membrane (Fig. 4C). To rule out potential misinterpretation due to volume variations at the cell periphery, we performed confocal stack acquisitions and z-reconstructions on MYC-ATG5-labeled cells (Fig. S4). We also quantified the amounts of plasma membrane-localized CDH2 (cadherin 2) and found that they were not significantly affected by CXCL12 or UTS2 treatments (Fig. 4D). Together, our data support the view that chemotactic GPCRs inhibit autophagy by hindering the ability of ATG5 to bind the plasma membrane. It should be noted that these effects were not dependent on the energy status of the cells, since they were observed in either DMEM (Fig. 4C), DMEM complemented with 10% serum or starvation (HBSS) medium (Fig. S5A and S5B).
In order to further evaluate this regulatory role of ATG5, we attempted to block the anti-autophagic effects of CXCR4 or UTS2R by overexpressing recombinant ATG5. We first evaluated the impact of ATG5 overexpression on autophagic flux, estimated by the quantification of SQSTM1 immunoreactivity. Interestingly, in the absence of chemotactic factors, SQSTM1 immunoreactivity was not modified by the overexpression of ATG5 (Fig. S6A), suggesting that ATG5 does not constitute a limiting factor under these conditions. However, CXCR4- or UTS2R-induced inhibition of autophagic flux, revealed by a marked increase in SQSTM1 immunoreactivity, was efficiently blocked by overexpression of ATG5. These data, obtained in either DMEM (Fig. S6A), DMEM complemented with 10% serum, or starvation (HBSS) medium (Fig. S6B and S6C), suggest that activation of chemotactic GPCRs hinders ATG5 function which then becomes limiting for autophagic activity.
We next investigated the effect of ATG5 overexpression on the biogenesis of autophagosomes, estimated by the accumulation of EGFP-LC3B puncta in the presence of CQ. Overexpression of ATG5 abolished the inhibitory effects of chemotactic GPCRs on the biogenesis of mature autophagosomes and, once again, these effects were observed in either DMEM (Fig. 4E), DMEM complemented with 10% serum or starvation (HBSS) medium (Fig. S7A and S7B). As a negative control, we performed experiments with a lysine mutant of ATG5 (ATG5K130R), which cannot be conjugated with ATG12, hence making it autophagy-incompetent. As expected, overexpression of the ATG5K130R mutant did not interfere with the inhibitory effects of chemotactic GPCRs on autophagosome biogenesis (Fig. 4E, S7A and S7B).
We next evaluated the effects of ATG5 overexpression on the formation of pre-autophagosomal vesicles from the plasma membrane and we found that the inhibitory effects of CXCL12 or UTS2 on the accumulation of ATG16L1-positive structures were completely abolished in cells overexpressing ATG5 (Fig. 4F).
Activation of CAPNs/calpains is a critical relay in the anti-autophagic effects of chemotactic GPCRs
Several CAPN substrates, such as the clathrin adaptors PICALM and the AP2 complex, participate in the formation of pre-autophagosomal vesicles from the plasma membrane.21,42 In vivo and in vitro experiments also identified ATG5,43,44 as well as ATG3, ATG4, ATG7, ATG9, ATG10, ATG12 and BECN1 as direct CAPN substrates.45,46 Moreover, CAPNs participate in many aspects of chemotactic migration, including cell spreading and formation of adhesion complexes,47 and CXCR4 has already been shown to activate CAPNs in neurons.48 This prompted us to evaluate whether the anti-autophagic effects of chemotactic GPCRs could be relayed by the activation of CAPNs. We first evaluated CAPN activity in HEK-293 cells using the BOC-LM-CMAC fluorescence assay. By using this approach, we detected an increase in CAPN activity after exposure of cells to either CXCL12 or UTS2 (Fig. 5A). This effect was blocked by co-incubation with the CAPN inhibitor ALLN, thus validating the specificity of the assay (Fig. 5A). We next examined the consequence of CAPN inhibition on the GPCR anti-autophagic effects described so far. The inhibitory effects of CXCL12 or UTS2 on the accumulation of ATG16L1-positive structures, or on MYC-ATG5 localization at the plasma membrane, were completely abolished by co-incubation with the CAPN inhibitor ALLN (Fig. 5B and 5C). We next investigated the involvement of CAPNs on the formation of mature autophagosomes by the use of the EGFP-LC3B marker. CXCL12 or UTS2 treatment significantly reduced the number of EGFP-LC3B puncta upon autophagy induction, and co-incubation with the CAPN inhibitor ALLN totally reversed these effects (Fig. 5D). Consistent with these results, siRNAs directed against either CAPN1 (calpain 1) or CAPN2 (calpain 2) also blocked the anti-autophagic actions of CXCL12 or UTS2 (Fig. S8, S9 and S10). Selective knockdown of the appropriate CAPN isoform with each siRNA was successfully confirmed by RT-qPCR and western blot analyses (Fig. S11).
Figure 5.
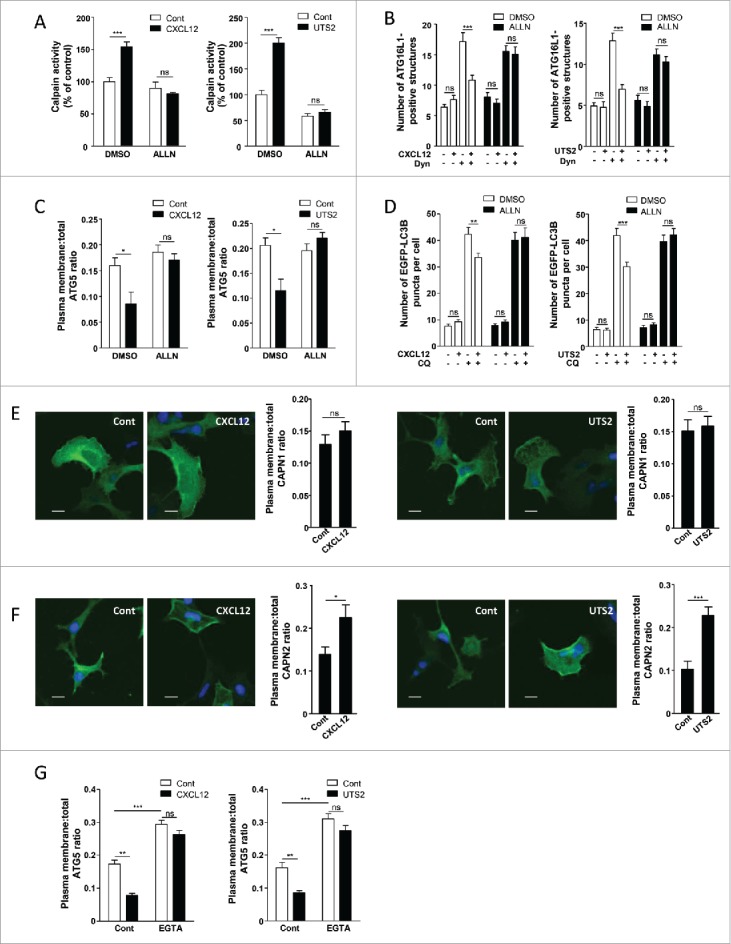
The anti-autophagic effects of chemotactic GPCRs depend on CAPN activation. (A) HEK-293 cells expressing CXCR4 (left panel) or UTS2R (right panel) were treated (1 h) with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M), with or without ALLN (10−6 M), as indicated. For each group, CAPN activity was measured using the BOC-LM-CMAC fluorescence assay. Data represent means ± SEM from 20 photographic fields per group. (B) HEK-293 cells expressing CXCR4 (left panel) or UTS2R (right panel) were starved in HBSS for 1 h in the absence or presence of Dynasore (Dyn; 10−4 M), ALLN (10−6 M) and CXCL12 (10−8 M, left panel) or UTS2 (10−9 M, right panel) as indicated. Cells were fixed and labeled with an anti-ATG16L1 antibody. For each photographic field, the number of ATG16L1-positive structures was quantified and normalized to the number of nuclei. Data represent means ± SEM from 15 photographic fields per group. (C) HEK-293 cells expressing MYC-ATG5 together with CXCR4 (left panel) or UTS2R (right panel) were placed in DMEM without serum for 1 h with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M), with or without ALLN (10−6 M), as indicated. Cells were fixed and labeled with an anti-MYC antibody. For each photographic field, the fraction of MYC-ATG5 protein localized at the plasma membrane was evaluated. Data represent means ± SEM from 15 photographic fields per group. (D) HEK-293 cells expressing EGFP-LC3B together with CXCR4 (left panel) or UTS2R (right panel) were placed in DMEM without serum for 6 h, with or without chloroquine (CQ, 5 × 10−5 M), ALLN (10−6 M) and CXCL12 (10−8 M, left panel) or UTS2 (10−9 M, right panel) as indicated. Cells were then fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified in confocal images. Data represent means ± SEM, from at least 100 cells per group. (E) HEK-293 cells expressing Flag-CAPN1 together with CXCR4 (left panels) or UTS2R (right panels) were placed in DMEM without serum for 1 h with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M). Cells were fixed and labeled with an anti-Flag antibody. For each photographic field, the fraction of Flag-CAPN1 protein localized at the plasma membrane was evaluated. Data represent means ± SEM from 15 photographic fields per group. Scale bars: 10 µm. (F) HEK-293 cells expressing Flag-CAPN2 together with CXCR4 (left panels) or UTS2R (right panels) were placed in DMEM without serum for 1 h with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M). Cells were fixed and labeled with an anti-Flag antibody. For each photographic field, the fraction of Flag-CAPN2 protein localized at the plasma membrane was evaluated. Data represent means ± SEM from 15 photographic fields per group. Scale bars: 10 µm. (G) HEK-293 cells expressing MYC-ATG5 together with CXCR4 (left panel) or UTS2R (right panel) were placed in DMEM without serum for 1 h with the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M), with or without EGTA (5 × 10−3 M), as indicated. Cells were fixed and the fraction of MYC-ATG5 protein localized at the plasma membrane was evaluated as in (C). Statistical significance was evaluated using a Mann and Whitney test (A, B, C, E, F, G) or an unpaired t test (D). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not statistically different.
