

ABSTRACT
Defective autophagy contributes to Alzheimer disease (AD) pathogenesis although evidence is conflicting on whether multiple stages are impaired. Here, for the first time, we have comprehensively evaluated the entire autophagic process specifically in CA1 pyramidal neurons of hippocampus from early and late-stage AD subjects and nondemented controls. CA1 neurons aspirated by laser capture microdissection were analyzed using a custom-designed microarray comprising 578 neuropathology- and neuroscience-associated genes. Striking upregulation of autophagy-related genes, exceeding that of other gene ontology groups, reflected increases in autophagosome formation and lysosomal biogenesis beginning at early AD stages. Upregulated autophagosome formation was further indicated by elevated gene and protein expression levels for autophagosome components and increased LC3-positive puncta. Increased lysosomal biogenesis was evidenced by activation of MiTF/TFE family transcriptional regulators, particularly TFE3 (transcription factor binding to IGHM enhancer 3) and by elevated expression of their target genes and encoded proteins. Notably, TFEB (transcription factor EB) activation was associated more strongly with glia than neurons. These findings establish that autophagic sequestration is both competent and upregulated in AD. Autophagosome-lysosome fusion is not evidently altered. Despite this early disease response, however, autophagy flux is progressively impeded due to deficient substrate clearance, as reflected by autolysosomal accumulation of LC3-II and SQSTM1/p62 and expansion of autolysosomal size and total area. We propose that sustained induction of autophagy in the face of progressively declining lysosomal clearance of substrates explains the uncommonly robust autophagic pathology and neuritic dystrophy implicated in AD pathogenesis.
KEYWORDS: Alzheimer disease, autophagosomes, autophagy, CA1 pyramidal neurons, dystrophic neurites, hippocampus, lysosomes, MTOR, TFE3, TFEB
Introduction
As neurons age, their dependence on mechanisms of protein clearance for survival increases even as the competence of these proteolytic systems progressively declines. In adult onset neurodegenerative diseases such as Alzheimer disease (AD), amyotrophic lateral sclerosis, and Parkinson disease, the responsible pathogenic protein accumulates only late in life, reflecting an emergence of aging- and disease-related deficits in eliminating damaged proteins.1-3 Autophagy, the principal lysosomal pathway for turning over intracellular constituents, is closely linked to the mechanisms underlying cellular aging and longevity4-6 and is upregulated during cellular stress.7 Several subtypes of autophagy are known, which are distinguished by how substrates are delivered to lysosomes.8 The major one of these subtypes, macroautophagy, hereafter referred to as autophagy, supports both the constitutive and selective turnover of a broad range of cell constituents and is also the only mechanism available to neurons for eliminating damaged organelles and large protein aggregates.9 Acting in concert with the BECN1-PIK3C3/VPS34 complex and several ubiquitin-ligase complexes,10,11 ULK1 initiates the formation of an elongating double-membrane structure that surrounds the substrate(s) and closes to form a vacuole, termed the autophagosome.12 Sequestration of substrates within an autophagosome involves their engagement with the phagophore membrane via LC3 proteins and other autophagy receptors such as SQSTM1/p62,13-15 which facilitate selective cargo recruitment and are themselves ultimately degraded by autophagy.
Autophagosome clearance is initiated upon fusion with lysosomes and/or late endosomes, which introduce into the resulting autolysosome the dozens of lysosomal acidic hydrolases and the acidification machinery necessary for enzyme activation and substrate digestion.1 The release of amino acids from autolysosomes and other factors maintain MTOR in its active state to inhibit autophagy induction in a feedback manner.16-18 Under conditions of nutrient deprivation or cellular/lysosomal stress, however, MTOR may become inhibited, leading to nuclear translocation of MiTF/TFE family proteins that positively regulate genes controlling lysosomal biogenesis and some aspects of autophagosome formation.19 These include members of the MiTF/TFE transcription factor family such as TFE3 (transcription factor binding to IGHM enhancer 3) and TFEB (transcription factor EB), which bind to a specific E-box (the “CLEAR sequence”) on the promoter of a large group of autophagic-lysosomal genes to promote their expression.20-22 Additional transcription factors from the FOXO (forkhead box) family, including FOXO1 and FOXO3, positively regulate smaller subsets of genes supporting autophagy in response to stress-responsive protein kinases such as AKT.23
Autophagy flux is markedly impaired in AD and AD models although information on the stage(s) of autophagy impaired and the molecular basis for deficits, especially in the authentic disease in humans, is incomplete and conflicting.24,25 The widespread appearance of dystrophic neurites filled mainly with autophagic vacuoles containing incompletely digested cellular constituents is a hallmark neuropathological feature of AD,26-28 along with the classical intraneuronal aggregates of MAPT/tau29 and extracellular deposits of β-amyloid.30 The storage of undigested substrates in AD brain, which is more extensive than in other adult-onset neurodegenerative disorders, reflects a major defect in lysosomal clearance mechanisms.26 That these lysosomal system deficits are key pathogenic factors in AD is strongly suggested by growing evidence that selectively restoring lysosomal proteolytic function in mouse AD models ameliorates diverse pathological, synaptic, and cognitive deficits.31,32 Although lysosomal function is evidently deficient in AD models,25,33 reports of additional alterations of earlier steps of autophagy (e.g. autophagy induction, autophagosome formation) have been conflicting,34-37 reflecting, in many cases, limited analysis of human samples and inferences made from a single or relatively few indices of autophagic function as well as the use of brain tissue composed of diverse cell types varying in their responses to disease.38 Here, we assessed all of the major steps of the autophagy pathway to evaluate the initiation as well as subsequent autophagy flux and the implications for the development of AD pathology.
In the present study, we used a unique approach involving the evaluation of autophagy alterations specifically within the vulnerable CA1 hippocampal neuronal population of AD brains by determining the expression profile of 44 selected autophagy-related genes in relation to 534 other degenerative disease-related genes comprising a custom-designed microarray platform.38-40 Expression changes in autophagy-related genes were interpreted in relation to morphological alterations of autophagy-related autophagic organelles in CA1 neurons combined with analyses of mRNA and levels, subcellular distributions, and activities of autophagy-related proteins in the hippocampus, which collectively reflect the functional activity of autophagy induction and autophagosome formation as well as the biogenesis and degradative capability of lysosomes. We also analyzed the changes in neuron-specific expression of major transcriptional regulators of autophagy and lysosomal biogenesis in AD brain. Our analysis reveals that autophagy is upregulated early and progressively in AD, despite an evolving impairment in the ability to clear autophagic substrates. This combination of pathological events provides a plausible basis for the well-described massive accumulations of substrate-laden autolysosomes within dystrophic neurites that are a hallmark of AD neuropathology.41
Results
Autophagosome formation is upregulated in CA1 neurons in AD brain
To obviate limitations of autophagy analyses at the regional brain tissue level, which includes the differential expression patterns of heterogeneous cell types, we re-analyzed a previously published microarray made on single case populations of CA1 pyramidal neurons microaspirated by laser capture microdissection (LCM), focusing on the mRNA expression of 44 autophagy-related genes (out of 578 total genes on the custom-designed array platform).40 The 44 genes selected as autophagy-related were classified by curating gene ontology databases using the most recent biological evidence supporting a relevant function in autophagy (Fig. 1A; see Materials and Methods). Considering Br. III/IV (n = 14) and V/VI (n = 15) together (Table 1), we observed that a disproportionately high percentage of the genes related to autophagy (34%, 15/44) were significantly upregulated relative to controls (n = 10) whereas only 2% of this group were downregulated (1/44) (Fig. 1A, B). By comparison, considering all 578 genes on the microarray, only 6% (38/578) were significantly upregulated while a relatively high percentage (15%, 85/578) were downregulated (Fig. 1C). Analysis of the Gene Ontology (GO) terms for highly expressed gene groups confirmed that “Autophagy” was the most highly represented biological process in our gene array (Fig. 1D). Notably, the majority of the upregulated autophagy genes were significantly altered in both Br. III/IV and V/VI cases (Fig. 1A), suggesting that autophagy responses in these neurons arise early and are then maintained during the progression of the disease. Upregulated autophagy genes included those involved in induction signaling (AKT1 and MAPK3), autophagosome formation (ATG3, ATG5, ATG12, and ATG8 orthologs GABARAP and MAP1LC3A), autophagosome-lysosome trafficking/fusion (RAB7A42 and RAB24), lysosomal biogenesis/proteolysis (CLCN7, LAMP1 and CTSD) and other RAB proteins involved in autophagy (RAB4A43 and RAB5A38,44). We performed additional qPCR measurements in the CA1 sector of the hippocampus on an independent cohort of controls, Br. III and Br. V cases shown in Table 3 (n = 9, 10, 10 respectively) that validated these microarray findings. As shown in Fig. 1E, 7 of 8 genes that were measured by qPCR analyses (the first 8 genes represented) for comparison to microarray analysis match in terms of the direction of change or absence of change, although qPCR analyses reflect expression from a mixed cell population in hippocampal tissue. The strong upregulation in AD brain of a particular set of ATG genes from our array (ATG3, ATG5, and ATG12) and 2 additional genes, ULK1 and PIK3C3/VPS34 measured by qPCR suggested the possible activation of FOXO transcription factors, which regulate a select group of autophagic genes,45 including the 5 from our study. Consistent with this possibility, FOXO1 was significantly upregulated by qPCR assessment (p ≤ 0.005) whereas FOXO3 expression was unchanged (Fig. 1E).
