

ABSTRACT
The regulation of plasma membrane (PM)-localized transmembrane protein/receptor trafficking has critical implications for cell signaling, metabolism and survival. In this study, we investigated the role of BECN1 (Beclin 1) in the degradative trafficking of PM-associated APP (amyloid β precursor protein), whose metabolism to amyloid-β, an essential event in Alzheimer disease, is dependent on divergent PM trafficking pathways. We report a novel interaction between PM-associated APP and BECN1 that recruits macroautophagy/endosomal regulatory proteins PIK3C3 and UVRAG. We found that BECN1 promotes surface APP internalization and sorting predominantly to endosomes and endolysosomes. BECN1 also promotes the targeting of a smaller fraction of internalized APP to LC3-positive phagophores, suggesting a role for BECN1-dependent PM macroautophagy in APP degradation. Furthermore, BECN1 facilitates lysosomal degradation of surface APP and reduces the secretion of APP metabolites (soluble ectodomains, sAPP). The association between APP and BECN1 is dependent on the evolutionarily conserved domain (ECD) of BECN1 (amino acids 267–337). Deletion of a BECN1 ECD subregion (amino acids 285–299) did not impair BECN1- PIK3C3 interaction, PtdIns3K function or macroautophagy, but was sufficient to impair the APP-BECN1 interaction and BECN1's effects on surface APP internalization and degradation, resulting in increased secretion of sAPPs. Interestingly, both the BECN1-APP association and BECN1-dependent APP endocytosis and degradative trafficking were negatively regulated by active AKT. Our results further implicate phosphorylation of the BECN1 Ser295 residue in the inhibition of APP degradation by AKT. Our studies reveal a novel function for BECN1 in the sorting of a plasma membrane protein for endolysosomal and macroautophagic degradation.
KEYWORDS: AKT, amyloid β, precursor protein, Beclin 1, plasma membrane, sorting
Introduction
Receptors/transmembrane proteins in the plasma membrane (PM) play critical roles in signal transduction, cell excitability, cell-cell and cell-matrix interactions, cellular metabolism regulation and numerous other vital cellular functions. Regulatory mechanisms that govern the localization of transmembrane proteins/receptors in the PM and that sort receptors for internalization and degradation have an impact on their downstream signaling effects. BECN1 (Beclin 1) has recently emerged as a key regulator of degradative trafficking pathways including the endolysosomal pathway1,2 and macroautophagy/autophagy.3,4 BECN1 and its rich network of interacting proteins (the BECN1 interactome) regulate the lipid kinase activity of the class III phosphatidylinositol-3-kinase (PtdIns3K). Interactions mediated by the coiled-coil domains of BECN1 and its interacting partners ATG14 and UVRAG are pivotal for regulating BECN1 function in autophagy and endosomal trafficking.5,6 Type I BECN1-PtdIns3K complexes containing ATG14 promote autophagy induction.7-10 Type II BECN1-PtdIns3K complexes containing UVRAG regulate endolysosome trafficking1,11 and play a role in autophagy maturation,8,9 which is dependent on interactions with the endosomal pathway.12 RUBCN/Rubicon binds to a subset of BECN1-PtdIns3K-UVRAG complexes and inhibits autophagy maturation and endocytic trafficking.8 In recent years, several integral membrane proteins have been discovered to regulate BECN1 complex activity and downstream autophagy.13-19 A role for BECN1 in endocytosis and sorting of PM receptors/transmembrane proteins from the cell surface for endolysosomal or autophagic degradation has not been reported.
APP (amyloid β precursor protein) is a type I transmembrane protein that is highly expressed in neuronal dendrites and axons. The functions of APP are incompletely understood; however, current evidence supports important roles for APP and its metabolites in the regulation of axon dynamics, synaptic adhesion and neurotrophism.20 Divergent APP sorting/trafficking pathways have a profound effect on APP metabolism. PM-localized APP can be cleaved by α-secretases to render soluble extracellular products including the sAPPα ectodomain. Alternatively, APP can be internalized into the endocytic pathway. Endocytosed APP may be trafficked for lysosomal degradation or may encounter BACE1/β-secretase and γ-secretase, which generate Aβ, the secreted metabolite that accumulates in extracellular neuritic plaques in Alzheimer disease (AD). APP adaptor/sorting proteins, including SORT1 (sortilin 1) and retromer,21-23 APBA2/X11/Mints24,25 and APBB1/Fe65,26 play important roles in regulating APP trafficking and metabolism and are potential therapeutic targets to reduce Aβ secretion.27 Novel PM-APP sorting mechanisms could provide new targets that might be leveraged for the prevention of Aβ neuropathology and AD. BECN1-dependent autophagy was previously shown to regulate APP levels and to mitigate Aβ neuropathology.28,29 We therefore hypothesized that BECN1 plays a role in the degradative sorting of PM-associated APP.
In this study, we examined the role of BECN1 in APP trafficking from the cell surface/plasma membrane. We found that BECN1 interacts with PM-localized APP to promote its internalization and sorting for lysosomal degradation in lieu of the secretion of APP metabolites (soluble ectodomains; sAPP). BECN1-mediated lysosomal sorting of PM-APP was predominantly via the endolysosomal pathway, although a smaller fraction was trafficked via the autophagy pathway. The interaction between APP and BECN1 is dependent on the evolutionarily conserved domain (ECD) of BECN1 (amino acids 267–337). Importantly, deletion of a subregion of the BECN1 ECD (amino acids 285–299) was sufficient to impair the APP-BECN1 interaction and BECN1's effects on surface APP internalization and degradation. The a.a. 285–299 BECN1 deletion increased the secretion of APP metabolites but did not significantly affect BECN1 PM association, BECN1-PIK3C3 interaction, PtdIns3K function or autophagy. Furthermore, both BECN1-APP association and BECN1-dependent APP endocytosis was negatively regulated by AKT. AKT regulation of BECN1-dependent APP trafficking appears to involve phosphorylation of the BECN1 Ser295 residue, a major AKT phosphorylation site in the BECN1 ECD region that regulates the APP-BECN1 interaction. Our studies reveal a novel function of BECN1 in the sorting of a plasma membrane protein from the cell surface for endolysosomal and autophagic degradation.
Results
BECN1 interacts with APP at the cell surface/plasma membrane
Recent studies have supported an important role for BECN1-dependent autophagy in regulating levels of APP and its metabolic byproducts. We hypothesized that BECN1-dependent regulation of APP levels involves interaction between APP and BECN1-PtdIns3K complexes. We performed co-immunoprecipitation (co-IP) experiments that demonstrated interaction between endogenous APP and BECN1 in whole cell extracts (WCE) and in synaptosomal fractions (P2) of cultured rat cortical neurons (Fig. 1A, top and bottom panels). We also detected the interaction between ectopic APP695 (predominant neuronal APP isoform) and FLAG-BECN1 via reciprocal co-IPs from HEK293 cells (Fig. 1B, right and left panels; Fig. S1). We next determined whether APP associates with components of BECN1-PtdIns3K complexes, namely, PIK3C3, UVRAG and ATG14. Immunoprecipitation (IP) of APP from HEK293 cells expressing APP695 and PIK3C3/VPS34, alone or in combination with FLAG-BECN1, followed by western blotting (WB) with PIK3C3 antibody, indicated that APP associates with PIK3C3 and that the interaction increases with BECN1 co-expression (Fig. 1C, top panel, lanes 1 vs. 2 and 3 vs. 4). IP of APP from HEK293 cells co-expressing APP695, FLAG-BECN1 and HA-UVRAG and/or MYC-ATG14 demonstrated that APP interacts with UVRAG to a much greater extent than with ATG14 under the conditions tested (Fig. 1D, lanes 3–5). APP association with UVRAG was markedly compromised in cells co-expressing a BECN1-ΔCC mutant, which does not bind UVRAG, compared to cells co-expressing wild-type (WT) BECN1 (Fig. 1D, lanes 5 vs. 6; Fig. S2). Collectively, our results show that APP is a novel BECN1-interacting protein and indicate that BECN1 is an important mediator of APP association with PIK3C3 and UVRAG.
Figure 1.