Since GPCR-induced activation of CAPNs reduces the pool of plasma membrane-localized ATG5, recruitment of CAPNs at plasma membrane subdomains may be critical in suppressing the formation of pre-autophagosomal vesicles. We therefore monitored the impact of CXCR4 or UTS2R activation on the subcellular localization of CAPN1 and CAPN2. In the absence of chemotactic factors, significant amounts of Flag-tagged CAPN1 and Flag-tagged CAPN2 were observed at the cell periphery (Fig. 5E and 5F). Although activation of CXCR4 or UTS2R did not seem to affect the subcellular distribution of Flag-CAPN1 (Fig. 5E), activation of either one of the receptors markedly increased the amounts of Flag-CAPN2 localized at the plasma membrane (Fig. 5F). Recruitment of CAPN2 at the plasma membrane through its PtdIns(4,5)P2 binding domain facilitates its access to specific substrates49-51 and allows its activation by local increases in calcium concentration resulting from calcium influxes.52 We therefore tested the effects of extracellular calcium chelation by bathing cells in 5 mM EGTA. In the absence of chemotactic factors, incubation of cells with EGTA significantly increased the amounts of plasma membrane-localized MYC-ATG5 (Fig. 5G). Moreover, EGTA treatment blocked the inhibitory effects of CXCL12 or UTS2 on the localization of MYC-ATG5 at the plasma membrane (Fig. 5G), reinforcing the hypothesis that local activation of CAPNs at the plasma membrane relays the anti-autophagic effects of the receptors.
We finally examined whether activation of CXCR4 or UTS2R could directly induce the CAPN-dependent cleavage of ATG5 in HEK-293 cells. By western blot analyses, we were not able to detect any ATG5 truncation product following activation of the receptors (data not shown), suggesting that CAPNs may reduce the pool of plasma membrane-localized ATG5 in an indirect manner, through the cleavage of yet unidentified proteins facilitating ATG5 anchoring.
The anti-autophagic properties of CXCR4 and UTS2R are required for chemotaxis and formation of adhesion complexes
We next evaluated the directional migration of HEK-293 cells using a transwell chemotaxis assay, in which cells loaded in the upper chamber of the transwell migrate toward the bottom chamber containing the chemoattractant. A gradient of CXCL12 or UTS2 significantly stimulated the migration of HEK-293 cells expressing CXCR4 or UTS2R, respectively (Fig. 6A; Fig. S12). We verified that these effects correspond to chemotaxis rather than stimulation of chemokinesis (random motility) by disrupting the gradients of ligands. Addition of equal concentrations of CXCL12 or UTS2 in the upper and lower chambers of the transwells abolished the stimulatory effects of the ligands (Fig. 6A).
Figure 6.
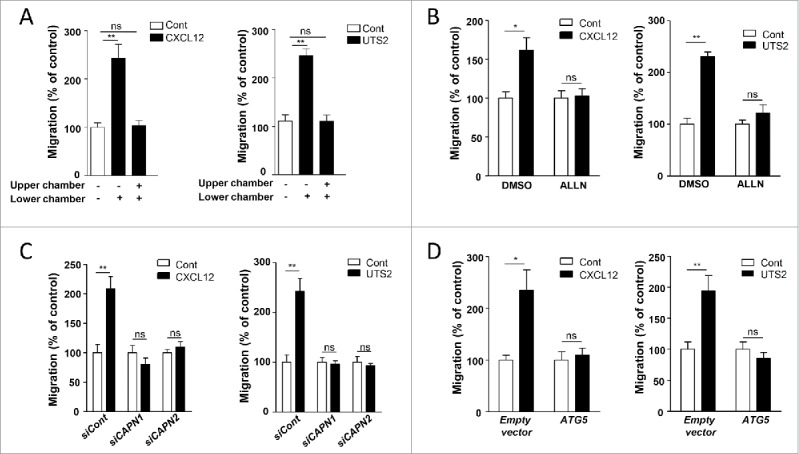
Inhibition of autophagy is required for CXCR4- or UTS2R-induced chemotaxis. (A) HEK-293 cells expressing CXCR4 (left panel) or UTS2R (right panel) were loaded in the upper chamber of transwells in DMEM without serum, containing or not the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M) in the upper or lower chamber, as indicated. After 24 h, cells that migrated onto the lower surface of the membrane were fixed, stained and counted. Data represent means ± SEM (n = 6). (B) HEK-293 cells expressing CXCR4 (left panel) or UTS2R (right panel) were pre-incubated (1 h) with ALLN (10−6 M), then loaded in the upper chamber of transwells, in DMEM without serum containing ALLN (10−6 M) in the upper and bottom chambers, and the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M) in the lower chamber. After 24 h, cell migration was quantified as in (A). (C) HEK-293 cells were transfected with CXCR4 (left panel) or UTS2R (right panel), together with either siRNA targeting CAPN1 (siCAPN1), CAPN2 (siCAPN2) or with nontargeting siRNA (siCont). Cells were then loaded in the upper chamber of transwells in DMEM without serum, containing or not the respective ligands (CXCL12, 10−8 M; UTS2, 10−9 M) in the lower chamber. After 24 h, cell migration was quantified as in (A). (D) HEK-293 cells were transfected with CXCR4 (left panel) or UTS2R (right panel), together with ATG5 or an empty vector. Cells were loaded in the upper chamber of transwells in DMEM without serum, containing or not the appropriate ligand (CXCL12, 10−8 M; UTS2, 10−9 M) in the lower chamber. After 24 h, cell migration was quantified as in (A). Statistical significance was evaluated in all experiments using a Mann and Whitney test. *P < 0.05; **P < 0.01; ns, not statistically different.
We next tested the effect of the CAPN inhibitor ALLN on cell migration. Treatment of cells with ALLN totally suppressed chemotaxis induced by CXCL12 or UTS2 (Fig. 6B). To confirm these results, we used siRNAs against CAPN1 or CAPN2 and we found that, similar to autophagy experiments, the knockdown of either one of these isoforms suppressed the pro-migratory properties of CXCL12 or UTS2 (Fig. 6C). Since CAPNs are well-known regulators of chemotactic migration47 and may exert their effects independently of autophagy modulation, we next wanted to selectively interfere with the anti-autophagic properties of CXCR4 or UTS2R by overexpressing ATG5. HEK-293 cells overexpressing recombinant ATG5 did not display any chemotactic behavior toward CXCL12 or UTS2 (Fig. 6D), suggesting that inactivation of ATG5 is required for GPCR-induced chemotaxis.