Figure 1.
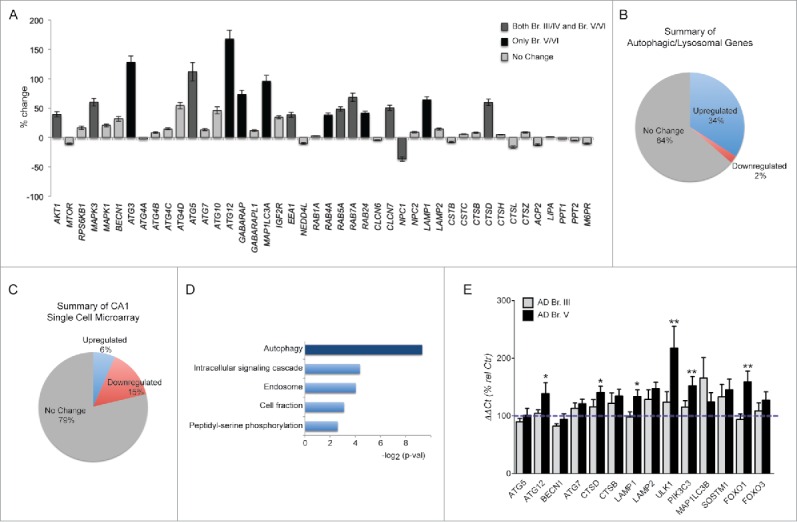
Changes in expression of autophagy-related genes in hippocampal CA1 pyramidal neurons. (A) Autophagy-related genes showing differential regulation in CA1 neurons via custom-designed microarray analysis of 578 genes. Significantly (p ≤ 0.05) altered in both Br. III/IV and Br. V/VI shown in dark gray; others significantly altered in Br. V/VI only shown in black, no change in gray. (B) Pie chart indicating proportions of significantly upregulated and downregulated autophagy genes on the array platform. (C) Pie chart illustrating a relative paucity of genes upregulated in all 578 transcripts contrasting with the higher proportion that are downregulated. (D) GO analysis of all highly expressed genes in Br. III/IV and Br. V/VI when compared to control group. Autophagy, the most represented term, is highlighted in darker blue. p value cut off 0.05. (E) qPCR validation of selected autophagy/lysosomal genes assayed in the CA1 sector of the hippocampus. Statistical significance is denoted by asterisks with p-values shown determined by one-way ANOVA or for 2 column analysis by Student t test. *p ≤ 0.05; **p ≤ 0.01.
Table 1.
Demographics of brain samples utilized in tissue microarray analysis from the Rush University Religious Orders Study (RROS) and the University of Pennsylvania Center for Neurodegenerative Disease Research (CNDR).
| Sample number | Braak stage | Brain bank | PMI (h) | Age at death | ApoE | Race/Sex |
|---|---|---|---|---|---|---|
| Ctr. 1 | I | RROS | 2.3 | 85.2 | E3/3 | WF |
| Ctr. 2 | II | RROS | 5.2 | 76.8 | E3/3 | WM |
| Ctr. 3 | II | RROS | 7.5 | 72.5 | E3/4 | WF |
| Ctr. 4 | I | RROS | 8.5 | 81.9 | E3/3 | WF |
| Ctr. 5 | I | RROS | 12.5 | 81.2 | E3/3 | WM |
| Ctr. 6 | I | RROS | 5 | 67.4 | E2/3 | WM |
| Ctr. 7 | ND | RROS | 4 | 87 | ND | ND |
| Ctr. 8 | I | CNDR | 8 | 81 | E3/3 | BF |
| Ctr. 9 | 0 | CNDR | 5.5 | 67 | E3/3 | WF |
| Ctr. 10 | I | CNDR | 15 | 64 | ND | ND |
| Avg/Tot | 7.4 + 3.9 | 76.4 + 8.2 | 5F/3M/2ND | |||
| Br. III/IV 1 | III | RROS | 11 | 92.8 | E3/3 | WM |
| Br. III/ IV 2 | III | RROS | 4 | 89.6 | E2/3 | WM |
| Br. III/ IV 3 | III | RROS | 3.6 | 88.7 | E3/3 | WF |
| Br. III/ IV 4 | III | RROS | 2.8 | 92.3 | E3/3 | WF |
| Br. III/ IV 5 | IV | RROS | 3 | 97.5 | E2,3 | WF |
| Br. III/IV 6 | IV | RROS | 13.4 | 89.2 | E3/4 | WM |
| Br. III/IV 7 | IV | RROS | 3.6 | 82.5 | E3/3 | WF |
| Br. III/IV 8 | IV | RROS | 4.8 | 80.5 | E3/3 | WF |
| Br. III/IV 9 | III | RROS | 4.3 | 82.1 | E3/3 | WM |
| Br. III/IV 10 | IV | RROS | 8 | 81 | E4/4 | WF |
| Br. III/IV 11 | IV | RROS | 13 | 81.5 | E3/3 | WF |
| Br. III/IV 12 | IV | RROS | 16 | 84.1 | E3/3 | WM |
| Br. III/IV 13 | IV | RROS | 6 | 83.5 | E3/4 | WF |
| Br. III/IV 14 | III | CNDR | 7 | 89 | E3/3 | BF |
| Avg/Tot | 7.2 + 4.4 | 86.7 + 5.3 | 9F/5M | |||
| Br. V/VI 1 | V | RROS | 6 | 87 | ND | M |
| Br. V/VI 2 | V | RROS | 5 | 80 | ND | M |
| Br. V/VI 3 | V | RROS | 8.5 | 81 | ND | F |
| Br. V/VI 4 | V | RROS | 10.7 | 84.2 | E3/3 | WF |
| Br. V/VI 5 | V | RROS | 3.5 | 84.8 | E3/4 | WM |
| Br. V/VI 6 | V | RROS | 2.2 | 82.7 | E4/4 | WM |
| Br. V/VI 7 | V | RROS | 4.5 | 80.1 | E3/4 | WM |
| Br. V/VI 8 | V | RROS | 3.1 | 92.8 | E3/3 | WF |
| Br. V/VI 9 | V | RROS | 4 | 94.1 | E2/4 | WF |
| Br. V/VI 10 | VI | CNDR | 6 | 69 | ND | WF |
| Br. V/VI 11 | VI | CNDR | 17 | 75 | ND | WM |
| Br, V/VI 12 | VI | CNDR | 8 | 62 | E2/3 | WM |
| Br. V/VI 13 | IV | CNDR | 12 | 91 | ND | WF |
| Br. V/VI 14 | VI | CNDR | 7 | 86 | ND | BF |
| Br. V/VI 15 | VI | CNDR | 7 | 80 | ND | WF |
| Avg/Tot | 7.0 + 3.9 | 82.0 + 8.6 | 8F/7M |
Abbreviations: W, white; B, black, ND, not divulged; PMI, Postmortem interval.
Table 3.