BECN1 interacts with APP on the cell surface/plasma membrane. (A) Interaction between endogenous APP and BECN1 was detected in whole cell extracts (WCE) of cultured rat cortical neurons (top panel) and in S2 (cytosol) and P2 (synaptosome) fractions (bottom panel) by immunoprecipitation (IP) with anti-BECN1 antibody. Normal mouse IgG (NmIgG) control was negative for BECN1. (B) Ectopic APP695 and FLAG-BECN1 association in HEK293 cells was demonstrated by IP with APP antibody CT-20 (left panel). Reciprocal co-IP of APP695 and FLAG-BECN1 in HEK293 cells was demonstrated by IP with FLAG antibody (right panel). (C) The interaction between ectopic APP695 and both endogenous and ectopic PIK3C3 was detected in HEK293 cells by IP with APP (CT-20) antibody. Co-expression of FLAG-BECN1 facilitated the interaction of APP with both endogenous and ectopic PIK3C3. (D) APP binding to the BECN1-UVRAG complex was detected in HEK293 cells by IP with APP antibody (CT-20). The interaction of APP with UVRAG was markedly impaired by coexpression of FLAG-BECN1 with a deletion of the coiled-coil (CC) domain (lanes 5 and 6). An interaction between APP and BECN1-ATG14 complexes is noted in the absence of UVRAG overexpression (lane 4). (E) BECN1 cofractionates with biotin-labeled surface proteins in cortical neurons (upper panel) and HEK293 cells (lower panel) as determined by immunoblot analyses of streptavidin-agarose bead affinity isolation with the indicated antibodies. The N-methyl-D-aspartate receptor subunits GRIN1/NR1 and GRIN2B/NR2B, and APP were used as markers for surface protein-enriched fractions in neurons and HEK293 cells, respectively. (F) BECN1 interaction with biotin-labeled, surface APP was detected in HEK293 cells by streptavidin-agarose bead affinity isolation of biotin-labeled surface proteins from FLAG-BECN1 (left panel, lane 3 versus 4) and APP (middle panel, lane 3 vs. 4) IPs probed with antibodies to the bait and prey proteins. These data confirm that BECN1 interacts with plasma membrane APP. Data shown in each figure panel (A-F) are representative of 2 to 4 independent experiments.
Emerging evidence suggests that BECN1 interactions occur in different subcellular locations, including the plasma membrane (PM), with distinct functional outcomes. In the context of APP-BECN1, we examined whether BECN1 associates with PM-localized APP. We first assessed if BECN1 is present in cell surface fractions of neurons and of HEK293 cells expressing APP and FLAG-BECN1. We labeled cell-surface proteins with cell impermeable NHS-S-S-biotin, precipitated biotinylated proteins with streptavidin-agarose beads and analyzed the surface protein precipitates with BECN1 antibody. We found that endogenous BECN1 co-precipitates with biotinylated surface proteins of neurons (Fig. 1E, upper panel, lane 4) and that ectopic BECN1 co-precipitates with surface proteins in HEK293 cells (Fig. 1E, bottom panel, lane 4).
We next employed cell surface biotinylation to determine whether BECN1 interacts with cell surface APP. Immunoprecipitation of total BECN1 or APP was first accomplished by incubating lysates from surface biotin-labeled cells with anti-FLAG or APP (CT20) antibody and protein A/G beads. The bound proteins were eluted with 1% SDS buffer and the eluates were then incubated with streptavidin beads to isolate PM-localized APP and BECN1. Western blot analysis of total and surface immunoprecipitates with FLAG and APP antibody clearly demonstrated BECN1 in association with cell surface APP in biotinylated samples but not in corresponding unlabeled samples (Fig. 1F, left panel, lanes 3 and 4; middle panel, lane 3 and 4). The extent of total APP-BECN1 co-IP was similar in biotin-labeled and unlabeled control samples (Fig. 1F, left panel, lanes 1 and 2; middle panel, lanes 1 and 2). We next addressed the importance of the APP endocytic motif, YENPTY, for the BECN1-APP surface interaction. It has been shown that proteins which bind to the endocytic motif of APP, such as APBA1/Mint1-APBA2/Mint2, DAB1, and APBB1/Fe65, regulate the internalization and intracellular degradative fate of APP. We co-expressed an endocytosis-defective mutant of APP, designated APP-AENATA, and FLAG-BECN1 in HEK293 cells and performed IP and streptavidin bead affinity isolation as outlined above. Our results indicate that the association of BECN1 with surface APP does not depend on the APP endocytic motif YENPTY (Fig. S3, right panel, lanes 2 and 4). Our results indicate that BECN1 interacts with surface proteins and identify a previously unknown interaction with plasma membrane APP.
The evolutionarily conserved domain (ECD) of BECN1 mediates the interaction with APP. We next sought to delineate the domains of BECN1 and APP that mediate their association. We co-expressed APP695 with FLAG-WT BECN1 or FLAG-BECN1 truncation mutants: the N terminus+BH3 domain (amino acids 1–150), the coiled-coiled (CC) domain (amino acids 151–241), N terminus+CC domain (amino acids 1–242), the BECN1 C terminus (amino acids 243–450) and BECN1 with a deletion of the CC domain (ΔCC) (Fig. 2A). Immunoprecipitation with FLAG antibody followed by immunoblotting with APP antibody revealed APP co-immunoprecipitation with full-length WT BECN1, recapitulating results in Fig. 1. We observed similar levels of co-immunoprecipitated APP with the BECN1 C terminus despite lower expression levels in the input (Fig. 2B, lanes 3 and 4; Fig. S4A, lanes 3 and 7), whereas APP binding by the BECN1 N terminus and CC-domain were nearly absent/considerably reduced in comparison to WT BECN1 (Fig. 2B, lanes 5 and 6; Fig. S4A, lanes 4 and 5). These results suggest that the C terminus of BECN1 primarily mediates the APP- BECN1 association.
Figure 2.

BECN1 associates with the intracellular C terminus of APP via the evolutionarily conserved domain (ECD). (A) Schematic representation of BECN1 deletion mutants used for mapping the interaction with APP. N-term, N terminus; BH3, BCL2 binding domain; CC, coiled-coil domain; C-term, C terminus (including evolutionarily conserved domain); ECD, evolutionarily conserved domain. (B and C) Analysis of BECN1-APP association in HEK293 cells cotransfected with APP and FLAG-BECN1 deletion mutants by FLAG IP demonstrate that the ECD of BECN1 is necessary and sufficient for the interaction with APP. (D) Interaction between APP C-terminal fragments (C99 and C60) and full-length BECN1 assessed by IP with APP (CT-20) antibody demonstrates that the C-terminal intracellular domain of APP interacts with BECN1. Data shown in each figure panel are representative of at least 2 independent experiments.
The C terminus of BECN1 harbors the evolutionarily conserved domain, which is pivotal for its autophagy and endosome regulatory functions. We next analyzed the effect of ECD deletion on APP-BECN1 interaction by co-expressing APP695 with a FLAG-tagged BECN1 ECD deletion mutant (Δ244–337) followed by IP with FLAG antibody and WB with APP antibody. Our results indicate that the ECD domain is indispensable for APP-BECN1 association as deletion of the domain nearly abolished BECN1 binding with APP (Fig. 2C, left panel, lane 3). We also confirmed that the APP C terminus is sufficient for the BECN1-APP interaction. IP with APP (CT20) antibody (Fig. 2D) or FLAG antibody (Fig. S4B) showed comparable binding of BECN1 with both full-length and APP C-terminal fragments (C99 and C60). Taken together, our results show that the interaction between APP and BECN1 is primarily mediated via the ECD of BECN1 and the C terminus of APP.
BECN1 promotes surface APP endocytosis and trafficking of internalized APP to endolysosomal and autophagosomal compartments
Based on our observations that BECN1 interacts with APP at the cell surface, we hypothesized that BECN1 regulates the endocytosis of surface APP and sorts it for lysosomal degradation. To test this hypothesis, we first assessed the effects of overexpressing or depleting BECN1 on APP internalization. To do so, we labeled N-terminal FLAG-APP695 on the surface of HeLa cells with FLAG antibody at 4°C. Internalization of FLAG-labeled APP was facilitated by incubating the cells in culture medium at 37°C for 5 min. We examined both the total amount of APP on the cell surface before internalization and the internalized pool of labeled APP (following acid treatment, which removes residual surface FLAG antibody) (Fig. 3A). Analysis by immunofluorescence microscopy revealed that BECN1 depletion induced a marked decrease in surface APP internalization (Fig. 3B, bottom-right image panels) compared to control cells (Fig. 3B, top-right image panels and graph). Western blot analysis with BECN1 antibody confirmed 70–80% knockdown in shBECN1-expressing cells compared to control shRNA-expressing cells (Fig. 3B, immunoblot).
Figure 3.