Chemokines are thought to regulate cell migration by coordinating the formation of adhesion complexes at the cell front. We therefore tested the effect of chemotactic GPCRs on the number of adhesion complexes, monitored by the immunodetection of the focal adhesion protein VCL. In control conditions, CXCL12 or UTS2 treatments induced the accumulation of VCL-positive adhesion complexes, mainly found at the periphery of cell protrusions (Fig. 7A and 7B). Similar to chemotactic experiments, overexpression of ATG5 (Fig. 7A and 7B), or CAPN inhibition using ALLN (Fig. 7C and 7D) totally abolished the formation of adhesion complexes induced by CXCL12 or UTS2. We also found that knockdown of ATG5 induced a significant increase in the number of VCL-positive adhesion complexes, therefore mimicking the effect of CXCL12 and UTS2 treatments (Fig. 7E). It should be noticed that adhesion experiments were performed in either DMEM (Fig. 7), DMEM with serum (Fig. S13) or starvation (HBSS) medium (Fig. S14), and showed almost identical results in these 3 conditions, indicating that these effects did not depend on the cells' energy status.
Figure 7.
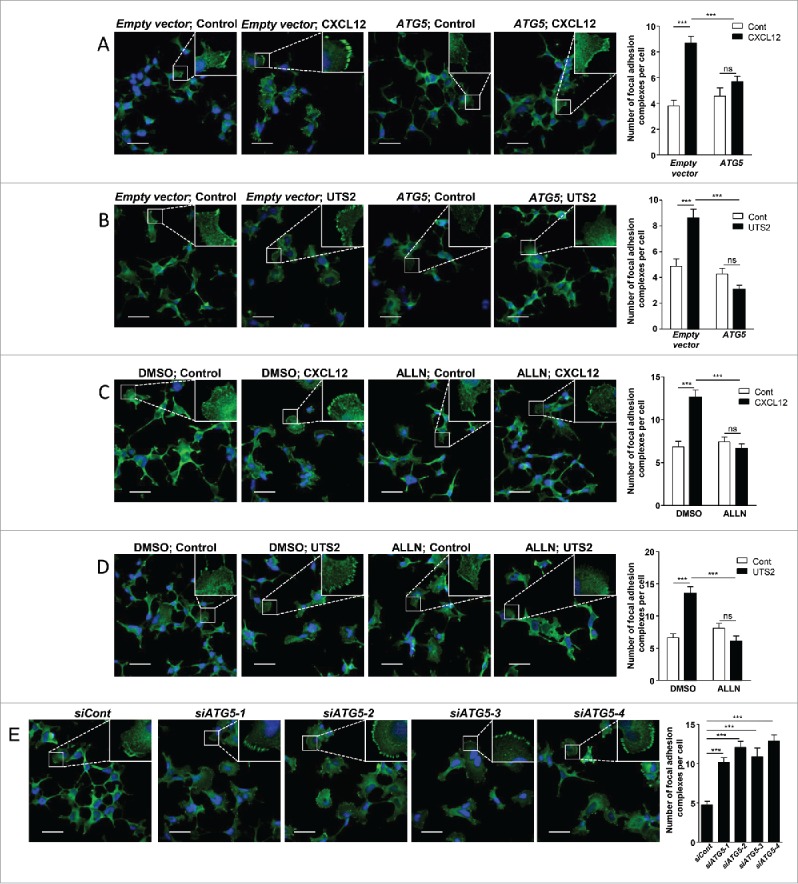
Inhibition of autophagy is required for CXCR4- or UTS2R-induced formation of adhesion complexes. (A) HEK-293 cells were transfected with CXCR4, together with ATG5 or an empty vector. Cells were then placed in DMEM without serum for 1 h in the absence (Control) or presence of CXCL12 (10−8 M). Cells were fixed and adhesion complexes were labeled with an anti-VCL antibody (green). For each photographic field, adhesion complexes were quantified and normalized to the number of nuclei (DAPI stained, blue). Data represent means ± SEM from 15 fields per group. (B) HEK-293 cells were transfected with UTS2R together with ATG5 or an empty vector. Cells were then placed in DMEM without serum for 1 h in the absence (Control) or presence of UTS2 (10−9 M). Cells were fixed and adhesion complexes were quantified as in (A). (C) HEK-293 cells expressing CXCR4 were placed in DMEM without serum for 1 h, in the absence (Control) or presence of CXCL12 (10−8 M) and ALLN (10−6 M), as indicated. Cells were fixed and adhesion complexes were quantified as in (A). (D) HEK-293 cells expressing UTS2R were placed in DMEM without serum for 1 h, in the absence (Control) or presence of UTS2 (10−9 M) and ALLN (10−6 M), as indicated. Cells were fixed and adhesion complexes were quantified as in (A). (E) HEK-293 cells were transfected with one of the 4 siRNAs targeting ATG5 (siATG5-1, siATG5-2, siATG5-3, siATG5-4) or a nontargeting siRNA (siCont). After being placed in DMEM without serum for 1 h, cells were fixed and adhesion complexes were quantified as in (A). Statistical significance was evaluated in all experiments using a Mann and Whitney test. ***P < 0.001; ns, not statistically different. Scale bars: 50 µm.
Collectively, these data suggest that inhibition of autophagy by CXCR4 or UTS2R favors the formation of adhesion complexes and efficient attachment of protrusions to the substratum, a necessary event for forward translocation of the cell body.
Inhibition of autophagy is required for chemotactic migration of glioma cells
Chemotactic migration is a key aspect of many physiological and pathological processes, including cancer progression. Among chemotactic GPCRs, CXCR4 is the most recognized in this process, and is involved in the growth, invasion and metastasis of a wide range of malignant tumors, including gliomas.4,5 Furthermore, it has recently been demonstrated that UTS2R and UTS2 are highly expressed in fresh glioblastoma explants as well as in glioma cell lines, and mediate chemotactic migration of these tumor cells.7 We therefore examined whether autophagy inhibition was, as in HEK-293 cells, a crucial step for GPCR-induced chemotaxis of glioma cells.
Using the U87 human glioblastoma cell line, we first tested the effects of CXCL12 or UTS2 on endogenous SQSTM1 expression levels. Similar to HEK-293 cells, treatment of U87 cells with CXCL12 or UTS2 significantly increased SQSTM1 immunolabeling, indicative of autophagic flux inhibition (Fig. 8A and 8B). We next tested the effects of CXCL12 or UTS2 on the number of autophagosomes in control conditions and under CQ, using the EGFP-LC3B puncta method. Once again, the low number of EGFP-LC3B-labeled autophagosomes in control conditions hindered accurate assessment of the effects of CXCL12 (Fig. 8C). However, we observed significant reduction in the number of EGFP-LC3B dots after a 6-h treatment with UTS2 (Fig. 8C). As expected, prevention of autophagosome degradation with CQ markedly increased the number of EGFP-LC3B dots per cell. In these conditions, treatment of cells with CXCL12 and UTS2 partially prevented autophagosome accumulation (Fig. 8C), indicating that activation of these chemotactic GPCRs also led to impaired autophagosome biogenesis in U87 cells.
Figure 8.
Inhibition of autophagy is required for chemotactic migration of U87 glioma cells. (A) U87 cells were treated (24 h) without (Cont) or with CXCL12 (10−8 M). Cells were fixed and labeled with an anti-SQSTM1/p62 antibody (green). For each photographic field, SQSTM1 immunoreactivity was quantified and normalized to the number of nuclei (DAPI stained, blue). Data represent means ± SEM from 20 photographic fields per group. Scale bars: 50 µm. (B) U87 cells were treated (24 h) without (Cont) or with UTS2 (10−8 M). Cells were fixed and SQSTM1 immunoreactivity was quantified as in (A). Scale bars: 50 µm. (C) U87 cells expressing the fluorescent protein EGFP-LC3B were treated (6 h) with or without CXCL12 (10−8 M), UTS2 (10−8 M) and chloroquine (CQ; 5 × 10−5 M), as indicated. Cells were fixed and the number of EGFP-LC3B fluorescent dots per cell was quantified in confocal images. Data represent means ± SEM, from at least 100 cells per group. Scale bars: 20 µm. (D) U87 cells were loaded in the upper chamber of transwells containing DMEM without serum, with or without CXCL12 (10−8 M) or UTS2 (10−8 M) in the lower or upper chamber, as indicated. After 24 h, cells that migrated onto the lower surface of the membrane were fixed, stained and counted. Data represent means ± SEM (n = 6). (E) U87 cells were transfected with an ATG5 expression vector or an empty vector. Cells were loaded in the upper chamber of transwells containing DMEM without serum, with or without CXCL12 (10−8 M) or UTS2 (10−8 M) in the lower chamber. After 24 h, cell migration was quantified as in (A). Data represent means ± SEM (n = 6). (F) U87 cells were transfected with ATG5 or an empty vector. Cells were then placed in DMEM without serum for 1 h in the absence (Control) or presence of CXCL12 (10−8 M) or UTS2 (10−8 M). Cells were fixed and adhesion complexes were labeled with an anti-VCL antibody (green). For each photographic field, adhesion complexes were quantified and normalized to the number of nuclei (DAPI stained, blue). Data represent means ± SEM from 15 fields per group. Scale bars: 50 µm. Statistical significance was evaluated using a Mann and Whitney test (A, B, D, E, F) or an unpaired t test (C). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not statistically different.