Demographics of Harvard Brain Tissue Resource Center (HBTRC) brain samples used in biochemical/molecular analyses.
| Sample | Case number | Braak Score | Age | Gender | PMI (h) | RIN |
|---|---|---|---|---|---|---|
| Control 1 | AN01404 | I | 74 | M | 18.3 | 6.2 |
| Control 2 | AN06429 | II | 77 | F | 23.3 | 5.1 |
| Control 3 | AN15823 | I | 76 | M | 24.2 | 4.4 |
| Control 4 | AN01717 | II | 79 | F | 20.6 | 4.2 |
| Control 5 | AN10180 | II | 73 | M | 24.0 | 5.6 |
| Control 6 | AN01234 | II | 85 | M | 20.8 | 5.4 |
| Control 7 | AN06221 | II | 82 | M | 24.1 | 5.3 |
| Control 8 | AN13219 | II | 93 | F | 12.3 | 6.7 |
| Control 9 | AN10936 | II | 86 | F | 23.8 | 6.5 |
| Avg/Tot | 80.6 + 6.5 | 5M/4F | 21.3 + 3.9 | |||
| Br. III 1 | AN18315 | III | 81 | M | 23.8 | 6.0 |
| Br. III 2 | AN01765 | III | 88 | F | 14.9 | 4.2 |
| Br. III 3 | AN04145 | III | 94 | M | 15.6 | 6.2 |
| Br. III 4 | AN01725 | III | 84 | F | 13.2 | 5.1 |
| Br. III 5 | AN05918 | III | 85 | M | 6.1 | 5.0 |
| Br. III 6 | AN05919 | III | 85 | M | 6.1 | 5.0 |
| Br. III 7 | AN08520 | III | 89 | F | 17.8 | 4.5 |
| Br. III 8 | AN15998 | III | 82 | F | 18.8 | 6.7 |
| Br. III 9 | AN18315 | III | 82 | F | 17.4 | 5.1 |
| Br. III 10 | AN19132 | III | 84 | F | 8.6 | 4.4 |
| Avg/Tot | 85.4 + 3.9 | 4M/6F | 14.2 + 5.8 | |||
| Br. V 1 | AN06801 | V | 76 | M | 23.7 | 5.3 |
| Br. V 2 | AN00391 | V | 89 | F | 15.0 | 2.5 |
| Br. V 3 | AN03479 | V | 83 | M | 26.6 | 5.3 |
| Br. V 4 | AN18658 | V | 80 | M | 20.6 | 3.8 |
| Br. V 5 | AN18751 | V | 76 | M | 12.5 | 6.9 |
| Br. V 6 | AN08533 | V | 84 | M | 12.6 | 5.2 |
| Br. V 7 | AN11070 | V | 86 | F | 15.3 | 6.3 |
| Br. V 8 | AN14636 | V | 89 | F | 15.8 | 5.4 |
| Br. V 9 | AN15437 | V | 90 | F | 20.5 | 5.2 |
| Br. V 10 | AN12565 | V | 76 | F | 21.4 | 5.9 |
| Avg/Tot | 83.2 + 5.6 | 5M/5F | 18.4 + 4.8 |
Abbreviations: RIN, RNA Integrity Number; PMI, Postmortem interval.
Additional support for the upregulation of autophagy in AD brain was provided by quantitative western blot analyses of hippocampal homogenates prepared from tissue used in qPCR analyses (Table 3). Results revealed no significant change of ULK1 levels, but elevated levels of PIK3C3/VPS34 (p ≤ 0.01), a component of the BECN1-PIK3C3/VPS34 complex that is positively regulated by ULK11 (Fig. 2A, B) in Br. V. Increased autophagosome formation in AD cases was also supported by elevated levels of the autophagic marker LC3B-II and LC3B-I in hippocampal total homogenate of Br. V samples (p ≤ 0.01) (Fig. 2A, B) and a marked increase in the total area/cell and in number of LC3-positive puncta in AD CA1 neurons (Fig. 5A, C, D) detectable already at Br. III. We also detected an increase of FOXO1 levels at Br. V (Fig. 2C, D).
Figure 2.
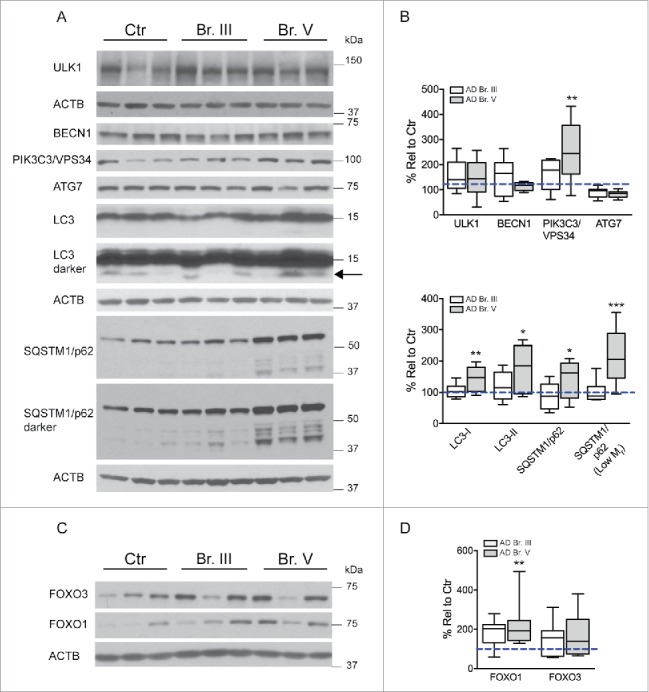
Evaluation of autophagy induction in AD. (A) Western blot analysis of indices of autophagic induction and autophagosome formation for ULK1, BECN1, PIK3C3/VPS34, ATG7, LC3, and SQSTM1/p62 and degradative products in AD hippocampus. (B) Histogram illustrating quantification of the findings in (A). (C–D) Western blot analysis of FOXO1 and FOXO3 levels in AD and control hippocampal tissue. Samples analyzed from patients as Control (n = 9), from AD Br. III (n = 10) and from AD Br. V (n = 10). Significance determined by one-way ANOVA with post-hoc Dunnett's Comparison to Control test. *p ≤ 0.05; **p ≤ 0.01. Western blots cropped as shown for purposes of clarity.
Figure 5.
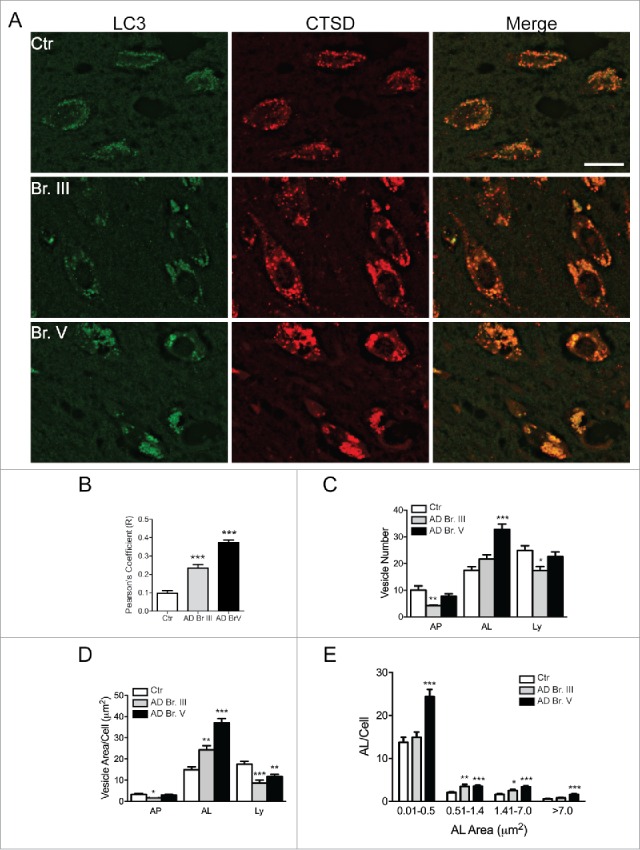
Double-immunofluorescent labeling of the CA1 region with LC3 and CTSD. (A) Immunofluorescent staining for LC3 (green channel) and CTSD (red) in CA1 hippocampal neurons from Control, Br. III and V. Scale bar: 20 μm. (B) Pearson's correlation coefficient of colocalization of CTSD with LC3 in hippocampal CA1 neurons. A minimum of 100 neurons were quantified per condition. (C–D) Characterization of vesicles in Control, Br. III and V CA1 neurons, identified as autophagosome (AP, LC3-positive, CTSD-negative vesicles), autolysosome (AL, LC3- and CTSD-positive vesicles) and lysosome (Ly, LC3-negative, CTSD-positive vesicles). Depicted is the total number and area covered per cell in (C) and (D) respectively. (E) Size distribution of autolysosomes in Control, Br. III and V CA1 neurons. Statistical significance denoted by asterisks with p-values shown determined by one-way ANOVA, with post-hoc Dunnett's Comparison to Control test for grouped analyses, and 2-way ANOVA for size distribution analyses. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Lysosomal biogenesis is mediated by TFE3 in CA1 neurons in AD brain
Upregulated lysosomal biogenesis in neurons in AD has previously been suggested by lysosomal morphometry46 and lysosomal protease CTSD (cathepsin D) expression patterns47 in affected neuronal populations of AD brain along with upregulation of CTSD gene and protein expression in neurofibrillary tangle-bearing CA1 pyramidal neurons.48 This earlier conclusion was strongly supported by microarray data obtained from hippocampal CA1 pyramidal neurons showing upregulated mRNA expression for 3 genes containing the CLEAR site in their promoter sequence, LAMP1, CLCN7, and CTSD, which was confirmed in the hippocampus by qPCR analysis of CTSD and LAMP1 (Fig. 1A, E). LAMP2, which is not a CLEAR family member,22 was not altered at the mRNA or protein levels. Consistent with mRNA findings, levels of CTSD and CTSB protein were also significantly elevated in hippocampus (p≤0.01, Fig. 3A, B) although levels of LAMP1 protein were not significantly increased. The discrepancy between the mRNA and protein levels of LAMP1 may be attributed to other levels of regulation between transcripts and protein products.49 Of the 44 genes related to autophagy analyzed by microarray, we also found 3 other genes containing the CLEAR site sequence (RAB5A, RAB7A and GABARAP) that were upregulated at the mRNA level. In light of these data, we next investigated in CA1 neurons the state of activation of the MiTF/TFE family of transcription factors known to regulate lysosomal biogenesis and CLEAR family proteins. To evaluate further roles for MiTF/TFE family members in autophagy upregulation in AD, we first measured TFEB and TFE3 mRNA levels by qPCR in a CA1 neuron population microdissected from hippocampus (Table 2, Ctr. Vs. Br. V, n = 5, 10 respectively) and also in whole hippocampal extracts from AD and control brains (Table 3) shown in Fig. 4A, B. Surprisingly, TFE3 mRNA levels were higher than those of TFEB in hippocampus, as indicated by an amplification plot showing an ≈1 cycle difference in CT values (Fig. 4C). Moreover, TFE3 mRNA expression was greater in Br. V CA1 neurons (p ≤ 0.0001; Fig. 4A) and also in whole AD hippocampal extracts (p ≤ 0.05; Fig. 4B) compared to controls.