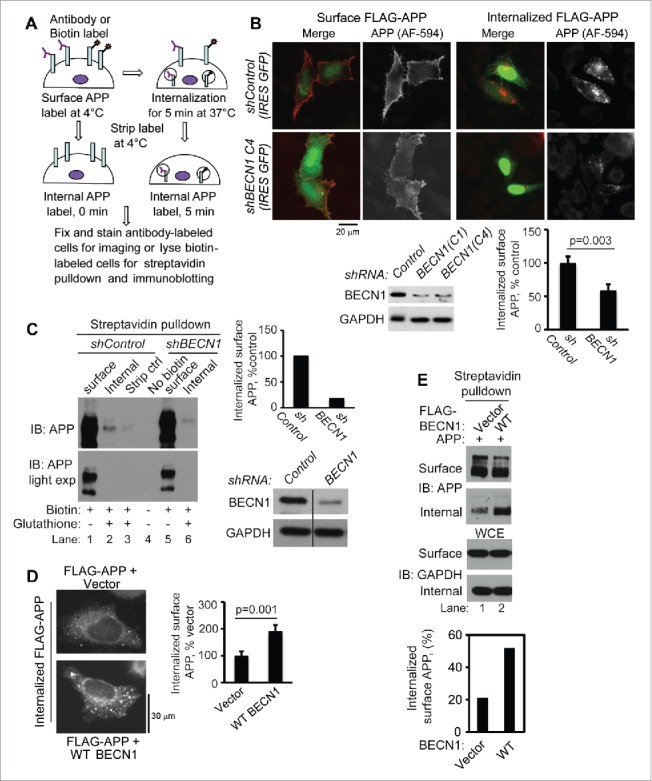
BECN1 promotes APP internalization. (A) Schematic of surface APP labeling and internalization assays. (B) BECN1 knockdown (shBECN1) reduced surface FLAG-APP695 internalization as demonstrated by FLAG antibody labeling of surface APP and monitoring internalized FLAG-APP by immunofluorescence in HeLa cells (p = 0.003, 2-tailed t-test; n = 80 cells). Internalized surface APP was normalized to total surface APP. IRES GFP, internal ribosome entry site. (C) BECN1 knockdown (shBECN1) reduced surface APP695 internalization in B103 APP neuronal cells (lane 2 versus lane 6) as measured by streptavidin affinity isolation of biotin-labeled, surface and internalized APP. Cells that were immediately treated with glutathione stripping buffer following biotin labeling showed a negligible amount of APP (lane 3), confirming that glutathione treatment efficiently removes surface biotin. Internalized surface APP was normalized to total surface APP. (D) Ectopic WT BECN1 enhances surface APP internalization as measured by immunofluorescence of internalized FLAG-APP (p = 0.001, 2-tailed t test; n = 55 cells). Internalized surface APP was normalized to total surface APP. (E) Ectopic WT BECN1 enhanced surface APP internalization in HEK293 cells as measured by streptavidin affinity isolation of biotin-labeled, surface and internalized APP. The graph represents densitometric analysis of the immunoblots in the upper panels, internalized APP normalized to surface APP. Data shown in each figure panel are representative of at least 2 independent experiments.
As a complementary approach, we labeled cell-surface APP with cell impermeable NHS-S-S-biotin on B103 APP neuroblastoma cells stably transfected with shBECN1 or control shRNA. Following internalization at 37°C, the remaining surface biotin was removed by treating cells with glutathione buffer. Biotinylated proteins were precipitated using streptavidin agarose beads and analyzed by WB with APP (22C11) antibody. Biochemical analyses showed that the total surface levels of APP were comparable between vector and BECN1-depleted cells (Fig. 3C, right panel, lanes 1 and 5) but that the internalized pool of APP was decreased approximately 80% in BECN1-depleted cells (with ∼80% BECN1 knockdown) compared to control cells (Fig. 3C, immunoblot lanes 2 versus 6 and graph).
In contrast to the effects of BECN1 depletion, ectopic BECN1-expressing cells demonstrated significantly more internalized APP than control cells as measured by immunofluorescence detection of internalized FLAG-APP in HeLa cells (Fig. 3D) and by western blot of internalized biotin-labeled APP in HEK293 cells (Fig. 3E). In all experiments, surface APP levels, measured by IF or ELISA following antibody labeling, and streptavidin-agarose bead affinity isolation following surface biotin labeling, were not significantly affected by BECN1 depletion (Fig. S5A, D and E) or by BECN1 overexpression (Fig. S5B, C and F).
To analyze the effect of BECN1 on post-endocytic traffic of internalized APP to endolysosomal and autolysosomal compartments, we assessed colocalization between internalized FLAG-APP and markers of early and late endosomes, autophagosomes and lysosomes in HeLa cells. We noted increases in trafficking of internalized FLAG-APP to EEA1-positive early endosomal compartments in GFP-EEA1 HeLa cells (data not shown), RAB7-positive late endosomal compartments in GFP-RAB7 HeLa cells (Fig. 4A and Fig. S6C; n = 23 and 241 cells, respectively) and LAMP1-positive lysosomal compartments in GFP-LAMP1 HeLa cells (Fig. 4B and Fig. S6A-B; n = 37 and 256 cells, respectively) with ectopic BECN1 expression (Fig. 4C). Concurrent treatment with the vacuolar-type H+-ATPase inhibitor bafilomycin A1 (50 nM) markedly impaired BECN1-driven trafficking of surface APP to late endosomes and lysosomes (Fig. S6). We also noted a statistically significant increase in the fraction of the internalized APP colocalizing with GFP-LC3-positive autophagosomes at 5 min post-internalization with BECN1 overexpression (Fig. 5A). With ectopic BECN1 expression, the colocalization of internalized APP with LC3-positive autophagosomes compared to EEA1-positive early endosomes was 1.44 +/− 0.37% vs. 21.35 +/− 2.49%, respectively, indicating that BECN1 facilitates APP sorting predominantly to endosomes. Interestingly, we also observed colocalization of internalized APP with the phagophore marker, GFP-ATG16L1 within 5 min of facilitating endocytosis in HeLa cells (Fig. 5B). Reciprocal IPs of ectopic APP and FLAG-ATG16L1 from HEK293 cells demonstrated that these 2 proteins interact (Fig. 5C, D). Streptavidin affinity isolation from APP immunoprecipitate, as outlined for BECN1-surface APP interaction, showed that a substantial fraction of PM-localized APP associated with ATG16L1 (Fig. 5E, lane 4). As seen with WT BECN1, the YENPTY internalization motif was dispensable for the APP-ATG16L1 interaction (data not shown). Collectively, our results support our hypothesis that BECN1 interaction with cell-surface APP facilitates surface APP endocytosis and sorting predominantly to endosomes but also to phagophores. Taken together with our APP-ATG16L1 interaction and colocalization data, our findings also suggest a role for PM autophagy in targeting surface APP to degradative compartments.
Figure 4.

BECN1 promotes surface APP trafficking to endolysosomes. (A-C) Surface FLAG-APP was labeled with FLAG antibody and the trafficking of internalized FLAG-APP to endosomal compartments was analyzed by immunofluorescence microscopy. Ectopic WT BECN1 enhanced the localization of internalized surface APP to GFP-RAB7-labeled late endosomes (A; p = 0.01039, 2-tailed t-test; n = 23 cells; representative of N=3 independent experiments) and to GFP-LAMP1-labeled lysosomes (B; p = 0.00035, 2-tailed t-test; n = 37 cells, representative of N = 4 independent experiments). Representative images are shown with regions of colocalization indicated by white arrowheads. Percent colocalization data (in A and B) are shown in the graphs (mean+/−SEM). Ectopic BECN1 expression was confirmed by immunoblotting whole cell extracts (C).
Figure 5.

BECN1 promotes surface APP internalization and trafficking to autophagosomes. (A) Ectopic BECN1 enhanced the localization of internalized surface APP to GFP-LC3-labeled autophagosomes. Surface APP was labeled with FLAG antibody and trafficking of internalized APP to GFP-LC3 compartments was analyzed by immunofluorescence microscopy. A representative image with regions of colocalization indicated by arrowheads and percent colocalization data is shown in the graph (mean+/−SEM) (*p < 0.05; 2-tailed t test; n = 287 cells; representative data from N = 2 experiments). (B) Internalized surface APP was also localized to ATG16L1-labeled autophagosomal precursors. (C) IP of FLAG-ATG16L1 from HEK293 cells co-immunoprecipitated ectopic APP (lane 3). (D) The reciprocal APP IP demonstrated interaction of FLAG-ATG16L1 with endogenous APP (lane 3) and ectopic APP (lane 4). (E) Interaction of surface APP with the phagophore marker ATG16L1 was demonstrated in HEK293 cells by streptavidin affinity isolation of biotin-labeled surface proteins in APP IP, and immunoblotting with the indicated antibodies.
BECN1 facilitates lysosomal degradation of surface APP and reduces APP metabolite secretion
Our finding that BECN1 sorts surface APP to endolysosomal compartments (Fig. 4) led us to hypothesize that BECN1 targets surface APP for lysosomal degradation with a resultant decrease in the generation of soluble APP ectodomains (sAPP) from surface APP (Fig. 6A). We next determined the effect of BECN1 on lysosomal degradation and metabolic processing of surface APP by performing biotin label chase experiments. Surface APP was labeled with cell-impermeable biotin on ice, and cell extracts and culture supernatants were collected at different time points post incubation of labeled cells at 37°C. Samples from streptavidin affinity isolation of biotin-labeled proteins were analyzed by immunoblotting with APP N-terminal antibody (22C11) to determine the levels of full-length, biotin-labeled cellular APP (FL-APP) and secreted, biotin-labeled sAPP. Our results show that co-expression of APP with WT BECN1 increased biotin-labeled full-length APP degradation (Fig. 6B, top panel and graph). Consistent with the effect on biotin-labeled full-length degradation, WT BECN1-expressing cells showed less accumulation of biotin-labeled sAPP in culture supernatants (Fig. 6B, bottom panel and graph). Conversely, BECN1 knockdown increased the extracellular accumulation of biotin-labeled sAPP compared to control cells (Fig. 6C, bottom panel and graph). Inhibition of the lysosomal pathway using ammonium chloride during the biotin label chase resulted in cellular accumulation of BECN1-driven internalized APP (Fig. 6D, top panel, lanes 6–8 compared to lanes 10–12 and graph) and caused a reversal in the BECN1-dependent reduction in sAPP secretion similar to levels seen in vehicle-treated cells (Fig. 6D, bottom panel, lanes 5–7 compared to lanes 9–11, and graph) and cells expressing APP interaction-deficient BECN1 Δ267–337 (Fig. 6D, bottom panel, lane 8). Ectopic BECN1 did not significantly facilitate APP degradation following mutation of the APP internalization motif (YENPTY) or the C-terminal ubiquitination sites (K649-651) under the conditions tested (Fig. S7A, B). Interestingly, co-expression of APP with UVRAG, ATG14 or ATG16L1 did not have the same effect as WT BECN1 on surface APP degradation as measured by extracellular sAPP levels underscoring the importance of BECN1 in APP degradative trafficking (Fig. S8). Our results thus indicate that BECN1 facilitates surface APP internalization and sorts this APP for lysosomal degradation with concomitant decrease in the secretion of APP metabolites.