We next tested whether overexpressing the ATG5 protein could impede CXCR4- and UTS2R-induced chemotactic migration of U87 cells. Using a transwell chemotaxis assay, we first demonstrated that CXCL12 and UTS2 significantly stimulated the migration of U87 cells (Fig. 8D), as previously described.7,53 We verified that these effects corresponded to chemotactic migration rather than chemokinesis, since addition of equal concentrations of CXCL12 and UTS2 in both chambers of the transwell abolished the stimulatory effects of the ligands (Fig. 8D). As was the case in HEK-293 cells, overexpression of recombinant ATG5 in U87 cells annulled their chemotactic behavior toward CXCL12 or UTS2 (Fig. 8E). We then tested the effects of ATG5 overexpression on the number of focal adhesion complexes in U87 cells. As expected, CXCL12 and UTS2 treatment induced the accumulation of VCL-labeled adhesion complexes at the cell periphery, and overexpression of ATG5 reversed these effects (Fig. 8F).
Discussion
Data presented in this study provide a new link between GPCR signaling and the autophagy machinery. We found that activation of 2 chemotactic GPCRs, CXCR4 or UTS2R, inhibits a very early phase of autophagosome biogenesis, i.e., the recruitment of ATG16L1 protein to endocytic vesicles. We demonstrated that this anti-autophagic role of CXCR4 and UTS2R represents a key step in the formation of adhesion complexes and efficient cell migration toward chemotactic stimuli. Moreover, experiments done in U87 glioma cells, endogenously expressing both CXCR4 and UTS2R, suggest that GPCR modulation of autophagy may have a profound impact on the invasive properties of cancer cells.
Few GPCRs have been shown to directly regulate autophagic activity.54 This includes the amino-acid responsive TAS1R1-TAS1R3 heterodimer, expressed in most tissues.55 Amino acid signaling through TAS1R1-TAS1R3 activates MTOR kinase, leading to inhibition of class III PtdIns3K activity and reduction of autophagosome biogenesis.56 AGTRs/angiotensin receptors were also found to inhibit autophagic activity in cardiomyocytes,57 podocytes58 and vascular smooth muscle cells,59 through activation of NADPH oxidase and generation of reactive oxygen species. Here, we found that CXCR4- and UTS2R-evoked inhibition of autophagy was not relayed by the modulation of MTOR kinase or class III PtdIns3K, but relied on impaired recruitment of ATG16L1 to endocytic vesicles. Accumulation of ATG16L1 at clathrin-coated endocytic structures and vesiculation of ATG16L1-positive precursors from the plasma membrane largely contribute to autophagosome biogenesis.21-23 Indeed, these ATG16L1-positive vesicles have been shown to undergo SNARE-mediated homotypic fusion or heterotypic fusion with ATG9-positive vesicles, to ultimately deliver an adequate source of phospholipids for the expansion of the phagophore.22,23,60 The importance of this phenomenon is underlined by our experiments with Dynasore showing that, in agreement with previous studies,21 endocytic activity was required for the efficient formation of autophagosomes. The exact nature and mode of formation of these precursor vesicles is not fully elucidated. Data from Ravikumar et al.21 demonstrated that CLTC was able to interact, via the AP2 complex, with the amino terminus of ATG16L1. Data obtained here indicate that ATG5 also participates in the recruitment of ATG16L1 to endocytic vesicles. We can speculate that the membrane-binding activity of ATG540 might allow the initial docking of an ATG5-ATG16L1 complex to the plasma membrane, in order to maximize the probability of interaction between ATG16L1 and AP2-CLTC. We demonstrated that activation of CXCR4 or UTS2R markedly reduced the pool of ATG5 proteins localized at the plasma membrane. Therefore, it can be envisaged that chemotactic GPCRs mainly act by precluding membrane docking of the ATG5-ATG16L1 complex. Alternatively, since ATG5 has been shown to co-immunoprecipitate from cell lysates with ATG16L1 and CLTC,21 and the N terminus of ATG16L1 allows both AP2-CLTC co-immunoprecipitation21 and direct ATG5 binding,15,61,62 it is conceivable that ATG5 may actually act as a “bridge” protein that connects ATG16L1 to the AP2-CLTC complex. Additional work is clearly needed to decipher the exact role of ATG5 in recruitment of ATG16L1 to endocytic vesicles.
CAPNs/calpains are well-characterized modulators of autophagy43,44,63 and CXCR4 has already been shown to activate CAPNs in neurons.48 We therefore investigated the involvement of these proteases in the anti-autophagic effects of chemotactic GPCRs. We observed that CAPN inhibition abrogated the autophagy-related effects of CXCR4 and UTS2R, i.e. their abilities to inhibit i) association of ATG5 with the plasma membrane, ii) recruitment of ATG16L1 to endocytic vesicles, and iii) formation of mature LC3B-positive autophagosomes. A previous report has shown that the CAPN-dependent cleavage of ATG5 could tightly control basal autophagy.44 Our attempts to demonstrate a direct cleavage of ATG5 in our model, following CXCR4 or UTS2R activation, were unsuccessful. This may be due to the fact that only a minor, plasma membrane-associated, fraction of ATG5 undergoes cleavage, or to the potential instability of the cleaved products. Alternatively, the anti-autophagic action of CAPN could depend on the cleavage of other proteins such as PICALM and/or the AP2 complex, since both proteins are critical for the formation of pre-autophagosomal vesicles from the plasma membrane21,42 and have already been described as CAPN substrates.64-66 The ATG7 protein, which is also cleaved by CAPNs67 and is essential for conjugation of ATG5 to ATG12, represents another candidate. Regardless of the CAPN substrate involved, data presented here constitute, to our knowledge, the first demonstration of GPCR-induced modulation of autophagy through the activation of these proteases. We can speculate from the literature that this anti-autophagic pathway, shared by UTS2R and CXCR4, may be relevant in chemotaxis induced by other GPCRs, as well as non-GPCR membrane receptors. Indeed, the spatial regulation of CAPN proteases is involved in many aspects of chemotactic migration, including cell spreading, formation of membrane protrusions, and adhesion complex formation and turnover.47 Moreover, cell migration triggered by several chemotactic receptors, including the GPCR for N-formylmethionine-leucyl-phenylalanine51 and the EGF- and VEGF-tyrosine kinase receptors,68-71 depends on CAPN activation. It will be informative to extend our study to a large panel of chemotactic receptors in order to test whether the CAPN-dependent repression of autophagy represents a hallmark of chemotaxis.