Figure 3.
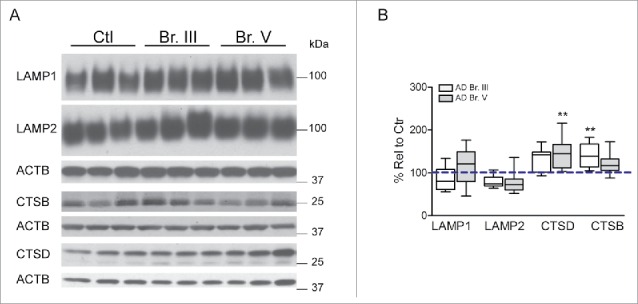
Analysis of lysosomal constituents in AD hippocampus. (A) Western blot analysis of structural and enzymatic constituents of lysosomes with quantification shown in (B). Samples analyzed from control patients (n = 9), from AD Br. III (n = 10) and from AD Br. V (n = 10). Significance determined by one-way ANOVA, with post-hoc Dunnett's Comparison to Control test. **p ≤ 0.01. Western blots cropped as shown for purposes of clarity.
Table 2.
Emory Alzheimer's Disease Resource Center (ADRC) demographics of single cell qPCR analysis of transcriptional activators.
| Sample No. | Case number | Braak stage | PMI (h) | Age at onset | Age at death | Duration | ApoE | Race/Sex |
|---|---|---|---|---|---|---|---|---|
| Ctr. 1 | OS99-08 | I | 3 | 74 | E3/3 | WF | ||
| Ctr. 2 | OS03-380 | II | 12 | 61 | E3/4 | BM | ||
| Ctr. 3 | OS03-390 | II | 7 | 74 | E3/3 | WF | ||
| Ctr. 4 | E10-142 | II | 5.5 | 94 | E3/3 | WM | ||
| Ctr. 5 | E14-06 | I | 12.5 | 56 | WM | |||
| AVG/Tot | . | 8.0 + 4.1 | 71.8 + 14.7 | 3M/3F | ||||
| Br. V 1 | OS00-11 | V | 4 | 49 | 55 | 6 | E3/3 | WM |
| Br. V 2 | E05-67 | V | 11.5 | 52 | 62 | 10 | E3/4 | WM |
| Br. V 3 | E07-36 | V | 22 | 91 | E3/3 | WM | ||
| Br. V 4 | E09-65 | V | 10 | 77 | 85 | 8 | E4/4 | WF |
| Br. V 5 | E10-56 | V | 15.5 | 76 | 82 | 6 | E4/4 | BF |
| Br. V 6 | OS01-10 | V | 8 | 79 | 87 | 8 | E3/4 | WF |
| Br. V 7 | E04-179 | V | 20 | 77 | 91 | 14 | E3/4 | WF |
| Br. V 8 | E13-134 | V | 17.5 | 75 | 79 | 4 | WF | |
| Br. V 9 | E08-97 | V | 19 | 78 | E3/4 | WF | ||
| AVG/Tot | 14.2 + 6.1 | 78.9 + 12.6 | 3M/6F |
Abbreviations: W, white; B, black; PMI, Postmortem interval.
Figure 4.

Analysis of TFE3 and TFEB transcription factors in hippocampal CA1 pyramidal neurons and glia. (A) qPCR analysis of CA1 neurons for TFE3 and TFEB. (B) qPCR for both TFEB and TFE3 in hippocampal total RNA. (C) Amplification plots for TFE3 (red) and TFEB (green) showing a 1.2 cycle difference equivalent to a greater than 10-fold higher level of TFE3 mRNA in hippocampal tissue. (D–E) Western blot analysis for TFE3 and TFEB in hippocampal tissue and relative quantification showing higher levels of TFE3. Samples analyzed from control patients (n = 9), from AD Br. III (n = 10) and from AD Br. V (n = 10). (F) Immunocytochemical localization of TFE3 in the hippocampal CA1 region. Cells immunostained for TFE3 and counterstained with Nissl. Black arrowheads indicate translocation of TFE3 in neuronal nuclei; blue arrowheads point to negative TFE3 nuclear staining. Scale bar: 10 μm. (G) Quantification of TFE3 nuclear translocation in glia and neurons. (H) Fluorescent immunolocalization of TFEB (yellow arrowheads for neuronal, blue for glia) with Nissl staining shows neuronal or glial nuclei and with decreasing Nissl staining on nuclear translocation in hippocampal CA1 region. Scale bar: 10 μm. (I) Quantification of TFEB nuclear translocation in glia and neurons. A minimum of 50 fields were quantified per condition. Statistical significance denoted by asterisks with p-values shown determined by one-way ANOVA, with post-hoc Dunnett's Comparison to Control test, or for 2 columns analysis by Student t test. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
In accord with mRNA levels, TFE3 protein levels were also increased in AD brain at both Braak stages (p ≤ 0.01; Fig. 4D, E). Most importantly, TFE3 immunocytochemistry (ICC) revealed that a significantly higher percentage of CA1 neurons exhibited translocation of TFE3 immunoreactivity from the cytoplasm to the nucleus (Fig. 4F, G), indicating activation of TFE3. Notably, TFEB mRNA levels in microdissected CA1 neurons were unchanged in AD, whereas levels were significantly increased in Braak stage V AD hippocampal tissue (p ≤ 0.05; Fig. 4A, B). The AD-related increase in hippocampus, but not in CA1 neurons, raised the possibility that the elevated TFEB expression was in glial cells. This observation is consistent with our preliminary evidence that TFEB is more highly expressed in glia than in neurons (unpublished data). Accordingly, TFEB protein levels did not differ in total extracts of hippocampus between AD and controls (Fig. 4D, E). However, fluorescence immunolabeling of TFEB in a semi-quantitative analysis of signal intensity confirmed that the frequency of glial cells with positive nuclear TFEB labeling was significantly higher in AD Braak V stage hippocampus (Fig. 4H, I). In contrast to glia, only a very small percentage of CA1 neurons exhibited nuclear translocation of TFEB, albeit at a significantly higher frequency in mild/moderate AD cases than in controls (Fig. 4I). Taken together, these data indicate a more prominent role for TFE3 in promoting lysosomal biogenesis and autophagy upregulation in CA1 neurons during AD pathogenesis and point to a relatively more important role for TFEB in glia of CA1 hippocampus.