Figure 6.

BECN1 facilitates lysosomal degradation of surface APP and reduces APP ecto-domain secretion. (A) Schematic of products of surface APP processing. AICD, amyloid precursor protein intracellular domain. (B) Surface APP degradation was assessed using biotin-label chase assay. The amount of biotin-labeled FL-APP at 2 h of label chase was calculated relative to surface APP at time 0 h. Ectopic WT BECN1 reduced biotin-labeled FL-APP (p = 0.0010, unpaired 2-tailed t test; n = 5) and secreted APP ectodomains (sAPP; p = 0.0257, unpaired 2-tailed t test; n = 4). (C) Knockdown of BECN1 increased secreted sAPP generated from biotin-labeled, surface APP (p = 0 .0377, unpaired 2-tailed t test; n = 3). (D) The lysosomal inhibitor ammonium chloride (25 mM) abrogated the effect of WT BECN1 on biotin-labeled FL-APP degradation (p = 0.032, unpaired 2-tailed t test; n = 4) and sAPP secretion (p = 0.0136, unpaired 2-tailed t-test; n = 4) as assessed by biotin-label chase assay.
BECN1 ECD mutant (Δ285–299) impairs surface APP degradation independently of BECN1-PIK3C3 interaction and function
Initial analysis of WCEs and culture supernatants from WT and BECN1 (Δ267–337; lacking the ECD)-expressing cells suggested that the BECN1 ECD is necessary for BECN1-induced reductions in FL-APP and sAPP levels (Fig. 6D, bottom panel, lanes 6 versus 8; Fig. S9). However, deletion of the entire ECD region compromises BECN1 endosomal and autophagy functions due to decreased PIK3C3/VPS34 binding3 (Fig. 7A, lane 4; Fig. S10). To further assess a role for the APP-BECN1 ECD interaction on surface APP degradation, we utilized BECN1 mutants with small deletions in the ECD30 in further experiments on surface APP trafficking. We first examined PIK3C3 binding, PM association and autophagy function of the different mutants. We chose the BECN1 Δ285–299 mutant for further experiments, as association of this mutant with PIK3C3 is intact (Fig. 7A; Fig. S11A). Furthermore, the BECN1 Δ285–299 mutant did not significantly reduce the levels of PM-associated BECN1 (Fig. S12A), did not alter the levels of autophagosomal LC3 or cause SQSTM1/p62 accumulation (Fig. S12B, lanes 1 vs. 6), and did not significantly attenuate the density of cytoplasmic FYVE puncta (a measure of phosphatidylinositol-3-phosphate [PtdIns3P] synthesis) (Fig. S12C). However, in reciprocal co-IP experiments, the BECN1 Δ285–299 mutant demonstrated markedly impaired APP binding (Fig. 7B, left and right panels), suggesting that amino acids 285–299 are an important mediator of the BECN1-APP interaction. Additional experiments revealed that the BECN1 Δ285–299 mutant was impaired in its ability to facilitate surface APP internalization (Fig. 7C) and to reduce secreted sAPP derived from surface APP compared to WT BECN1 (Fig. 7D). Taken together, these data demonstrate that amino acids 285–299 of the BECN1 ECD are important for the BECN1-APP interaction, surface APP internalization and sorting of internalized APP for degradation in lieu of sAPP secretion.
Figure 7.

A subregion of the BECN1 ECD interacts with and sorts surface APP for degradation independently of BECN1's PIK3C3 regulatory function. (A) PIK3C3-BECN1 association was analyzed in HEK293 cells cotransfected with FLAG-tagged BECN1 ECD deletion mutants and MYC-PIK3C3 by IP with MYC antibody followed by immunoblotting with antibodies, as indicated. Small BECN1 ECD deletions, including Δ267–284, Δ285–299, Δ300–321and Δ322–337, did not impair the interaction of BECN1 with PIK3C3. (B) Deletion of amino acids 285–299 from the BECN1 ECD impaired APP-BECN1 association as detected by FLAG IP (left panel) and by APP IP (right panel). (C) WT BECN1, but not the BECN1 ▵285–299 mutant, enhanced surface APP internalization as demonstrated by APP antibody (6E10)-labeling of surface APP and monitoring internalized APP by immunofluorescence in HeLa cells. Representative image panels and quantification of internalized APP puncta are shown (ANOVA with post-hoc Tukey's honestly significant difference test; n = 190 cells; representative data of N = 2 experiments). Internalized surface APP was normalized to total surface APP. Expression of WT and mutant BECN1 was confirmed by immunoblotting with FLAG antibody. (D) BECN1-driven surface APP degradation is impaired by deletion of the BECN1 ECD region 285–299, as assessed by biotin-label chase assay. Quantification of sAPP generated from biotin-labeled APP is shown and was used as a measure of APP degradation (ANOVA with post-hoc Tukey's honestly significant difference test; N = 3).
AKT regulates BECN1-dependent surface APP degradative sorting
Recent studies have shown BECN1 to be a substrate for AKT phosphorylation. AKT phosphorylates BECN1 residues Ser234 and Ser295 and inhibits its autophagy function.31 Considering that the BECN1-APP interaction region we found in the BECN1 ECD, amino acids 285–299 harbors the AKT phosphorylation site, Ser295, we examined if AKT phosphorylation of BECN1 regulates the APP-BECN1 interaction and BECN1-dependent surface APP endocytosis and degradative trafficking. Co-expression of myr-AKT, a constitutively active form of AKT with markedly enhanced Ser473 phosphorylation31,32 (Fig. S13), caused a marked reduction in the interaction of WT FLAG-BECN1 with APP (Fig. 8A, lanes 4 and 5). Quantification of surface APP internalization by immunofluorescence demonstrated that myr-AKT co-expression significantly impaired WT BECN1-facilitated APP endocytosis but not APP endocytosis facilitated by the BECN1S295A mutant (Fig. 8B, left panel). Furthermore, analysis of sAPP levels in culture supernatants from WT and BECN1S295A-expressing cells revealed that active AKT co-expression increased secreted sAPP levels from WT BECN1-expressing cells (Fig. 8C, lanes 2 versus 4; 2 separate experiments shown), but not from BECN1S295A cells (Fig. 8C, lanes 3 vs. 5). To specifically examine the effect of active AKT on BECN1-mediated surface APP degradation and sAPP generation, we performed a 2-h biotin-label chase assay (Fig. 8D). In the absence of active AKT co-expression, both BECN1 WT and the S295A mutant showed similar decreases of biotin labeled surface APP (14–16% labeled APP remaining) and similar levels of sAPP, 2 h post chase (Fig. 8D, lanes 1 and 2). In the presence of active AKT, surface APP degradation was markedly inhibited in WT BECN1-expressing cells (35% labeled APP remaining; Fig. 8D, lane 3 versus lane 1) but not in BECN1S295A-expressing cells (17% labeled APP remaining; Fig. 8D, lane 4 vs. lane 2). The levels of sAPP were correspondingly increased in WT BECN1 cells but remained unchanged in S295A cells (Fig. 8D, bottom immunoblot). Our results demonstrate regulation of the BECN1-APP interaction and BECN1-mediated surface APP endocytosis and degradative trafficking by AKT.
Figure 8.

AKT attenuates the BECN1-APP interaction, BECN1-driven surface APP internalization and degradation via phosphorylation of BECN1S295. (A) Expression of constitutively active AKT (myr-AKT) markedly reduced the BECN1-APP interaction in HEK293 cells as demonstrated by IP with APP (CT-20) antibody and immunoblotting with FLAG antibody (lanes 4 and 5). Data shown are representative of N = 4 independent experiments. (B) The effect of Myr-AKT on BECN1-driven APP internalization was determined by labeling surface APP (antibody 6E10) and monitoring of APP internalization by immunofluorescence in HeLa cells. Myr-AKT expression attenuated WT BECN1-driven APP internalization but not APP internalization driven by phosphonull mutant BECN1S295A expression. Quantification of internalized APP puncta is shown in the graph (left panel; mean+/−SEM). Immunoblotting with BECN1 showed comparable expression of WT and phosphomutant (right panel). (C) Myr-AKT expression attenuated WT BECN1-, but not BECN1S295A-, mediated effects on APP ectodomain (sAPP) secretion in HEK293 cells (2 different experimental replicates shown). (D) Active AKT inhibited WT BECN1-driven surface APP degradation but did not affect BECN1S295A (SA)-driven surface APP degradation as assessed by a biotin-label chase assay. (E) Model of BECN1-mediated lysosomal degradation of surface APP. The BECN1 ECD interacts with PM-localized APP to facilitate APP internalization and trafficking to autophagosomal and endolysosomal compartments for degradation in lysosomes (green arrows). This results in reduction in levels of surface APP and sAPP derived from surface APP. The BECN1-APP interaction, BECN1-driven surface APP internalization and sorting for lysosomal degradation are impaired by deletion of the BECN1 ECD and reduced by AKT-dependent BECN1 Ser295 phosphorylation (red bars), resulting in cellular APP stabilization and increased secretion of APP ectodomains (red arrows). Data shown in each figure panel are representative of at least 2 independent experiments.