Chemotactic receptors engage signaling pathways that coordinate the formation of adhesion complexes at the cell front. We found here that induction of this process through UTS2R or CXCR4 activation was dependent on the repression of autophagy since i) formation of adhesions was totally blocked by pharmacological inhibition of CAPNs or overexpression of ATG5, and ii) the effect of chemokines on adhesions was mimicked by siRNA targeting of ATG5. Several proteins involved in formation of adhesions were found to localize in autophagosomes. In particular, the SRC kinase, an essential protein for integrin signaling, rapidly shuttles from focal adhesions to autophagosomes when inducing cell-matrix detachment.30 Proteomic profiling also identified RHOB as an autophagosome-associated protein in a pancreatic tumor cell line.72 RHOB is a small GTPase that has recently been described as a key player in cell adhesion and directional migration. Indeed, Vega et al.73 demonstrated that RHOB stabilizes lamellipodial protrusions by increasing ITGB1 (integrin subunit β 1) surface levels and activity, and that RHOB-depleted cells have impaired directional migration in a chemotactic gradient of serum.73,74 Recently, autophagic activity has been shown to be essential for selective degradation of adhesion proteins such as PXN, VCL and ZYX.28,31 More specifically, Kenific et al.31 found that the GFP-LC3B protein colocalized with focal adhesions primarily during disassembly. It can therefore be envisaged that inhibition of autophagy by chemotactic GPCRs may act as a master switch that prevents autophagic degradation of key proteins involved in the formation/maturation of adhesion complexes and/or directional migration. Further work is needed to understand how, mechanistically, these proteins could be targeted to autophagy degradation. If membrane-associated, proteins involved in cell adhesions could be internalized in early endosomes, which could later fuse with mature autophagosomes.75 Alternatively, cell-adhesion components could specifically associate with ATG16L1-containing pre-autophagosomal vesicles and thus be targeted to the growing phagophore. In this scenario, inhibition of the formation of pre-autophagosomal vesicles by chemotactic GPCRs could increase the localization of cell-adhesion proteins at the cell front and in this manner directly participate in the growth and stabilization of the lamellipodium. It is interesting to note that a proteomic analysis of ATG16L1-positive precursor vesicles already identified proteins (CTTN [cortactin], CFL1 [cofilin 1]) involved in actin dynamics and lamellipodium expansion.23
Global inhibition of autophagosome biogenesis using interfering RNAs against ATG proteins results in increased focal adhesion size and reduced migration rate.28,31 Although these migration studies could appear to conflict with our present data, indicating that autophagy inhibition by CXCR4 or UTS2R stimulates migration, they actually stress the fact that efficient chemotactic migration may imply compartmentalized rather than general inhibition of the autophagy machinery. It can therefore be proposed that activation of chemotactic GPCRs at the front of polarized cells would allow, through inhibition of autophagy, the efficient formation of adhesion complexes, while autophagy would still remain active at a distance from the sites of GPCR activation/signaling in order to enable focal adhesion disassembly. Caution should thus be taken in the interpretation of migration studies using interfering RNAs targeting ATG proteins. Global inhibition of autophagy using this method may indeed reflect the effects of cell exposure to a homogeneous rather than a gradient concentration of chemokine, i.e. induction of numerous, large and unproductive focal adhesions at the entire cell periphery that blunt migration. Demonstration of a compartmentalized regulation of autophagy in cells exposed to a gradient of ligand may require dynamic imaging of forming ATG5-ATG16L1-containing pre-autophagosomal endosomes, as previously investigated.21
Additional autophagy-related mechanisms may amplify the pro-migratory properties of CXCR4 and UTS2R. We found that inhibition of the autophagic flux following treatments with CXCL12 or UTS2 induced, as expected, the cytosolic accumulation of the SQSTM1 protein. Through its LC3B-interacting region, SQSTM1 acts as a receptor protein that contributes to the autophagic degradation of various cargo, such as ubiquitinated protein aggregates35 and mitochondria.76 SQSTM1 has recently been shown to play a critical role in the migration processes. Indeed, SQSTM1 can directly bind to and prevent the proteasomal degradation of the transcription factor TWIST1,25,77 a core regulator in both early embryonic morphogenesis and cancer metastasis.78,79 Through this SQSTM1-TWIST1 axis, autophagy-defective cancer cells engage a transcriptional program, called the epithelial-to-mesenchymal transition (EMT), that strengthens their invasive phenotype.25,77 Moreover, recent data obtained in glioblastoma indicate that autophagy inhibition, through the knockdown of ATG5 or ATG7, stimulates the expression of the EMT regulators SNAI1/SNAIL and SNAI2/SLUG,80 as well as cell invasion. It can therefore be envisaged that inhibition of autophagy by chemotactic GPCRs contributes to EMT-like events during tumor progression. Further studies will be needed to address this point.
In summary, data from this study indicate that, in addition to extracellular nutrients such as amino acids, detection of extracellular guidance cues by GPCRs can tightly control autophagic activity in order to optimize lamellipodial adhesions and chemotactic migration. Based on the numerous studies highlighting a key role of the CXCL12-CXCR4 and UTS2-UTS2R systems in cancer cell invasion,4,7,11,12,81 and on the well-characterized action of the CXCL12-CXCR4 axis during immune response,82 our data may have broad implications and may help to envisage new therapeutic strategies for cancer or immunological disorders.
Materials and methods
Antibodies and reagents
Antibodies were as follows: mouse monoclonal anti-SQSTM1/p62 (Abcam, AB56416), mouse monoclonal anti-VCL/vinculin (Sigma-Aldrich, V9264), mouse monoclonal anti-FLAG (Sigma-Aldrich, F1804), mouse monoclonal anti-MYC (Santa Cruz Biotechnology, sc-40), rabbit polyclonal anti-ATG16L1 (Cell Signaling Technology, 8089), sheep polyclonal anti-CDH2/cadherin 2 (R&D Systems, AF6426). Secondary antibodies used were Alexa Fluor 488-conjugated antibody against mouse IgG (Invitrogen, A21202), Alexa Fluor 488-conjugated antibody against rabbit IgG (Invitrogen, A21206), and Alexa Fluor 633-conjugated antibody against sheep IgG (Invitrogen, A21100). Other reagents in this study were 3-methyladenine (3-MA; Sigma-Aldrich, M9281), human UTS2 (Polypeptide Laboratories, SC1355), human CXCL12 (Bio-Techne, 350-NS-050), N-acetyl-Leu-Leu-Norleu-al (ALLN; Sigma-Aldrich, A6186), chloroquine (Sigma-Aldrich, C6628), DAPI (Invitrogen, D3571), Dynasore (Sigma-Aldrich, D7693), hematoxilin (Sigma-Aldrich, HHS32), ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA; Sigma-Aldrich, E4378), FN1/fibronectin (Sigma-Aldrich, F0895), Mowiol (Merck Millipore, 475904), normal donkey serum (Sigma-Aldrich, D9663), paraformaldehyde (Sigma-Aldrich, 252549), PP242 (Sigma-Aldrich, P0037), Dulbecco's phosphate-buffered saline (DPBS; Sigma-Aldrich, D1408), and Triton X-100 (Sigma-Aldrich, T9284).
Plasmid constructs and small interfering RNA (siRNA)
The expression vector encoding the fusion protein EGFP-LC3B (24920, deposited by Toren Finkel) and the expression vectors encoding the fusion proteins Flag-CAPN1 and Flag-CAPN2 (60941 and 60942, deposited by Yi Zhang) are available from Addgene. The expression vector encoding the fusion protein hrGFP-WIPI1 was obtained by PCR amplification of cDNA prepared from HEK-293 cells, using the WIPI1 specific primers 5′-GATCCTCGAGCGATGGAGGCCGAGGCCGC-3′ and 5′-GATCGAATTCTCATGACTGCTTCGTTTTGCCC-3′. The PCR fragment was subsequently digested with XhoI (Promega Corporation, R6161) and EcoRI (Promega Corporation, R6011), and cloned in-frame with humanized recombinant GFP in the phrGFP-N1 expression vector (Agilent Technologies, 240145). The expression vector encoding human ATG5 was obtained by PCR amplification of cDNA prepared from HEK-293 cells, using the ATG5-specific primers 5′-GATCGGATCCATGACAGATGACAAAGATGTGCT-3′ and 5′-GATCCTCGAGTCAATCTGTTGGCTGTGGGATG-3′. The PCR fragment was digested with BamHI (Promega Corporation, R6021) and XhoI, and cloned into pcDNA3.1+ (Invitrogen, V790-20). The expression vector encoding MYC-tagged human ATG5 was obtained by PCR amplification using the ATG5-specific primers 5′-GATCGTCGACCATGACAGATGACAAAGATGTGC-3′ and 5′-GATCCTCGAGTCAATCTGTTGGCTGTGGGATG-3′. The PCR fragment was digested with SalI (Promega Corporation, R6051) and XhoI, and cloned into pCMV-MYC-N (BD-Biosciences, PT3282-5). To produce the conjugation-defective ATG5 mutant (ATG5K130R), the plasmid encoding MYC-ATG5 was mutated to replace Lys130 by Arg using the QuikChange Site-directed Mutagenesis Kit (Agilent Technologies, 200523) using the specific primers 5′-TTTATGTCATGTATGAGAGAAGCTGATGCTTTA-3′ and 5′-TAAAGCATCAGCTTCTCTCATACATGACATAAA-3′). Human CXCR4 and UTS2R cDNAs are inserted into the pCMV-MYC-N vector, in frame with the MYC epitope. For experiments utilizing the MYC-tagged ATG5 construct, we expressed untagged receptors by transfecting cells with pcDNA3.1+ vectors containing human CXCR4 or UTS2R cDNAs. All constructs were verified by sequencing. Control (D-001810-03), ATG5-1-4 (J-004374-07, J-004374-08, J-004374-09, J-004374-10), CAPN1 (L-005799) and CAPN2 (L-005804) siRNA were purchased from Dharmacon.