Clearance of autophagic substrates by lysosomes is defective in CA1 neurons in AD
Despite evidence that lysosomal biogenesis is more activated in AD brain, including our earlier studies,46,47 additional observations indicated that lysosomal degradative efficiency is impaired. To evaluate lysosomal function, we assessed LC3 clearance specifically in CA1 pyramidal cells using double-immunofluorescence labeling analysis with antibodies to the autophagosome marker LC3 and the lysosomal hydrolase CTSD (Fig. 5A). Qualitatively, the presence of LC3 in many CTSD-positive neurons was readily apparent in Br. III CA1 hippocampus and quantitative image analysis confirmed a significant increase of Pearson's correlation coefficient for LC3-CTSD colocalization, with further increase to nearly 4-fold in Br. V CA1 neurons compared to controls (p ≤ 0.001; Fig. 5B). Most LC3 signal colocalized within CTSD-positive vesicles; in fact, the relative area and number of LC3-positive puncta containing no CTSD signal (i.e. autophagosomes) was decreased in CA1 neurons in AD Br. III and was not changed in AD Br. V, suggesting that the formation of autolysosomes (ALs), derived by the fusion of an autophagosome (AP) and lysosome (CTSD-positive vesicles negative for LC3), within the neuronal perikarya, was not compromised in AD. Interestingly, at Br. III stage, we observed a significant increase of area occupied by ALs associated with a decrease of area and number of APs and lysosomes (Fig. 5C, D), as a sign of an early upregulation of autophagy and possibly of more efficient AP-lysosome fusion. With the progression of the disease, there was an accumulation of ALs indicated by a significantly increased number and total area of these structures (p ≤ 0.001; Fig. 5C, D), reinforcing the notion of progressively impaired turnover of LC3 in CA1 neurons at late stages of AD. In further support of progressively dysfunctional autophagic clearance, size analysis revealed significantly increased numbers of ALs between 0.51–1.41 μm2 and 1.41–7.0 μm2 (p ≤ 0.01 and p ≤ 0.05, respectively, Fig. 5E) at Br. III stage while at Br. V, all sizes categories of ALs were abnormally enlarged, presumably with accumulated substrate (Fig. 5E). Consistent with these data, immunoblots revealed increased levels of LC3-II and a second autophagic substrate SQSTM1/p62 and its intermediate breakdown products (p ≤ 0.05-0.001; Fig. 2C, D), at Br. V. Collectively, these findings indicate that autolysosomal proteolytic function becomes progressively impaired in CA1 neurons in AD.
Autophagy deficits in AD can be interrogated further by analysis of the prominent neuritic dystrophy characteristic of this disease.26 In the hippocampal CA1 region of Br. V samples, immunostaining for LC3 and CTSD revealed dystrophic neurites in the neuropil, which were strongly positive for both antibodies (Fig. 6A). Dystrophic neurites are more numerous adjacent to senile plaques that are identified as large amorphous granular deposits that are weakly positive for CTSD, consistent with earlier established neuropathological patterns.50 Examination of z-stack images of a typical dystrophic swelling (Fig. 6B) revealed abundant autophagic vesicles of varying subtypes positive for LC3 co-existing with ones positive for CTSD, consistent with ultrastructural analyses showing that dystrophic neurites in AD brain are filled nearly completely with undigested autophagic vacuoles rather than with mixtures of different organelles.26 Autolysosomes containing LC3 (CTSD- and LC3-positive) are abundant, as in the perikarya. Large LC3-positive, CTSD-negative autophagosomes, however, are also common raising a possibility that, in axons, fusion between these 2 compartments may be less efficient; however, the numbers of lysosomes in axons is limited and the abundance of APs in neurites is more likely due to limited availability of neurite lysosomes rather than inefficient AP-lysosome fusion. The pattern in dystrophic neurites reinforces the conclusion that autolysosomal proteolysis is impeded, which has been previously shown to selectively slow the retrograde transport of autophagic vacuoles that can promote dystrophic swellings.51
Figure 6.
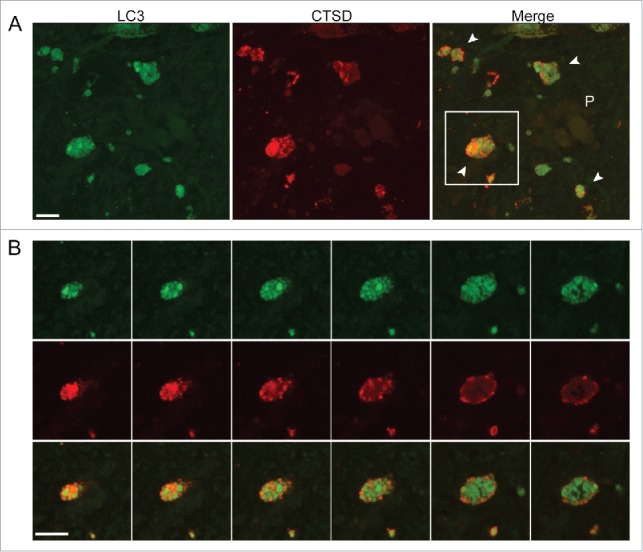
Neuritic dystrophy adjacent to senile plaques in the CA1 region in AD brains. (A) The image of AD Br. V hippocampal CA1 depicts a population of putative dystrophic neurites (white arrowheads in the merged image) surrounding a weakly CTSD-positive senile plaque (P). (B) Z-stack images of consecutive optical slices through the single profile indicated by the box in (A) confirms the typical appearance of a dystrophic neurite, which is shown to contain mainly autophagic vacuoles including LC3-positive AP (green), LC3- and CTSD-positive AL, (yellow-orange vesicles) and less frequent CTSD-positive-only lysosomes (red). Scale bar: 10 μm.
Since neuronal cell loss is widely reported in AD52-54 and may influence analyses at a tissue level, we also performed quantitative immunoblot analyses of the CA1 sector of the hippocampus, which revealed a 24% reduction of neuron-specific TUBB3 (p ≤ 0.05) in Braak stage V hippocampus, indicative of a mild-to-moderate loss of neurons as expected (Fig. 7). Similar analyses of GFAP (glial fibrillary acidic protein) indicated a significant 85% increase in GFAP (p ≤ 0.01; Fig. 7), consistent with the expected reactive astrogliosis. These data highlight the importance of the cell population-specific analyses in this study, which are substantially less influenced by these changes in cellular composition in the AD hippocampus.
Figure 7.
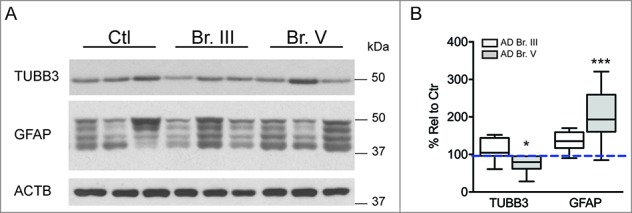
Expression of cell-type markers in AD hippocampus shows alterations in neuronal and glial content. (A) Representative western blots illustrating neuronal ΤUBB3 and glial GFAP markers in AD and control hippocampus. (B) Relative quantification of markers in hippocampus indicates downregulation of neuronal-specific TUBB3 and upregulation of GFAP, consistent with neuronal cell loss and concomitant astrogliosis. Statistical significance denoted by asterisks with p-values shown determined by one-way ANOVA, Dunnett's test. *p ≤ 0.05; **p ≤ 0.01. Western blots cropped as shown for purposes of clarity.
Discussion
Our study is the first analysis of multiple stages of autophagy specifically in a single vulnerable population of neurons during the progression of AD. The cell-specific approaches used were designed to overcome notorious limitations of molecular neuropathological analysis in brain tissue, where cell populations are heterogeneous and responses of individual neuronal and/or non-neuronal populations, including differential cell losses, are admixed.40 By assessing autophagy broadly, we could obtain a relatively full picture of the variables contributing to the robust autophagy pathology in AD brain.
We establish that autophagy induction and autophagosome formation in CA1 pyramidal neurons are competent and, in fact, are upregulated at the transcriptional level early in AD development and remain upregulated even as the clearance of autophagic substrates by lysosomes becomes progressively impaired. An activation of neuronal autophagy in AD is supported by independent lines of evidence established in microaspirated CA1 neurons and regional hippocampal dissections, including: (a) increase in the total area/cell and in number of LC3-positive puncta, (b) increased expression of key components involved in autophagosome formation; and (c) activation of TFE3 and TFEB transcription factors regulating lysosomal biogenesis and additional genes that support autophagy.20 Notably, analysis of significantly altered transcripts by DAVID software55 identified the autophagy pathway as the most broadly upregulated gene family among the gene ontology groups represented on our microarray. Most other gene ontology groups altered in CA1 neurons were downregulated, notably transcripts encoding NTFs/neurotrophins and neurotrophin receptors, synaptic-related proteins, and components related to glutamatergic neurotransmission.38,40 Many of the autophagy subgroup of genes are targets of the MiTF/TFE and FOXO families of transcription factors. MiTF/TFE family members are activated upon induction of autophagy56,57 or prolonged mitochondrial stress58 in order to sustain both autophagosome formation and clearance by lysosomes (lysosome biogenesis). Our results establish TFE3 as the pre-eminent MiTF/TFE family member in hippocampal neurons. By contrast, TFEB, often considered the most relevant master transcriptional regulator of autophagy and lysosome function,20,59 showed greater nuclear translocation in glial cells, perhaps to support reactive gliosis and the scavenging of dying neuronal processes and/or aggregated proteins. The changing cellular composition observed in Br. V hippocampus, including astrogliosis and moderate loss of neuron-specific markers underscores the importance of our cell-specific analytical approaches. Autophagy assessment in tissue is complicated by these changes as well as by evidence that some autophagy-related components are differentially expressed in neurons and glia, and responses of these different cell types to disease and pathology are likely to differ.