Discussion
BECN1 clearly has important roles in the regulation of endolysosomal and autophagy pathways, and our developing knowledge of BECN1 trafficking mechanisms has suggested a role in the regulation of trafficking of PM proteins. Our study demonstrates that BECN1 localizes to the PM, interacts with an integral PM protein (APP) and facilitates the association of APP with PIK3C3 and UVRAG. We show that the BECN1 ECD (amino acids 267–337) is sufficient to interact with APP and to promote surface APP internalization and degradation via early/late endosomes and, to a lesser extent, autophagosomes. BECN1 promotes the lysosomal degradation of surface APP in lieu of sAPP ectodomain secretion. We found that the amino acid 285–299 subregion of the BECN1 ECD is critical for the interaction and sorting of surface APP, but this is not due to reduced BECN1 membrane localization or impairment of the BECN1- PIK3C3 interaction and regulatory function. Finally, we demonstrated that AKT, which phosphorylates BECN1 in the amino acid 285–299 subregion of the ECD at Ser295, reduces BECN1-APP interaction and BECN1-driven APP endocytosis and degradation. Our data identify APP as a previously unknown protein in the BECN1 interactome. Furthermore, our study demonstrates a novel, PtdIns3K-independent function of the BECN1 ECD to interact with and promote endocytosis and degradative sorting of a PM-localized cargo (Fig. 8E).
Levine and colleagues previously demonstrated that a BECN1 deletion mutant lacking a large region encompassing the ECD (amino acids 244–337) is markedly impaired in its ability to interact with PIK3C3.3 A smaller region of the ECD, amino acids 267–284, mediates the interaction of BECN1 with the autophagy-inhibiting proteins GLIPR2/GAPR-1 and HIV-derived Nef.30 Interestingly, deletion of amino acids 267–284 does not impair the BECN1-PIK3C3 interaction.30 In agreement with these published findings, our data similarly show that a large ECD deletion (amino acids 267–337) markedly impairs the BECN1-PIK3C3 interaction, whereas small ECD deletions, including Δ267–284 and Δ285–299, do not impair the BECN1-PIK3C3 interaction or function. Overall, deletion of the entire BECN1 ECD domain perturbs the core function of BECN1 in the regulation of PIK3C3, whereas smaller ECD regions regulate BECN1 interactions with other proteins (e.g., Nef, GLIPR2, EGFR [epidermal growth factor receptor], APP, etc).
Integral PM proteins, including EGFR, have previously been shown to interact with BECN1 to regulate downstream autophagy. Wei and colleagues demonstrated that activated EGFR interacts with the BH3 domain and ECD of BECN1 to inhibit autophagy via recruitment of RUBCN and dissociation of PtdIns3K.13 Lapatinib, an inhibitor of the ERBB2/Her2 tyrosine kinase receptor, was reported to disrupt the interaction of ERBB2 with the BECN1 ECD, resulting in the release of BECN1 to the cytosol to activate autophagy.19 Tan and colleagues showed conversely that inactive EGFR promotes autophagy through its interaction with RUBCN, which dissociates the inhibitory RUBCN from BECN1.14 In response to Aβ, PRNP (prion protein) recruits BECN1 to CAV/caveolin-rich lipid rafts to induce autophagy.33 Autophagy promoting interactions of BECN1 with the integral membrane proteins EMC616 and VMP1 (vacuole membrane protein 1),17 and autophagy inhibiting interactions with ITPR/IP3R15 have also been reported. Plasma membrane interacting proteins such as SH3GLB1/Bif-134 can interact with BECN1complexes to activate downstream autophagy. BECN1 localization to cell membranes is facilitated by its C terminus35 and by hydrophobic amino acids of its ECD.36 The possibility that APP regulates endolysosomal and/or autophagy function through its interactions with BECN1 and/or ATG16L1 will require additional investigation.
The aforementioned studies provide substantial evidence for the modulation of BECN1-dependent autophagy by integral membrane proteins and extracellular signals. In contrast, the potential role for BECN1 in the degradative sorting of plasma membrane proteins has not been addressed. GJA1/Cx43 (gap junction protein α 1), interacts with BECN1-PtdIns3K complexes to inhibit autophagy. It is not clear if the GJA1-BECN1 interaction is important for the autophagic degradative sorting of GJA1, which is driven by NEDD4 ubiquitination37 and which helps to sustain autophagy activation.18 Interestingly, recent work has shown that the BECN1 homolog BECN2 interacts with GPRASP1/GASP1 to facilitate post-endosomal degradative sorting of G-protein coupled receptors.38 Functionally, BECN2 prevents receptor recycling similar to the inhibition of APP recycling by BECN1 to reduce APP metabolism at the plasma membrane. BECN1 similarly might modulate signal transduction via other plasma membrane receptors by regulating their internalization and promoting their degradation in lieu of receptor recycling.
A role for BECN1 in the degradative sorting of PM-APP may have important implications for AD. Deficiencies in endolysosomal trafficking and autophagy have long been implicated in the pathogenesis of sporadic AD.39 Human AD brains and brains from mouse AD models show reduced levels of endosomal and autophagy regulators BECN1,29,40 PIK3C3 and PtdIns3P.41 Morel and colleagues recently showed that sorting of APP into intraluminal vesicles of multivesicular bodies depends on PIK3C3 and the ESCRT complex.41 BECN1 reduces cellular FL-APP levels and Aβ in cell culture models and in mouse brain.28,29 Furthermore, BECN1 facilitates microglial Aβ clearance by promoting exocytic recycling of the phagocytic receptor CD36 through the retromer pathway.42 Interestingly, no interaction between BECN1 and CD36 was identified, but a critical role for BECN1-PtdIns3K in maintaining PtdIns3P levels for recruiting the retromer machinery was demonstrated. Our data suggest the possibility that BECN1 deficiency in AD impairs degradative sorting of PM-APP, the precursor of toxic Aβ. Therapeutic strategies to restore BECN1 levels, perhaps through transcription factors like GABP,43 or to restore BECN1 function through signaling pathways that affect BECN1 phosphorylation, might therefore be pursuable goals.
Aberrant AKT signaling has also been implicated in AD pathogenesis.44,45 A prior cell culture study demonstrated that activated AKT reduces lysosomal APP degradation,46 however the mechanism(s) remained unclear. AKT was recently shown to interact with and phosphorylate BECN1 at S234 and S295 to suppress autophagy and drive tumorigenesis.31 Our data show that AKT phosphorylation of BECN1 at S295 suppresses BECN1-driven degradative sorting of surface APP and results in increased secretion of APP metabolites. Our findings suggest that AKT regulates BECN1-dependent APP sorting and degradation, and indicate the possibility of a link between BECN1 and dysregulated AKT-associated pathways in aging, metabolic syndrome and AD.
Our data demonstrate that a small proportion of surface APP interacts with BECN1-PIK3C3-ATG14 complexes and, upon internalization, is rapidly trafficked to LC3-labeled autophagosomes. We also observed that surface APP interacts with ATG16L1 and is trafficked to ATG16L1-positive phagophores. These observations suggest the possibility of APP internalization directly in PM-derived autophagosomal precursors.47 Interestingly, BECN1 and ATG16L1 interactions with APP did not require the YENPTY internalization motif of APP which interacts with AP2-clathrin and which is necessary for APP internalization and internalization of PM-derived autophagosomal precursors. These data suggest the possibility that ATG16L1 might interact through PM receptors like APP to divert clathrin-coated endosomes to the autophagy pathway.
In summary, we have demonstrated a novel function of BECN1 in the degradative sorting of cell surface APP. This sorting function is dependent on an ECD subregion that interacts with APP and is distinct from other known BECN1 ECD functions in PIK3C3 interaction, namely PtdIns3K lipid kinase activity regulation and BECN1 PM localization. Furthermore, we demonstrated that activated AKT negatively regulates BECN1-mediated degradative sorting of PM-APP, likely through phosphorylation of the BECN1 Ser295 residue. Our study has important implications for the role of BECN1 in PM cargo sorting for endolysosomal and autophagy trafficking and for our understanding of APP trafficking and metabolism.
Materials and methods
All animal procedures were designed in accordance with the Guidelines for Animal Use of Stanford University and were approved by the Institutional Animal Care and Use Committee (Protocol 26911).