Cell culture and transfections
The multiform glioblastoma cell line U87-MG (WHO grade IV) was purchased from American Type Culture Collection (ATCC, HTB-14™). The HEK-293 cell line (ATCC, CRL1573™) was generously provided by Dr. L. Prézeau, C. Barrère and I. Bidaud (IGF, Montpellier, France). These cell lines were routinely maintained in our laboratory according to the instructions from ATCC. For all experiments, cells were cultured in either DMEM (Gibco, 41965-039), supplemented with 10% fetal bovine serum (Gibco, 10270-106) or HBSS (Gibco, 14025). Transient transfections were performed using either the Amaxa® Cell Line Nucleofector® Kit V (Lonza, VCA-1003) or FuGene HD (Promega Corporation, E2311) according to the manufacturer's protocol. For experiments done with siRNAs targeting CAPN1 and CAPN2, HEK-293 cells were subject to 2 rounds of transfection in order to ensure adequate knockdown of the long-lived proteins CAPN1 and CAPN2.83
Immunocytochemistry and image analysis
Cells were fixed in paraformaldehyde (4%, 10 min), permeabilized with Triton X-100 (0.05%, 5 min) and blocked with normal donkey serum (2%, 1 h). Cells were incubated overnight with the appropriate primary antibody at 4°C followed by incubation for 2 h at room temperature with the appropriate secondary antibody. Cells were counterstained with DAPI (1 µg/mL, 10 min) to label nuclei, and imaged by confocal microscopy (Leica TCS SP2 confocal laser scanning microscope). Focal adhesions: in VCL-labeled cells, the number of focal adhesion complexes was calculated using a particle analysis plug-in after successive image treatment by ImageJ software. MYC-ATG5 and Flag-CAPN1/CAPN2 localization: the amount of immunoreactive signal localized at the plasma membrane was measured by defining a region of interest using the ImageJ software, and expressed as a ratio to whole-cell immunoreactivity. ATG16L1-positive structures: in ATG16L1-labeled cells, the number of ATG16L1-positive structures was assessed with the Imaris software (Bitplane), using the automated spot detection function.
Autophagy assays
Cyto-ID labeling and Flow cytometry: HEK-293 cells were harvested in Accutase solution (Merck Millipore, SF006), rinsed in DPBS and incubated for 30 min at room temperature in DMEM without phenol red, containing 5% fetal bovine serum and the fluorescent Cyto-ID probe (Cyto-ID® Autophagy detection kit; Enzo Life Sciences, ENZ-51031K200). After incubation, cells were washed and analyzed on a FACScalibur flow cytometer (BD Biosciences) operated with the CellQuest software. hrGFP-WIPI1 and EGFP-LC3B redistribution: cells expressing either hrGFP-WIPI1 or EGFP-LC3B were fixed in paraformaldehyde (4 %, 10 min) and imaged by confocal microscopy (Leica TCS SP2 confocal laser scanning microscope). The number of hrGFP-WIPI1 and EGFP-LC3B dots was assessed using the Imaris software. SQSTM1/p62 accumulation: autophagic flux was assessed by immunocytochemical analysis of endogenous SQSTM1 levels as described above. The integrated densities obtained from the green channel in 20 different images were normalized to the number of nuclei (DAPI stained) and represent levels of the SQSTM1 protein.
Analysis of CAPN activity
CAPN activity was determined by using a fluorescent CAPN substrate, t-butoxycarbonyl-Leu-L-Met-chloromethylaminocoumarin (BOC-LM-CMAC; Molecular Probes, A6520). Cleavage by CAPNs of this substrate produces fluorescence with an emission maximum around 430 nm. Cells cultured on glass slides (Menzel-Gläser, 0985) were incubated with the fluorescent substrate (50 µM) for 20 min at 37°C. Cells were then fixed and imaged by confocal microscopy. The integrated densities per area obtained from 20 different images were normalized to the number of cells in the area and are representative of CAPN activity.
Migration assay
Cells (100,000/filter) were seeded on FN1 (fibronectin 1)-coated transwell filters (8-μm pores, 24 wells; Corning Incorporated, 3422). The gradient of ligands was generated by addition of CXCL12 or UTS2 in the lower chamber. After 24 h, cells on the upper surface of the filter were removed using a cotton swab. Cells that migrated onto the lower surface were fixed, stained with hematoxilin and filters were mounted on glass slides with Mowiol and randomly photographed with a Nikon microscope (Champigny-sur-Marne, France; 10 fields/filter). Cells on photographic fields were counted by using the “Cell counter” plug-in of ImageJ software.
Statistical analysis
Data are presented as the mean ± SEM for each group of samples. Statistical analyses were performed using the GraphPad Prism 5.00 software (GraphPad Software). Comparisons between 2 groups were performed using unpaired t test or Mann and Whitney test.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- ACTB
actin β
- BECN1
Beclin 1
- CAPN1
calpain 1
- CAPN2
calpain 2
- CDH2
cadherin 2
- CLTC
clathrin heavy chain
- CQ
chloroquine
- CXCL12
C-X-C motif chemokine ligand 12
- CXCR4
C-X-C motif chemokine receptor 4
- EGFP
enhanced green fluorescent protein
- DMEM
Dulbecco's modified Eagle's medium
- EMT
epithelial-to-mesenchymal transition
- HBSS
Hanks balanced salt solution
- GPCR
G protein-coupled receptor
- hrGFP
humanized recombinant green fluorescent protein
- MAP1LC3B
microtubule-associated protein 1 light chain 3 β
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- PICALM
phosphatidylinositol binding clathrin assembly protein
- PtdIns3K
phosphatidylinositol 3-kinase
- PtdIns3P
phosphatidylinositol 3-phosphate
- PtdIns(4,5)P2
phosphatidylinositol 4,5-biphosphate
- PXN
paxillin
- SQSTM1
sequestosome 1
- VCL
vinculin
- UTS2
urotensin 2
- UTS2R
urotensin 2 receptor
- WIPI1
WD repeat domain phosphoinositide interacting 1
- ZYX
zyxin
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Funding
This work was supported by Inserm, Géfluc, TC2N network, the Ligue contre le Cancer Normandie, the French Agence Nationale de la Recherche and the University of Rouen. P.C. is recipient of a fellowship from the French ministry, and V.L.J. is recipient of a fellowship from Haute-Normandie.