Because autophagosome clearance is impaired in AD,24,60,61 it has previously been difficult to determine whether increased induction/autophagosome formation also contributes to the massive buildup of autophagy substrates in AD brain. Earlier investigations of autophagy induction in AD brain and AD models have yielded conflicting results, related partly to differences in experimental models used, confounding effects on autophagy of expressing mutant proteins at high levels,24 and tissue-level analyses often involving insufficient numbers of autophagy measures to confirm the functionality of a given step in the autophagic process.62 Lipinski and coauthors36 found in AD cortical tissue that transcription of genes promoting autophagy was generally upregulated and that of negative regulators was downregulated, consistent with our findings. Moreover, MTOR kinase activity is reduced in PSEN1/PS1-APP mouse brain and cells treated with Aβ1-42,63 although it is reported to be increased in several other AD mouse models,64,65 in inferior parietal lobule35 in neurons exhibiting neurofibrillary degeneration in the AD brain,66 and in cell models stimulated by Aβ oligomers67 or mutant APP transfection.64 In view of these controversial conclusions about the net direction of autophagy change in AD, we used a broad range of functional autophagy indices, involving molecular, biochemical, and imaging techniques and a focus on individual CA1 vulnerable neuron populations. The multidimensional approach in our study, which includes autophagy gene expression analysis, allowed us to conclude that autophagy is upregulated in AD brain.
Although autophagy gene expression is already upregulated at early AD stages, lysosomal degradative function becomes strikingly impaired beginning early (Br. III stage), but especially by the later Br. V stage as evidenced by buildup of SQSTM1/p62 and LC3-II, and the accumulation and increased colocalization of LC3 with CTSD within elevated numbers and area of enlarged autolysosomes. An MTORC1 downregulation due to impaired lysosomal proteolysis16-18 may also be one cause for the observed activation and nuclear translocation of MiTF/TFE family transcription factors. Notably, the increased colocalization of LC3-II with CTSD in autolysosomes demonstrates that autophagosome-lysosome fusion is relatively preserved. Along the neurites of the most affected neurons, however, enlarged LC3-positive CTSD-negative autophagosomes were frequently seen although their presence is less likely a reflection of impaired AP-Ly fusion than to the fact that lysosomes are inefficiently transported into axons,51 and their availability for fusion events may be limiting, especially when autophagosome formation is upregulated as in AD. Expansion of autolysosomal compartments, as seen in AD brain68 is also seen upon inhibiting lysosomal proteolysis in vivo or in vitro,51,69 and in mouse models of β-amyloidosis.32 In the latter case, targeted enhancement of lysosomal proteolysis rescues deficits of synaptic plasticity, cognition, and substantially ameliorates amyloid deposition and tauopathy32 underscoring the pathogenic importance of these lysosomal deficits. Moreover, when additional proteolytic dysfunction is superimposed on the brain in AD, such as that exerted by mutant PSEN1 (presenilin 1) on V-ATPase function and lysosomal acidification,25,31,70-72 lysosomal pathology and disease onset are substantially accelerated. Such alterations of lysosomal pH, if present in AD brain, will negatively affect cathepsin maturation and activity,73 explaining why CTSD upregulation alone would not be sufficient to maintain adequate clearance of lysosomal substrates.
Collectively, our findings suggest that in AD, early upregulation of autophagy is an initial neuroprotective response to cell stress,74 which ultimately becomes counterproductive as lysosomal function becomes progressively impaired. Indeed, an increased substrate load in the presence of a developing AD-related impairment of lysosomal clearance,69 serves as a ‘double hit’ on lysosomes. Failing lysosomes are further overburdened by an accelerated delivery of endocytic substrates driven by elevated levels of the APP β-C-terminal fragment in AD and mediated by an APP β-C-terminal fragment-APPL1-RAB5 mechanism.75 Notably, endocytic pathway genes were the third most upregulated gene family in CA1 neurons of AD brain38 and their upregulation early in disease60 has been linked to abnormally accelerated endocytosis and accumulation of substrate in distal compartments in AD and DS.46,76 The pathobiology in AD is reminiscent of Niemann-Pick Type C, a lysosomal storage disease sharing various neuropathological features with AD.77 In Niemann-Pick Type C patient cells, primary lysosomal dysfunction is partially relieved by experimentally lowering an abnormally elevated level of autophagy induction.78
In conclusion, this multidimensional analysis of autophagy at different levels of the pathway in a specific vulnerable neuronal population highlights, for the first time, multiple alterations of autophagy in AD that provide new insight into the development of AV-laden dystrophic neurites that are a hallmark of AD neuropathology.26 The novel observation that transcriptional factors regulating lysosomal biogenesis and function may be operating differentially in individual cell types in the brain is one of the various observations that underscore the importance of evaluating the autophagy pathway in single populations of cells in disease states, such as AD.
Materials and methods
Tissue
For the custom-designed microarray analysis, tissue was procured from the Rush Religious Orders Study (RROS, Rush University Medical Center, Chicago, IL, USA) and the Center for Neurodegenerative Disease Research (CNDR, University of Pennsylvania, Philadelphia, PA, USA) as described previously by Ginsberg et al. with demographics shown in Table 1.38 Pathology as determined by the methodology of Braak and Braak was the primary criterion used to type tissues grouped as Controls (Br. 0-II, n = 10), Br. III/IV, n = 14, and Br. V/VI, n = 15).79 Based on results from these analyses, subsequent studies were typed solely on the basis of the primary neuropathological assessment as described by Braak and Braak.79 For qPCR and western blot analyses, fresh-frozen postmortem human brain tissue was obtained from the Harvard Brain Tissue Resource Center (HBTRC, McLean Hospital, Belmont, MA, USA) with demographic information outlined in Table 3. For these studies, we used hippocampus from cases assigned an AD diagnosis at either Braak stage III (n = 10) or Braak stage V (n = 10) (see Table 3). Nondemented, age-matched subjects with little or no AD pathology in these regions were analyzed as controls (Controls, n = 9). Groups were matched for age and postmortem interval (Table 3). For single-cell analysis of transcriptional activators via qPCR, frozen samples of CA1 region were obtained from Emory University Alzheimer's Disease Research Center (ADRC), Atlanta, GA, USA with patient demographics shown in Table 2. Finally, for ICC analysis of autophagic and lysosomal determinants and transcriptional activators, formalin-fixed hippocampi were procured from HBTRC and ADRC as shown in Table 4 and grouped as age-matched controls and Braak III or V as outlined above. Tissue was examined and neuropathological designations were based on NIA Reagan Recommendation, CERAD, and Braak staging criteria.79-81 All studies were performed under the guidelines of the Institutional Review Board of the NYU Langone Medical Center and NKI. Clinical and neuropsychological evaluation criteria for the RROS/CNDR and ADRC cohorts have been published previously.82-84 Antemortem cognitive assessment tests were performed, including the Mini-Mental State Exam (MMSE).82,85 Scores were available within the last year of death. For the ADRC cohort used in single-cell qPCR analysis, patients were categorized as Control (n = 5) or moderate AD (n = 9) (Table 2).
Table 4.
Formalin-fixed hippocampal sections utilized in immunocytochemical analyses from Emory ADRC and Harvard HBTRC.
| Sample No. | Case number | Brain bank | Braak stage | PMI (h) | Age at onset | Age at death | Duration | ApoE | Race/Sex |
|---|---|---|---|---|---|---|---|---|---|
| Ctr. 1 | E08-101 | ADRC | II | 17 | 78 | E3/3 | WF | ||
| Ctr. 2 | 6865 | HBTRC | ND | 18.7 | 78 | F | |||
| Ctr. 3 | 6573 | HBTRC | ND | 15 | 79 | F | |||
| Ctr. 4 | 7862 | HBTRC | ND | 18.0 | 73 | M | |||
| Ctr. 5 | 8341 | HBTRC | ND | 15.7 | 82 | F | |||
| Ctr. 6 | 5326 | HBTRC | ND | 12.1 | 75 | F | |||
| Ctr. 7 | 5859 | HBTRC | 0 | 12.5 | 60 | F | |||
| Ctr. 8 | 5919 | HBTRC | II | 18.7 | 80 | M | |||
| Ctr. 9 | 5452 | HBTRC | ND | 7.6 | 73 | M | |||
| Ctr. 10 | 5832 | HBTRC | I | 16.7 | 75 | F | |||
| Ctr. 11 | 5619 | HBTRC | ND | ||||||
| Ctr. 12 | 5582 | HBTRC | ND | 74 | |||||
| AVG/Total | 15.2 + 3.5 | 75.2 + 5.8 | 7F/3M | ||||||
| Br. III 1 | OS03-76 | ADRC | III | 17.5 | 70 | 78 | 8 | E3/3 | WM |
| Br. III 2 | E08-127 | ADRC | III | 20 | 76 | 82 | 6 | E4/4 | BF |
| Br. III 3 | E10-30 | ADRC | III | 2 | 77 | 90 | 13 | E3/3 | WF |
| Br. III 4 | 6776 | HBTRC | III | 17.2 | 80 | F | |||
| AVG/Total | 14.2 + 8.2 | 82.5 + 5.3 | 3F/1M | ||||||
| Br. V 1 | E08-97 | ADRC | V | 19 | 78 | E3/4 | WF | ||
| Br. V 2 | E06-15 | ADRC | V | 13.5 | 67 | 72 | 5 | E3/4 | WM |
| Br. V 3 | 5979 | HBTRC | VI | 17.9 | 80 | M | |||
| Br. V 4 | 5035 | HBTRC | V | 20 | 75 | F | |||
| Br. V 5 | 6209 | HBTRC | V | 13 | 75 | F | |||
| Br. V 6 | 6917 | HBTRC | V | 24.1 | 79 | F | |||
| Br. V 7 | 6297 | HBTRC | V | 6.5 | 75 | F | |||
| Br. V 8 | 6265 | HBTRC | V | 7.9 | 78 | F | |||
| Br. V 9 | 6215 | HBTRC | V | 15 | 82 | M | |||
| Br. V 10 | 6164 | HBTRC | V | 14.1 | 73 | M | |||
| Br. V 11 | 5850 | HBTRC | V | 5 | 73 | M | |||
| AVG/Total | 14.2 + 6.0 | 76.4 + 3.4 | 6F/5M |
Abbreviations: ND, not divulged; B, black; W, white; PMI, postmortem interval.