Plasmids
The following plasmids were procured as gifts from the following providers via Addgene: Dennis Selkoe and Tracy Young-Pearse, Harvard University: pCAX APP695 (30137),48 pCAX FLAG-APP (30154), pCAX APP AENATA (30144), pCAX APP C99 (30146), pCAX APP C60 (30152), pCAX APP delta CT (30143); Qing Zhong, University of Texas Southwestern: pcDNA4 FLAG-BECN1 (24388),10 pcDNA4 FLAG-BECN1 1–150 (24389), pcDNA4 FLAG-BECN1 151–241 (24390), pcDNA4 FLAG-BECN1 1–242 (24391), pcDNA4 FLAG-BECN1 Δ151–241 (24393), pcDNA4 FLAG-BECN1 243–450 (24392); Esteban Dell'Angelica, UCLA: LAMP1-mGFP (34831);49 Silvia Corvera, University of Massachusetts: GFP-EEA1 WT (42307);50 Richard Pagano: GFP-RAB7 WT (12605),51 RAB11-GFP (12674); Noboru Mizushima, University of Tokyo: pCI-neo-HA-hUVRAG (24290),5 pCI-neo-MYC-hATG14 (24293); William Sellers, Harvard University: pcDNA3-myr-HA-AKT1 (9008),32 pcDNA3 T7 AKT1 K179M T308A S473A (9031).32 The following investigators kindly provided additional plasmids: Beth Levine, University of Texas Southwestern: pBICEP-BECN1 ECD deletion mutants and pcDNA4-MYC-PIK3C3;30 Tamotsu Yoshimori, Osaka University: p3X FLAG-CMV10 ATG16L1, pEGFP-C1-ATG16L1, pEGFP-LC3;52 Sergio Grinstein, University of Toronto: 2X FYVE-mRFP;53 Richard Wang, University of Texas Southwestern: pBABE FLAG-BECN1 WT and pBABE FLAG-BECN1 S295A mutant.31 pCAX KR-APP695 mutant was generated by site-directed mutagenesis where lysine residues 649–651 in pCAX APP695 were replaced by alanine. pCI-neo/WT and S295A BECN1 were constructed by cloning corresponding FLAG-BECN1 fragments between ECoRI sites in the pCI-neo vector (Promega, E1841). All constructs were verified by sequencing.
Cell culture and transfections
HeLa and HEK293 human embryonic kidney cells were obtained from ATCC (CCL-2 and CRL-1573, respectively) and maintained in ATCC-recommended medium at 37°C and 5% CO2. B103 neuroblastoma cells and B103 APP cells stably expressing human APP695 were kindly provided by Dr. Tony Wyss-Coray (Stanford University) and maintained in DMEM (Cellgro, 10-017-CV) containing 10% fetal bovine serum (Invitrogen, 26140–079), 1% penicillin-streptomycin (Cellgro, 30-002-CI) and 100 μg /ml G418 (Invitrogen, 10131). Primary cortical neurons were established from cortices of E18 embryos and plated on poly-D-lysine (Sigma, P1024)-coated tissue culture plates at a density of 7 × 104/cm2 in Neurobasal medium (Thermo Fisher, 21103049) containing 1 mM GlutaMAX-I (Thermo Fisher, 35050061) and 2% B-27 (Thermo Fisher, 17504044).
To establish HeLa cells stably expressing GFP-LC3, cells were transfected with pEGFP-LC3 using Lipofectamine 2000 reagent (Invitrogen, 11668019), per the manufacturer's protocol. Transfected cells were selected with 1 mg/ml G418 and stable lines were maintained in 100 μg/ml G418. To deplete endogenous BECN1, B103 APP or HeLa cells were transfected with shRNA for BECN1 or control nontargeting shRNA (Qiagen, 336311) using Lipofectamine 2000 transfection reagent. Cells were harvested at 48 h or 72 h, as indicated, or selected with puromycin (Cellgro, MT61385RA) to derive cells with stable knockdown of BECN1.
Cell lysis and immunoblot analyses
Cells grown to ∼80% confluence were lysed directly on the plate with RIPA buffer (50 mM Tris-HCl, pH 7.6, 100 mM NaCl, 2mM EDTA,1% Triton X-100 [Sigma, T8787], 0.5% sodium deoxycholate [Sigma, D6750] and 0.1% SDS [Bio-Rad Laboratories, 161–0418]) containing protease inhibitors (Sigma, P8340), 1 mM sodium orthovanadate (Sigma, S6508) and 1 mM sodium fluoride (Sigma, S1504). The lysates were transferred to microcentrifuge tubes and rotated for 20 min. Lysates were cleared by centrifugation at 16,200 x g for 30 min and protein estimation was done using Bradford assay reagent (Bio-Rad Laboratories, 500–0205). Equal amount of protein was analyzed by immunoblotting with appropriate antibodies: APP 22C11 (Millipore, MAB348), APP CT-20 (Millipore, 171610), FLAG M2 (Sigma, F1804), GAPDH (Cell Signaling Technology, 2118S), BECN1 (BD Biosciences, 612113 and Cell Signaling Technology, D40C5), GFP (B2; Santa Cruz Biotechnology, sc-9996), ATG14 (Cell Signaling Technology, 5504) UVRAG (MBL International, M160-3), ATG16L1 (MBL International, PM040), PIK3C3/VPS34 (Cell Signaling Technology, 4263), AKT and phosphorylated-Ser473 AKT (Cell Signaling Technology, 9272 and 4058). Primary antibodies were detected using horseradish peroxidase-labeled secondary antibodies (Bio-Rad, NXA931 and NA934) and ECL prime chemiluminescence (Thermo Fisher, 45002401). X-ray film images were scanned and densitometry was performed using NIH ImageJ software.
Cell surface biotinylation
Cells grown to confluency in 35-mm, poly-D-lysine-coated tissue culture dishes were placed on ice and washed with phosphate-buffered saline (PBS; Cellgro, 21031-CV). Cell surface proteins were labeled with biotin using 0.5 mg/ml of EZ-link sulpho-NHS-S-S-biotin (Pierce Thermo Scientific, 21331) in PBS on ice for 25 min. Excess unreacted biotin was quenched with 50 mM ammonium chloride (Sigma, A9434) followed by 3 washes with PBS. To assess total surface APP, labeled cells were left on ice. To analyze the amount of internalized APP, biotin-labeled cells were incubated at 37°C for the desired time and then returned to ice. The biotin label remaining on the surface after internalization was removed by incubating cells with cold 60 mM glutathione-PBS buffer (glutathione: Sigma, G4251; PBS: Cellgro, 21031-CV; 1% bovine serum albumin: Fisher Scientific, BP1600-100; 0.83 M NaCl, 0.83 M NaOH) followed by 3 washes with PBS. Untreated cells (no biotin control), and biotin-labeled cells left on ice and treated with glutathione buffer (stripping control) were used to estimate the efficiency of addition and removal of biotin, respectively. Cells were lysed with RIPA lysis buffer with inhibitors as outlined above. Biotinylated proteins were recovered by incubating cell lysates with streptavidin agarose beads (EMD Millipore, 16–126) at 4°C for 2–3 h. The beads were washed with buffer (50 mM Tris-HCl, pH 7.6, 100 mM NaCl, 2 mM EDTA, 1% Triton X-100 and 0.1% SDS) and finally with Tris-buffered saline (20 mM Tris-HCl, pH 7.6, 150 mM NaCl). The precipitated proteins were eluted with 2X SDS sample buffer, boiled and analyzed by western blotting with anti-APP antibody (22C11) to detect total surface APP (input) and internalized APP as well as anti-FLAG or anti-BECN1 antibody to detect BECN1 associated with surface proteins.
Immunoprecipitations
For immunoprecipitation experiments, transfected cells in 10-cm tissue culture dishes or cortical neurons at 16–22 days in vitro were washed with PBS (Sigma, D8537) and lysed in plate with 400–500 μl of cold Triton X-100 or RIPA buffer, as indicated. Lysates were transferred to 1.5 ml tubes, rotated for 20 min, and clarified by centrifugation at 16,200 x g for 30 min. The supernatants (500 μg–2 mg total protein) were incubated with appropriate primary antibodies overnight at 4°C. Twenty μl of protein A (EMD Millipore, 16–156) or protein G agarose beads (EMD Millipore, 16–266) were added and the tubes rotated at 4°C for 2 h. The beads were washed sequentially with buffers containing 0.5 M sodium chloride+1% Triton X-100, 0.1% SDS+1% Triton X-100, 1% Triton X-100 and TBS. Samples were resuspended in 2x Laemmli sample buffer (Bio-Rad Laboratories, 161–0737) containing 2-mercaptoethanol (Sigma, M3148), boiled for 5 min at 95°C and centrifuged briefly. Supernatants were resolved on 4–12% TGX gels (Bio-Rad Laboratories, 456–1084), transferred to PVDF membrane (EMD Millipore, IPVH00010) and probed with appropriate primary antibodies.