References
- [1].Lauffenburger D, Horwitz AF. Cell migration: A physically integrated molecular process. Cell 1996; 84:359-69; PMID:8608589; http://dx.doi.org/ 10.1016/S0092-8674(00)81280-5 [DOI] [PubMed] [Google Scholar]
- [2].Salcedo R, Zhang X, Young HA, Michael N, Wasserman K, Ma WH, Martins-Green M, Murphy WJ, Oppenheim JJ. Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood 2003; 102:1966-77; PMID:12791666; http://dx.doi.org/ 10.1182/blood-2002-11-3400 [DOI] [PubMed] [Google Scholar]
- [3].Tiveron MC, Cremer H. CXCL12/CXCR4 signalling in neuronal cell migration. Curr Opin Neurobiol 2008; 18:237-44; PMID:18644448; http://dx.doi.org/ 10.1016/j.conb.2008.06.004 [DOI] [PubMed] [Google Scholar]
- [4].Chatterjee S, Behnam Azad B, Nimmagadda S. The Intricate Role of CXCR4 in Cancer. Adv Cancer Res 2014; 124:31-82; PMID:25287686; http://dx.doi.org/ 10.1016/B978-0-12-411638-2.00002-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gagliardi F, Narayanan A, Reni M, Franzin A, Mazza E, Boari N, Bailo M, Zordan P, Mortini P. The role of CXCR4 in highly malignant human gliomas biology: Current knowledge and future directions. Glia 2014; 62:1015-23; PMID:24715652; http://dx.doi.org/ 10.1002/glia.22669 [DOI] [PubMed] [Google Scholar]
- [6].Tan W, Martin D, Gutkind JS. The Galpha13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J Biol Chem 2006; 281:39542-9; PMID:17056591; http://dx.doi.org/ 10.1074/jbc.M609062200 [DOI] [PubMed] [Google Scholar]
- [7].Lecointre C, Desrues L, Joubert JE, Perzo N, Guichet PO, Le Joncour V, Brulé C, Chabbert M, Leduc R, Prézeau L, et al.. Signaling switch of the urotensin II vasosactive peptide GPCR: prototypic chemotaxic mechanism in glioma. Oncogene 2015; 34:5080-94; PMID:25597409; http://dx.doi.org/ 10.1038/onc.2014.433 [DOI] [PubMed] [Google Scholar]
- [8].Zhang YG, Li J, Li YG, Wei RH. Urotensin II induces phenotypic differentiation, migration, and collagen synthesis of adventitial fibroblasts from rat aorta. J Hypertens 2008; 26:1119-26; PMID:18475149; http://dx.doi.org/ 10.1097/HJH.0b013e3282fa1412 [DOI] [PubMed] [Google Scholar]
- [9].Xu S, Jiang H, Wu B, Yang J, Chen S. Urotensin II induces migration of endothelial progenitor cells via activation of the RhoA/Rho kinase pathway. Tohoku J Exp Med 2009; 219:283-8; PMID:19966526; http://dx.doi.org/ 10.1620/tjem.219.283 [DOI] [PubMed] [Google Scholar]
- [10].Segain JP, Rolli-Derkinderen M, Gervois N, Raingeard de la Blétière D, Loirand G, Pacaud P. Urotensin II is a new chemotactic factor for UT receptor-expressing monocytes. J Immunol 2007; 179:901-9; http://dx.doi.org/ 10.4049/jimmunol.179.2.901 [DOI] [PubMed] [Google Scholar]
- [11].Grieco P, Franco R, Bozzuto G, Toccacieli L, Sgambato A, Marra M, Zappavigna S, Migaldi M, Rossi G, Striano S, et al.. Urotensin II receptor predicts the clinical outcome of prostate cancer patients and is involved in the regulation of motility of prostate adenocarcinoma cells. J Cell Biochem 2011; 112:341-53; PMID:21080343; http://dx.doi.org/ 10.1002/jcb.22933 [DOI] [PubMed] [Google Scholar]
- [12].Federico A, Zappavigna S, Romano M, Grieco P, Luce A, Marra M, Gravina AG, Stiuso P, D'Armiento FP, Vitale G, et al.. Urotensin-II receptor is over-expressed in colon cancer cell lines and in colon carcinoma in humans. Eur J Clin Investig 2014; 44:285-94; PMID:24372535; http://dx.doi.org/ 10.1111/eci.12231 [DOI] [PubMed] [Google Scholar]
- [13].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Proikas-Cezanne T, Ruckerbauer S, Stierhof YD, Berg C, Nordheim A. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Letts 2007; 581:3396-404; PMID:17618624; http://dx.doi.org/ 10.1016/j.febslet.2007.06.040 [DOI] [PubMed] [Google Scholar]
- [15].Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679-88; PMID:12665549; http://dx.doi.org/ 10.1242/jcs.00381 [DOI] [PubMed] [Google Scholar]
- [16].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mizushima N, Yoshimorim T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/ 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11:1433-7; PMID:19898463; http://dx.doi.org/ 10.1038/ncb1991 [DOI] [PubMed] [Google Scholar]
- [20].Hailey DW, Kim PK, Satpute-Krishnan P, Rambold AS, Sougrat R, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656-67; PMID:20478256; http://dx.doi.org/ 10.1016/j.cell.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre- autophagosomal structures. Cell 2010; 12:747-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013; 154:1285-99; PMID:24034251; http://dx.doi.org/ 10.1016/j.cell.2013.08.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Morozova K, Sidhar S, Zolla V, Clement CC, Scharf B, Verzani Z, Diaz A, Larocca JN, Hajjar KA, Cuervo AM, et al.. Annexin A2 promotes phagophore assembly by enhancing Atg16L+ vesicle biogenesis and homotypic fusion. Nat Commun 2015; 6:5856-66; PMID:25597631; http://dx.doi.org/ 10.1038/ncomms6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tuloup-Minguez V, Hamaï A, Greffard A, Nicolas V, Codogno P, Botti J. Autophagy modulates cell migration and β1 integrin membrane recycling. Cell Cycle 2013; 12:3317-28; PMID:24036548; http://dx.doi.org/ 10.4161/cc.26298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Qiang L, He YY. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy 2014; 10:1864-5; PMID:25126736; http://dx.doi.org/ 10.4161/auto.32171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, Lin YJ, Wu CT, Liu HS. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014; 59:505-17; PMID:23913306; http://dx.doi.org/ 10.1002/hep.26659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Belaid A, Ndiaye PD, Cerezo M, Cailleteau L, Brest P, Klionsky DJ, Carle GF, Hofman P, Mograbi B. Autophagy and SQSTM1 on the RHOA(d) again: emerging roles of autophagy in the degradation of signaling proteins. Autophagy 2014; 10:201-8; PMID:24300375; http://dx.doi.org/ 10.4161/auto.27198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, Mui S, Macleod KF. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Reports 2016; 15:1660-72; PMID:27184837; http://dx.doi.org/ 10.1016/j.celrep.2016.04.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep 2012; 13:733-40; PMID:22732841; http://dx.doi.org/ 10.1038/embor.2012.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M, et al.. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 2012; 14:51-60; http://dx.doi.org/ 10.1038/ncb2386 [DOI] [PubMed] [Google Scholar]
- [31].Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, Wittmann T, Debnath J. NBR 1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 2016; 212:577-90; PMID:26903539; http://dx.doi.org/ 10.1083/jcb.201503075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chan LLY, Shen D, Wilkinson AR, Patton W, Lai N, Chan E, Kuksin D, Lin B, Qiu J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy 2012; 8:1371-82; PMID:22895056; http://dx.doi.org/ 10.4161/auto.21028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1-12; PMID:19200872; http://dx.doi.org/ 10.1016/S0076-6879(08)03601-X [DOI] [PubMed] [Google Scholar]
- [35].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [36].Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283:22847-57; PMID:18524774; http://dx.doi.org/ 10.1074/jbc.M802182200 [DOI] [PubMed] [Google Scholar]
- [37].Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLOS Biol 2009; 7:0371-83; http://dx.doi.org/ 10.1371/journal.pbio.1000038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 2006; 10:839-50; PMID:16740485; http://dx.doi.org/ 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- [40].Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, Martens S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J 2012; 31:4304-17; PMID:23064152; http://dx.doi.org/ 10.1038/emboj.2012.278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6:764-76; PMID:20639694; http://dx.doi.org/ 10.4161/auto.6.6.12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, et al.. PICALM modulates autophagy activity and tau accumulation. Nat Commun 2014; 5:4998-5016; PMID:25241929; http://dx.doi.org/ 10.1038/ncomms5998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124-32; PMID:16998475; http://dx.doi.org/ 10.1038/ncb1482 [DOI] [PubMed] [Google Scholar]
- [44].Xia HG, Zhang L, Chen G, Zhang T, Liu J, Jin M, Ma X, Ma D, Yuan J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 2010; 6:61-6; PMID:19901552; http://dx.doi.org/ 10.4161/auto.6.1.10326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metabolism 2010; 11:467-78; PMID:20519119; http://dx.doi.org/ 10.1016/j.cmet.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Norman JM, Cohen GM, Bampton ETW. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010; 6:1042-56; PMID:21121091; http://dx.doi.org/ 10.4161/auto.6.8.13337 [DOI] [PubMed] [Google Scholar]