ICC
Formalin-fixed, human hippocampus (Table 4) was sectioned at 40 μm on a vibratome (Leica Biosystems, Buffalo Grove, IL, USA) for LC3 (MBL Int., M152-3; mouse monoclonal) and CTSD (Scripps Laboratories, RC245; rabbit polyclonal) colocalization, or paraffin embedded and sectioned at 7 μm for TFE3 and TFEB (Cell Signaling Technology, 14779 and 4240, respectively; polyclonal rabbit) immunolabeling. Where applicable, paraffin sections were deparaffinized. Antigen-retrieval was performed by heating tissue sections in sodium citrate buffer (10 mM, pH 6.0) at 95°C for 20 min. Sections were blocked and incubated in primary antibody in 1xTris-buffered saline (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) containing 0.1% Tween-20 (v:v; Fisher Scientific, BP337) and 3% normal goat serum (v:v; Vector Laboratories, S-1000) for up to 3 days at 4°C. Alexa Fluor-conjugated secondary antibodies (ThermoFisher/Life Technologies, A-11029, A-11010, A-11008, A-11003) were used for immunofluorescence and ABC detection was used for DAB (Vector Laboratories, PK-4005). Autofluorescence was quenched with 1-2.5 mM CuSO4 in 50 mM ammonium acetate, pH 5.0 for 60 min at RT. For CTSD/LC3 stained imaging, sections were stained in 600 nM DAPI (Sigma, D9542) for 15 min at room temperature prior to mounting. DAB labeling of TFE3 following cresyl violet staining for 15 min was inspected on a Zeiss AxioSkop II equipped with an HrM digital camera (Zeiss USA, Thornwood, NY, USA). Sections labeled for TFEB were counterstained for Nissl using cresyl violet as described by Alvarez-Buylla.86 In CA1, glia were differentiated from pyramidal neurons on the basis of morphology and nuclear staining by DAPI or cresyl violet Nissl staining.86 Immunofluorescent images for CSTD, LC3, or TFEB were collected on a confocal microscope (Zeiss LSM510 or LSM810). Pearson's correlation coefficient was quantified using ZEN V. 2.1 software from Zeiss. Quantifications for LC3- and CTSD-immunoreactive puncta were performed using FIJI software.87 Fluorescence images were adjusted for brightness, contrast and color balance by using Adobe Photoshop CS (San Jose, CA, USA).
Single population microarray
Immunocytochemistry to identify neurofilament-immunoreactive CA1 neurons for laser capture microdissection88 and terminal continuation (TC) RNA amplification procedures have been described in detail previously.38,40,89,90 Individual CA1 pyramidal neurons from fresh-frozen hippocampi (Table 1) were microaspirated via LCM (Arcturus PixCell IIe, MDS, Sunnyvale, California). Fifty cells were captured for population cell analysis. A total of 212 custom-designed arrays were performed on the cohort of subjects as described previously.38,39 The TC RNA amplification procedure has been employed previously using human CA1 pyramidal neurons accessed via LCM as input sources of RNA.38,40,90 The TC RNA amplification protocol is available at http://cdr.rfmh.org/pages/ginsberglabpage.html. Array platforms consisted of 1 μg of linearized cDNA purified from plasmid preparations adhered to high-density nitrocellulose (GE Healthcare, Hybond XL). 578 cDNAs/ESTs were utilized on the current array platform. All of the autophagic and endosomal-lysosomal genes were derived from human sequences. Procedures for custom-designed microarray analysis and patients' demographics have been described in detail.38,40,89,90 Microarray analysis was performed on CA1 pyramidal neurons obtained from Controls (Br. 0-II, n = 10), Br. III/IV (n = 14), and Br. V/VI (n = 15) cases (Table 1). Briefly, expression of TC-amplified RNA bound to each linearized cDNA minus background was expressed as a ratio of the total hybridization signal intensity of the array, enabling a profile of relative changes in mRNA levels. Relative changes in total hybridization signal intensity were analyzed by mixed models analysis for repeated measures, with random intercept, fixed effect for diagnosis, Kenward-Roger denominator degrees of freedom, and unequal variance assumption as warranted.38,40,89 The level of statistical significance was set at (p < 0.01); trend level changes were characterized as (p < 0.02) – (p < 0.05) as described previously.90-92
RNA preparation
For routine qPCR analyses 100 mg of gray matter from hippocampus procured from the Harvard Brain Tissue Resource Center (HBTRC), part of the NeuroBioBank organism (Table 3), was dissected and extracted in 1.5 ml Trizol reagent (ThermoFisher/Life Technologies, 15596026) using a hand-held homogenizer (6 × 15 s bursts; Pro-Scientific, Oxford, CT, USA) followed by mixing with 300 μL of chloroform. Samples were centrifuged at 12000 g for 15 min at 4°C. The aqueous phase was collected and 750 μL isopropanol was added; samples were spun again at 12000 g for 10 min at 4°C. Supernatant was removed and the pellet washed 2 times with 75% ice-cold ethanol and centrifuged at 7500 g for 5 min at 4°C. The pellet was redissolved in 100 μL of RNAase-free distilled water. Total RNA quality (RNA Integrity Number) was assessed on an Agilent Bioanalyzer using an RNA Nanochip (2100; Agilent Technologies; Santa Clara, CA).
For analysis of transcriptional activators, slides were prepared from 40-μm sections cut from fresh-frozen samples of CA1 region obtained from Emory University Alzheimer's Disease Research Center (see Table 2) in a cryotome (MICROM, Walldorf, Germany). Slides were placed on a precooled metal plate and regions enriched in CA1 pyramidal neurons were visualized under a dissecting microscope using the Human Brain Atlas to reconnoiter landmarks. Cells were scraped using an ice-cold scalpel and collected in microcentrifuge tubes. Cells were extracted for total RNA using the TaqMan Gene Expression Cells-To-CT kit (ThermoFisher/Life Technologies, 439902) as per the manufacturer's instructions with the following modifications: Cells were lysed in 50 μl Lysis Buffer containing DNAse1 to remove contaminating genomic DNA for 5 min at room temperature, followed by the addition of 5 μl Stop solution for 2 min. Two reactions per sample were pooled and 22.5 μl subjected to reverse transcription in a final volume of 40 μl. Following reverse transcription, aliquots of 10 μl in triplicate were used for qPCR analysis as described below.
Preparation of cDNA and qPCR
cDNA was prepared from total RNA using TaqMan Reverse Transciption Reagent kit (ThermoFisher/Life Technologies, N808-0234) according to the manufacturer's instructions. Following reverse transcription, 20 ng of sample cDNA was loaded in triplicate into wells of a 96-well optical reaction plate (ThermoScientific, AB-2100) containing appropriate target gene primer (Table 5; ThermoFisher/Life Technologies). Three housekeeping genes were run and assessed for stability: GAPDH (glyceraldehyde 3-phosphate dehydrogenase), ACTB (actin β), and HPRT1 (hypoxanthine phosphoribosyltransferase 1). Total reaction volume per well was 20 μL. In the case of single cell transcriptional activators, the housekeeping genes used were GAPDH and SDHA (succinate dehydrogenase complex flavoprotein subunit A) and total reaction volume was 10 μl. qPCR was performed in the ABI Prism 7900HT Sequence Detection System (Applied Biosystems Branchburg, NJ, USA) as described previously.91,92
Table 5.