To detect interaction between BECN1 and surface APP, HEK293 cells transfected with APP695 and FLAG-BECN1 were incubated with 0.5 mg/ml EZ-link sulpho NHS-S-S-biotin to label surface proteins or left untreated. Cells were lysed in RIPA buffer. Lysates were diluted 8- to 10-fold with 1% Triton X-100 lysis buffer and precleared with 20 μl of protein A/G agarose beads prior to immunoprecipitation with FLAG or APP antibody and protein G beads (Protein A beads: Millipore 16156; Protein G beads: Millipore, 16266). The beads were washed sequentially with 1% Triton X-100 buffer containing 0.5 M NaCl, 1% Triton X-100 buffer with 0.1% SDS, 1% Triton X-100 buffer and TBS. The immunoprecipitates were resuspended in Tris-SDS buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1% SDS, containing protease inhibitors), boiled at 95°C for 5 min, and chilled on ice, followed by centrifugation at 16,200 x g for 5 min. Twenty percent of the eluate (total IP) was mixed with an equal volume of 2X SDS sample buffer. Eighty percent of the eluate was diluted with Triton X-100 buffer and incubated with streptavidin agarose beads to affinity isolate the biotin-labeled proteins present in the FLAG or APP immunoprecipitates (surface IP). The beads were washed with Triton X-100 buffer containing 0.1% SDS and the bound proteins eluted with SDS sample buffer. The eluates (from total and streptavidin affinity isolation) were analyzed by immunoblotting as described above.
Live cell labeling of surface APP, internalization and sorting
Cells expressing N-terminal FLAG tagged-APP or untagged APP and relevant BECN1, ATG16L1, EEA1, RAB7, LAMP1 or GFP-LC3 plasmids were cultured in 8-well chambered coverglass (Nalge Nunc International, 155409) at a density of 5 × 104 cells/well), as indicated. Cells were washed with PBS, incubated on ice for 1 h with FLAG-M2 (Sigma, F1804) or APP (6E10) antibodies (BioLegend, 803001) at 1:500 dilution and washed several times with PBS to remove unbound antibody. To facilitate APP internalization and sorting to endolysosomal compartments, cells were incubated at 37°C in culture medium for 5 and 20 min, respectively. To remove the FLAG or APP antibody bound to APP remaining on the cell surface post internalization, cells were washed with ice-cold buffer containing 0.5 M CH3COOH (Fisher Scientific, BP2401) and 0.5 M NaCl (acid stripping). Unlabeled cells and cells labeled with FLAG antibody with or without acid stripping, were fixed with 4% paraformaldehyde (Thermo Scientific, 28908) in PBS for 20 min at room temperature. Antibody-labeled APP in untreated, fixed, nonpermeabilized cells (total surface APP) and in permeabilized cells after acid stripping (internalized APP) was detected by immunofluorescence microscopy using anti-mouse Alexa Flour 594 secondary antibody (ThermoScientific, A21203). The following control experiments were performed: 1) To estimate the total amount of FLAG-labeled APP on the cell surface, cells were fixed for immunofluorescence immediately following labeling and washing. 2) To ensure complete stripping of surface-bound FLAG antibody and to avoid possible contamination of the internal signal, labeled cells were immediately subject to acid stripping and fixed prior to analysis. 3) As a negative control, cells were incubated in PBS without FLAG or APP antibody. In select experiments, the vacuolar-type H+-ATPase inhibitor bafilomycin A1 (Baf A1, 50 nM; Sigma, B1793) was employed to inhibit endolysosomal acidification. Control cells were treated with equal volumes of dimethyl sulfoxide vehicle (Sigma, D8418).
For additional analysis of the effects of BECN1 on surface APP levels, we performed ELISA-based measurements. The cell surface level of APP was measured in HeLa cells transfected with N-terminal FLAG tagged-APP and relevant BECN1 plasmids (vector, WT BECN1, shCTRL and shBECN1). Forty-eight h post transfection, cells were placed on ice and labeled with FLAG antibody (live labeling) and fixed. Alternatively, cells were fixed and then labeled with FLAG antibody. Antibody labeled FLAG-APP was detected by horseradish peroxidase -conjugated anti-mouse secondary antibody using O-phenylenediamine (Sigma, P9029) as a substrate. The colorimetric reaction was allowed to proceed for 45 min. Absorbance readings (OD 450 nm) were recorded every 15 min with an Epoch plate reader (BioTek, Winooski, VT, USA). Color development was linear at all time points tested. The experiment was done in triplicate. The absorbance readings at 45 min were used for quantification. The following controls were used for background subtraction: cells transfected with vector control and labeled with FLAG, FLAG-APP-expressing cells incubated with only primary or secondary antibody, or labeled with FLAG and acid stripped.
Twelve-bit images were captured with a Leica DMI6000B epifluorescence microscope (JH Technologies, Fremont, CA, USA) under a 40X or 63X objective using LAS-AF software (Leica). Exposure times and image acquisition parameters were equal between samples within an experiment. The amount of internalized APP was quantified using ImageJ software in original images and normalized to the amount of total APP surface label at time 0 min. For colocalization analysis, Z-stack images were first deconvolved using the 3D-deconvolution module in the LAS-AF software. After setting thresholds and background, the colocalization rate, area of colocalization and Pearson's correlation measurements were made in a defined area of interest using the colocalization tool of the LAS-AF software. All quantifications were performed on original deconvolved images. For presentation, images were transferred to Adobe Photoshop (version CS5) for resizing, brightness/contrast and labeling. Image scaling was identical for all images and treatment groups within an experiment.
Surface APP degradation and processing
To assess surface APP degradation and processing, cells expressing APP695 and FLAG-tagged BECN1 were subject to cell-surface biotinylation and incubated in DMEM for different time periods. Streptavidin affinity isolation of biotinylated proteins from cellular extracts and culture supernatant followed by immunoblotting with APP (22C11) antibody was performed to determine the level of full-length cellular APP (FL-APP) and soluble APP (sAPP), respectively. The level of biotin-labeled surface APP at time point 0 h was considered 100%. To evaluate the involvement of the lysosomal pathway in surface APP degradation, cells were treated with the lysosomal inhibitor 25 mM ammonium chloride (Sigma, A9434), for the duration of the biotin label chase. Densitometric analysis of immunoblots was done using ImageJ software. The amount of cellular and soluble labeled APP recovered at different times of the chase was expressed relative to the amount of labeled cellular APP at time 0 h, which was considered 100%. All values were normalized to GAPDH to ensure equal input for the streptavidin affinity isolation.
Statistical analysis
Data in graphs are expressed as mean +/− standard error (SEM). Statistical analyses were performed using Graphpad Prism or the SYSTAT 13 statistical software suite (Systat Software Inc., San Jose, CA, USA). Statistical significance was determined using the Student unpaired t test when comparing 2 experimental groups. One-way or 2-way ANOVAs and multiple comparison post-tests (Tukey's honestly significant difference test or Bonferroni test) were used when comparing multiple groups.
Supplementary Material
Abbreviations
- Aβ
amyloid-β
- AD
Alzheimer disease
- AKT/PKB
AKT serine/threonine kinase
- APP
amyloid β precursor protein
- ATG14
autophagy-related 14
- ATG16L1
autophagy-related 16 like 1
- BECN1
Beclin 1
- co-IP
co-immunoprecipitation
- ECD
evolutionarily conserved domain
- FL-APP
full-length APP
- GFP
green fluorescent protein
- IF
immunofluorescence
- IP
immunoprecipitation
- LC3
microtubule-associated protein 1 light chain 3
- Myr
myristoylated
- PIK3C3/VPS34
phosphatidylinositol 3-kinase, catalytic subunit type 3
- PM
plasma membrane
- PtdIns3K
class III phosphatidylinositol 3-kinase complex
- P2
synaptosomal fraction
- sAPP
soluble APP ectodomains
- SQSTM1/p62
sequestosome 1
- UVRAG
UV radiation resistance associated
- WB
western blot
- WCE
whole cell extract
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors especially thank Dr. Richard Wang for insightful discussion of the data and Drs. Beth Levine, Tamotsu Yoshimori and Richard Wang for aforementioned plasmids.
Funding
This work was supported by a K08 grant from the National Institute of Neurologic Disorders and Stroke (NS085324; PI: EDP) and a New Investigator in Alzheimer Disease Award (RAG12401; PI: EDP) from the American Federation for Aging Research.