- [47].Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci 2005; 118:3829-38; PMID:16129881; http://dx.doi.org/ 10.1242/jcs.02562 [DOI] [PubMed] [Google Scholar]
- [48].Lysko DE, Putt M, Golden JA. SDF1 reduces interneuron leading process branching through dual regulation of actin and microtubules. J Neurosci 2014; 34:4941-62; PMID:24695713; http://dx.doi.org/ 10.1523/JNEUROSCI.4351-12.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Leloup L, Shao H, Bae YH, Deasy B, Stolz D, Roy P, Wells A. M-calpain activation is regulated by its membrane localization and by its binding to phosphatidylinositol 4,5-bisphosphate. J Biol Chem 2010; 285:33549-66; PMID:20729206; http://dx.doi.org/ 10.1074/jbc.M110.123604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shao H, Chou J, Baty CJ, Burke NA, Watkins SC, Stolz DB, Wells A. Spatial localization of m-calpain to the plasma membrane by phosphoinositide biphosphate binding during epidermal growth factor receptor-mediated activation. Mol Cell Biol 2006; 26:5481-96; PMID:16809781; http://dx.doi.org/ 10.1128/MCB.02243-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nuzzi PA, Senetar MA, Huttenlocher A. Asymmetric localization of calpain 2 during neutrophil chemotaxis. Mol Biol Cell 2007; 18:795-805; PMID:17192410; http://dx.doi.org/ 10.1091/mbc.E06-09-0876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Saraiva N, Prole DL, Carrara G, Johnson BF, Taylor CW, Parsons M, Smith GL. HGAAP promotes cell adhesion and migration via the stimulation of store-operated ca2+ entry and calpain 2. J Cell Biol 2013; 202:699-713; PMID:23940116; http://dx.doi.org/ 10.1083/jcb.201301016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yang S, Chen J, Jiang X, Wang Q, Chen Z, Zhao W, Feng Y, Xin R, Shi J, Bian X. Activation of chemokine receptor CXCR4 in malignant glioma cells promotes the production of vascular endothelial growth factor. Biochem Biophys Res Commun 2005; 335:523-8; PMID:16084492; http://dx.doi.org/ 10.1016/j.bbrc.2005.07.113 [DOI] [PubMed] [Google Scholar]
- [54].Wauson EM, Dbouk HA, Ghosh AB, Cobb MH. G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol Metab 2014; 25:274-82; PMID:24751357; http://dx.doi.org/ 10.1016/j.tem.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Foster SR, Roura E, Thomas WG. Extrasensory perception: odorant and taste receptors beyond the nose and mouth. Pharmacol Ther 2014; 142:41-61; PMID:24280065; http://dx.doi.org/ 10.1016/j.pharmthera.2013.11.004 [DOI] [PubMed] [Google Scholar]
- [56].Wauson EM, Zaganjor E, Lee AY, Guerra ML, Ghosh AB, Bookout AL, Chambers CP, Jivan A, McGlynn K, Hutchison MR, et al.. The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol Cell 2012; 47:851-62; PMID:22959271; http://dx.doi.org/ 10.1016/j.molcel.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Porrello ER, D'Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG, Delbridge LMD. Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension 2009; 53:1032-40; PMID:19433781; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.108.128488 [DOI] [PubMed] [Google Scholar]
- [58].Yadav A, Vallabu S, Arora S, Tandon P, Slahan D, Teichberg S, Singhal PC. ANG II promotes autophagy in podocytes. Am J Cell Physiol 2010; 299:488-96; http://dx.doi.org/ 10.1152/ajpcell.00424.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yu KY, Wang YP, Wang LH, Jian Y, Zhao XD, Chen JW, Murao K, Zhu W, Dong L, Wang GQ, et al.. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res Cardiol 2014; 109:416-31; PMID:24847907; http://dx.doi.org/ 10.1007/s00395-014-0416-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell 2011; 146:303-17; PMID:21784250; http://dx.doi.org/ 10.1016/j.cell.2011.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12-ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 2013; 20:59-66; PMID:23202584; http://dx.doi.org/ 10.1038/nsmb.2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kim JH, Hong SB, Lee JK, Han S, Roh KH, Lee KE, Kim YK, Choi EJ, Song HK. Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 2015; 11:75-87; PMID:25484072; http://dx.doi.org/ 10.4161/15548627.2014.984276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4:295-305; PMID:18391949; http://dx.doi.org/ 10.1038/nchembio.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rudinskiy N, Grishchuk Y, Vaslin A, Puyal J, Delacourte A, Hirling H, Clarke PGH, Luthi-Carter R. Calpain hydrolysis of α- and β2-adaptins decreases clathrin-dependent endocytosis and may promote neurodegeneration. J Biol Chem 2009; 284:12447-58; PMID:19240038; http://dx.doi.org/ 10.1074/jbc.M804740200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ando K, Brion JP, Stygelbout V, Suain V, Authelet M, Dedecker R, Chanut A, Lacor P, Lavaur J, Sazdovitch V, et al.. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathologica 2013; 125:861-78; PMID:23589030; http://dx.doi.org/ 10.1007/s00401-013-1111-z [DOI] [PubMed] [Google Scholar]
- [66].Kim JA, Kim HL. Cleavage of purified neuronal clathrin assembly protein (CALM) by caspase 3 and calpain. Exp Mol Med 2001; 33:245-50; PMID:11795487; http://dx.doi.org/ 10.1038/emm.2001.40 [DOI] [PubMed] [Google Scholar]
- [67].Kim JS, Nitta T, Mohuczy D, O'Malley KA, Moldawer LL, Dunn WA Jr, Behrns KE. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology 2008; 47:1725-36; PMID:18311843; http://dx.doi.org/ 10.1002/hep.22187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Glading A, Chang P, Lauffenburger DA, Wells A. Epidermal growth factor receptor activation of calpain Is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem 2000; 275:2390-8; PMID:10644690; http://dx.doi.org/ 10.1074/jbc.275.4.2390 [DOI] [PubMed] [Google Scholar]
- [69].Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol 2004; 24:2499-512; PMID:14993287; http://dx.doi.org/ 10.1128/MCB.24.6.2499-2512.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shiraha H, Glading A, Chou J, Wells A, Jia Z. Activation of m-Calpain (Calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m-Calpain. Mol Cell Biol 2002; 22:2716-27; PMID:11909964; http://dx.doi.org/ 10.1128/MCB.22.8.2716-2727.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Su Y, Cui Z, Li Z, Block ER. Calpain-2 regulation of VEGF-mediated angiogenesis. FASEB J 2006; 20:1443-51; PMID:16816119; http://dx.doi.org/ 10.1096/fj.05-5354com [DOI] [PubMed] [Google Scholar]
- [72].Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014; 509:105-9; PMID:24695223; http://dx.doi.org/ 10.1038/nature13148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Vega FM, Colomba A, Reymond N, Thomas M, Ridley AJ. RhoB regulates cell migration through altered focal adhesion dynamics. Open Biol 2012; 2:120076-85; PMID:22724071; http://dx.doi.org/ 10.1098/rsob.120076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J Microscopy 2013; 251:242-9; PMID:23488932; http://dx.doi.org/ 10.1111/jmi.12025 [DOI] [PubMed] [Google Scholar]
- [75].Razi M, Chan EYW, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol 2009; 185:305-21; PMID:19364919; http://dx.doi.org/ 10.1083/jcb.200810098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/ 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- [77].Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S, Thorburn A, He YY. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A 2014; 111:9241-6; PMID:24927592; http://dx.doi.org/ 10.1073/pnas.1322913111 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [78].Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA, et al.. Twist , a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927-39; PMID:15210113; http://dx.doi.org/ 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- [79].Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011; 19:372-86; PMID:21397860; http://dx.doi.org/ 10.1016/j.ccr.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Catalano M, D'Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V, Limatola C, Cecconi F, et al.. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 2015; 9:1612-25; PMID:26022108; http://dx.doi.org/ 10.1016/j.molonc.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Franco R, Zappavigna S, Gigantino V, Luce A, Cantile M, Cerrone M, Facchini G, Perdonà S, Pignata S, Lorenzo G Di, et al.. Urotensin II receptor determines prognosis of bladder cancer regulating cell motility / invasion. J Exp Clin Cancer Res 2014; 33:1-10; PMID:24383517; http://dx.doi.org/ 10.1186/1756-9966-33-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Nagasawa T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J Mol Med 2014; 92:433-9; PMID:24722947; http://dx.doi.org/ 10.1007/s00109-014-1123-8 [DOI] [PubMed] [Google Scholar]
- [83].Zhang W, Lane RD, Mellgren RL. The major calpain isozymes are long-lived proteins. J Biol Chem 1996; 271:18825-30; PMID:8702541; http://dx.doi.org/ 10.1074/jbc.271.31.18825 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.