References
- [1].McKnight NC, Zhong Y, Wold MS, Gong S, Phillips GR, Dou Z, Zhao Y, Heintz N, Zong WX, Yue Z. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS genetics 2014; 10:e1004626; PMID:25275521; http://dx.doi.org/ 10.1371/journal.pgen.1004626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ruck A, Attonito J, Garces KT, Nunez L, Palmisano NJ, Rubel Z, Bai Z, Nguyen KC, Sun L, Grant BD, et al.. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy 2011; 7:386-400; PMID:21183797; http://dx.doi.org/ 10.4161/auto.7.4.14391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005; 1:46-52; PMID:16874027; http://dx.doi.org/ 10.4161/auto.1.1.1542 [DOI] [PubMed] [Google Scholar]
- [4].Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/ 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- [5].Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360-72; PMID:18843052; http://dx.doi.org/ 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li X, He L, Che KH, Funderburk SF, Pan L, Pan N, Zhang M, Yue Z, Zhao Y. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun 2012; 3:662; PMID:22314358; http://dx.doi.org/ 10.1038/ncomms1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010; 190:511-21; PMID:20713597; http://dx.doi.org/ 10.1083/jcb.200911141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al.. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Natre Cell Biol 2009; 11:385-96; http://dx.doi.org/ 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- [9].Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468-76; PMID:19270693; http://dx.doi.org/ 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Nail Acad Sci U S A 2008; 105:19211-6; http://dx.doi.org/ 10.1073/pnas.0810452105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thoresen SB, Pedersen NM, Liestol K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res 2010; 316:3368-78; PMID:20643123; http://dx.doi.org/ 10.1016/j.yexcr.2010.07.008 [DOI] [PubMed] [Google Scholar]
- [12].Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 2007; 179:485-500; PMID:17984323; http://dx.doi.org/ 10.1083/jcb.200702115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al.. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269-84; PMID:24034250; http://dx.doi.org/ 10.1016/j.cell.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tan X, Thapa N, Sun Y, Anderson RA. A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015; 160:145-60; PMID:25594178; http://dx.doi.org/ 10.1016/j.cell.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al.. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Different 2009; 16:1006-17; http://dx.doi.org/ 10.1038/cdd.2009.34 [DOI] [PubMed] [Google Scholar]
- [16].Li Y, Zhao Y, Hu J, Xiao J, Qu L, Wang Z, Ma D, Chen Y. A novel ER-localized transmembrane protein, EMC6, interacts with RAB5A and regulates cell autophagy. Autophagy 2013; 9:150-63; PMID:23182941; http://dx.doi.org/ 10.4161/auto.22742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Molejon MI, Ropolo A, Re AL, Boggio V, Vaccaro MI. The VMP1-Beclin 1 interaction regulates autophagy induction. Scient Rep 2013; 3:1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bejarano E, Yuste A, Patel B, Stout RF Jr., Spray DC, Cuervo AM. Connexins modulate autophagosome biogenesis. Nat Cell Biol 2014; 16:401-14; PMID:24705551; http://dx.doi.org/ 10.1038/ncb2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Han J, Hou W, Lu C, Goldstein LA, Stolz DB, Watkins SC, Rabinowich H. Interaction between Her2 and Beclin-1 proteins underlies a new mechanism of reciprocal regulation. J Biol Chem 2013; 288:20315-25; PMID:23703612; http://dx.doi.org/ 10.1074/jbc.M113.461350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Muller UC, Zheng H. Physiological functions of APP family proteins. Cold Spring Harbor Perspect Med 2012; 2:a006288; http://dx.doi.org/ 10.1101/cshperspect.a006288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Caglayan S, Takagi-Niidome S, Liao F, Carlo AS, Schmidt V, Burgert T, Kitago Y, Füchtbauer EM, Füchtbauer A, Holtzman DM, et al.. Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer's disease mutation. Scie Translat Med 2014; 6:223ra20; PMID:24523320 [DOI] [PubMed] [Google Scholar]
- [22].Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al.. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 2005; 102:13461-6; PMID:16174740; http://dx.doi.org/ 10.1073/pnas.0503689102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, Militz D, Schmidt V, Madsen P, Nyengaard JR, et al.. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci 2012; 32:1467-80; PMID:22279231; http://dx.doi.org/ 10.1523/JNEUROSCI.2272-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chaufty J, Sullivan SE, Ho A. Intracellular amyloid precursor protein sorting and amyloid-beta secretion are regulated by Src-mediated phosphorylation of Mint2. J Neurosci 2012; 32:9613-25; PMID:22787047; http://dx.doi.org/ 10.1523/JNEUROSCI.0602-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rogelj B, Mitchell JC, Miller CC, McLoughlin DM. The X11/Mint family of adaptor proteins. Brain Res Rev 2006; 52:305-15; PMID:16764936; http://dx.doi.org/ 10.1016/j.brainresrev.2006.04.005 [DOI] [PubMed] [Google Scholar]
- [26].Pietrzik CU, Yoon IS, Jaeger S, Busse T, Weggen S, Koo EH. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J Neurosci 2004; 24:4259-65; PMID:15115822; http://dx.doi.org/ 10.1523/JNEUROSCI.5451-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, Small SA. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol 2014; 10:443-9; PMID:24747528; http://dx.doi.org/ 10.1038/nchembio.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T. Regulation of amyloid precursor protein processing by the beclin 1 complex. PloS one 2010; 5:e11102; PMID:20559548; http://dx.doi.org/ 10.1371/journal.pone.0011102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al.. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118:2190-9; PMID:18497889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, et al.. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013; 494:201-6; PMID:23364696; http://dx.doi.org/ 10.1038/nature11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. AKT-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/ 10.1126/science.1225967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/AKT pathway. Proc Natl Acad Sci U S A 1999; 96:2110-5; PMID:10051603; http://dx.doi.org/ 10.1073/pnas.96.5.2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nah J, Pyo JO, Jung S, Yoo SM, Kam TI, Chang J, Han J, Soo A An S, Onodera T, Jung YK. BECN1/Beclin 1 is recruited into lipid rafts by prion to activate autophagy in response to amyloid beta 42. Autophagy 2013; 9:2009-21; PMID:24145555; http://dx.doi.org/ 10.4161/auto.26118 [DOI] [PubMed] [Google Scholar]
- [34].Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mulé JJ, et al.. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:1142-51; PMID:17891140; http://dx.doi.org/ 10.1038/ncb1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol 2013; 33:3675-88; PMID:23878393; http://dx.doi.org/ 10.1128/MCB.00079-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M, Liu M, Tian Y, Lu P, Wang FL, et al.. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res 2012; 22:473-89; PMID:22310240; http://dx.doi.org/ 10.1038/cr.2012.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell 2012; 23:2156-69; PMID:22496425; http://dx.doi.org/ 10.1091/mbc.E11-10-0844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].He C, Wei Y, Sun K, Li B, Dong X, Zou Z, Liu Y, Kinch LN, Khan S, Sinha S, et al.. Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell 2013; 154:1085-99; PMID:23954414; http://dx.doi.org/ 10.1016/j.cell.2013.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harbor Perspect Med 2012; 2:a006361; http://dx.doi.org/ 10.1101/cshperspect.a006361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer's disease brain. Neurobiol Dis 2011; 43:68-78; PMID:21081164; http://dx.doi.org/ 10.1016/j.nbd.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall'Armi C, Simoes S, Point Du Jour KS, McCabe BD, et al.. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 2013; 4:2250; PMID:23907271; http://dx.doi.org/ 10.1038/ncomms3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, et al.. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 2013; 79:873-86; PMID:24012002; http://dx.doi.org/ 10.1016/j.neuron.2013.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhu W, Swaminathan G, Plowey ED. GA binding protein augments autophagy via transcriptional activation of BECN1-PIK3C3 complex genes. Autophagy 2014; 10:1622-36; PMID:25046113; http://dx.doi.org/ 10.4161/auto.29454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rickle A, Bogdanovic N, Volkman I, Winblad B, Ravid R, Cowburn RF. AKT activity in Alzheimer's disease and other neurodegenerative disorders. Neuroreport 2004; 15:955-9; PMID:15076714; http://dx.doi.org/ 10.1097/00001756-200404290-00005 [DOI] [PubMed] [Google Scholar]
- [45].Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C. Activation of AKT/PKB, increased phosphorylation of AKT substrates and loss and altered distribution of AKT and PTEN are features of Alzheimer's disease pathology. J Neurochem 2005; 93:105-17; PMID:15773910; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02949.x [DOI] [PubMed] [Google Scholar]
- [46].Shineman DW, Dain AS, Kim ML, Lee VM. Constitutively active AKT inhibits trafficking of amyloid precursor protein and amyloid precursor protein metabolites through feedback inhibition of phosphoinositide 3-kinase. Biochemistry 2009; 48:3787-94; PMID:19236051; http://dx.doi.org/ 10.1021/bi802070j [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747-57; PMID:20639872; http://dx.doi.org/ 10.1038/ncb2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci 2007; 27:14459-69; PMID:18160654; http://dx.doi.org/ 10.1523/JNEUROSCI.4701-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Falcon-Perez JM, Nazarian R, Sabatti C, Dell'Angelica EC. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci 2005; 118:5243-55; PMID:16249233; http://dx.doi.org/ 10.1242/jcs.02633 [DOI] [PubMed] [Google Scholar]
- [50].Lawe DC, Patki V, Heller-Harrison R, Lambright D, Corvera S. The FYVE domain of early endosome antigen 1 is required for both phosphatidylinositol 3-phosphate and Rab5 binding. Critical role of this dual interaction for endosomal localization. J Biol Chem 2000; 275:3699-705; PMID:10652369; http://dx.doi.org/ 10.1074/jbc.275.5.3699 [DOI] [PubMed] [Google Scholar]
- [51].Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest 2002; 109:1541-50; PMID:12070301; http://dx.doi.org/ 10.1172/JCI0215420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Bio Cell 2008; 19:2092-100; http://dx.doi.org/ 10.1091/mbc.E07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol 2001; 155:19-25; PMID:11581283; http://dx.doi.org/ 10.1083/jcb.200107069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.