Primers used for qPCR analyses.
| Function | Gene name | Protein name | Probe I.D. |
|---|---|---|---|
| Housekeeping | ACTB | actin β | Hs99999903_m1 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | Hs00266705_g1 | |
| HPRT1 | hypoxanthine phosphoribosyltransferase 1 | Hs00266705_g1 | |
| SDHA | succinate dehydrogenase complex flavoprotein subunit A | Hs00417200_m1 | |
| Transcriptional activator | TFEB | transcription factor EB | Hs01065086_m1 |
| TFE3 | transcription factor binding to IGHM enhancer 3 | Hs00232406_m1 | |
| FOXO1 | forkhead box O1 | Hs01054576_m1 | |
| FOXO3 | forkhead box O3 | Hs00818121_m1 | |
| Autophagosome formation-elongation | ULK1 | unc-51 like autophagy activating kinase 1 | Hs00177504_m1 |
| ATG5 | autophagy related 5 | Hs00169468_m1 | |
| ATG12 | autophagy related 12 | Hs01047860_g1 | |
| ATG7 | autophagy related 7 | Hs00894898_m1 | |
| MAP1LC3B/LC3B | microtubule associated protein 1 light chain 3 β | Hs00917683_m1 | |
| BECN1 | beclin 1 | Hs00186838_m1 | |
| PIK3C3/VPS34 | phosphatidylinositol 3-kinase catalytic subunit type 3 | Hs00176908_m1 | |
| SQSTM1/p62 | sequestosome 1 | Hs00177654_m1 | |
| Lysosome | CTSD | cathepsin D | Hs00157205_m1 |
| CTSB | cathepsin B | Hs00947433_m1 | |
| LAMP1 | lysosomal associated membrane protein 1 | Hs00174766_m1 | |
| LAMP2 | lysosomal associated membrane protein 2 | Hs00174474_m1 |
Taq-Man validated primers purchased from Life Technologies/ThermoFisher.
Gene Ontology (GO) analysis
The GO analysis was done using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 as described previously.55 A unique list of gene symbols was uploaded including all significantly upregulated genes in MCI and AD compared to control, detected by the single population microarray.
Calculation of qPCR results
Following qPCR, the target genes were normalized against the housekeeping genes. Results were calculated using the ΔΔCt method (Applied Biosystems, Branchburg, NJ, USA, Bulletin #2). Control values were averaged as a geometric mean and sample values were recalculated and expressed as percent control.93,94 Outliers were recognized as values falling beyond 2 standard deviations of the mean, and were discarded from the analyses.
Preparation of tissue extracts
100 mg of brain tissue procured from HBTRC corresponding to samples used for qPCR (Table 3) was homogenized as previously described95 in a Tissue Homogenization buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM each EDTA, EGTA and DTT, 250 mM sucrose (EMD Millipore, SX1075, 1 mM β-glycerophosphate, 1 mM NaF, 0.2 mM NaVO4, 5 μg/ml each leupeptin (Sigma, L5793, antipain (Sigma, A6191) and pepstatin (Enzo Life Sciences, 260-009) and 1 mM each benzamidine (Sigma, 434760) and PMSF (Sigma, P7626)). For enzymatic activity assays, extracts were acidified with 0.4 M NaAc, pH 5 and supernatants used as described previously.31 Protein content was determined by the BCA method.96 The lysates were solubilized in Laemmli buffer for western blotting.
SDS-PAGE and western blotting
SDS-polyacrylamide gel electrophoresis followed by western blotting was performed as described previously.97 The immunoreactive bands were visualized with ECL reagent (Amersham, RPN2209) and the bands were quantified using MultiGauge V. 3.0 (Fuji Film) software. Target proteins were normalized against ACTB/β-actin, unless otherwise noted.
Antibodies for western analyses
TUBB3 (Clone TU-20; Sigma, SAB4700544), GFAP (Sigma, SAB4501162), TFE3 (Sigma, HPA023881), ATG7 (Cell Signaling Technology, 2631), TFEB (Cell Signaling Technology, 4240), FOXO1 (Cell Signaling Technology, 2880), FOXO3 (Cell Signaling Technology, 2497) ULK1 (Cell Signaling Technology, 4773), BECN1 (BD Biosciences, 612113), SQSTM1/p62 (BD Biosciences, 610832), LC3 (Novus Biologics, NB100-2220), LAMP1 and LAMP2 (Developmental Studies Hybridoma Bank, University of Iowa, H4A3 and H4B4, respectively), CTSD D-2-3,98 CTSB (Cortex Biochemicals, CR6009RP) and TFEB (Bethyl Laboratories, A303-672 and -673).
Statistical analysis of qPCR and western blot analyses
Statistical significance was tested using one-way analysis of variance (ANOVA) with post-hoc analysis of Dunnett's Comparison to Control test, assessed with Graphpad Prism software, V. 6.0. If the ANOVA was found significant (*p ≤ 0.05), then the Family-Wise Error Rate is protected at 0.05 using Student t test as reported in the text or shown in Figures. Alternatively, if the ANOVA is not significant, it is necessary to use the Dunnett's Multiple Comparison Test of the Family-Wise Error Rate at 0.05 as reported in the text or shown in Figures. For size distribution analyses, significance was determined by 2-way ANOVA assessed with Graphpad Prism software.
Abbreviations
- AD
Alzheimer disease
- ALs
autolysosomes
- AP
autophagosome
- CTSD
cathepsin D
- ICC
immunocytochemistry
- LCM
laser capture microdissection
- TC
terminal continuation
- TFE3
transcription factor binding to IGHM enhancer 3
- TFEB
transcription factor EB
Acknowledgments
Tissues were kindly provided by the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R24 MH068855 and Emory Medical School Alzheimer's Disease Research Center, supported in part by PHS grant 2P50AG025688-11, as well as the University of Pennsylvania Center for Neurodegenerative Disease Center. We are indebted to the altruism and support of the participants in the Religious Orders Study and the Center for Neurodegenerative Research at UPENN. A list of participating groups and funding can be found at the website: http://www.rush.edu/rumc/page-R12394.html. We thank Arthur Saltzman for his expert technical support; we also wish to thank Eugene Laska, Ph.D., for support in Statistical analyses.
Funding
This work was supported by the following grants from the National Institute on Aging: P01AG017617 (RAN), AG043375 and AG014449 (SDG).
References
- [1].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/ 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- [2].Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal 2012; 17:766-74; PMID:22098160; http://dx.doi.org/ 10.1089/ars.2011.4405 [DOI] [PubMed] [Google Scholar]
- [3].Menzies FM, Moreau K, Rubinsztein DC. Protein misfolding disorders and macroautophagy. Curr Opin Cell Biol 2011; 23:190-7; PMID:21087849; http://dx.doi.org/ 10.1016/j.ceb.2010.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4:176-84; PMID:18059160; http://dx.doi.org/ 10.4161/auto.5269 [DOI] [PubMed] [Google Scholar]
- [5].Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol 2010; 12:842-6; PMID:20811357; http://dx.doi.org/ 10.1038/ncb0910-842 [DOI] [PubMed] [Google Scholar]
- [6].Knuppertz L, Hamann A, Pampaloni F, Stelzer E, Osiewacz HD. Identification of autophagy as a longevity-assurance mechanism in the aging model Podospora anserina. Autophagy 2014; 10:822-34; PMID:24584154; http://dx.doi.org/ 10.4161/auto.28148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 2013; 23:310-22; PMID:23726895; http://dx.doi.org/ 10.1016/j.semcancer.2013.05.008 [DOI] [PubMed] [Google Scholar]
- [8].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/ 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 2008; 4:590-9; PMID:18497567; http://dx.doi.org/ 10.4161/auto.6259 [DOI] [PubMed] [Google Scholar]
- [10].Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/ 10.1038/ncb2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al.. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10:776-87; PMID:18552835; http://dx.doi.org/ 10.1038/ncb1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bernard A, Klionsky DJ. Autophagosome formation: tracing the source. Dev Cell 2013; 25:116-7; PMID:23639440; http://dx.doi.org/ 10.1016/j.devcel.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nakatogawa H, Ohbayashi S, Sakoh-Nakatogawa M, Kakuta S, Suzuki SW, Kirisako H, Kondo-Kakuta C, Noda NN, Yamamoto H, Ohsumi Y. The autophagy-related protein kinase Atg1 interacts with the ubiquitin-like protein Atg8 via the Atg8 family interacting motif to facilitate autophagosome formation. J Biol Chem 2012; 287:28503-7; PMID:22778255; http://dx.doi.org/ 10.1074/jbc.C112.387514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165-78; PMID:17632063; http://dx.doi.org/ 10.1016/j.cell.2007.05.021 [DOI] [PubMed] [Google Scholar]
- [15].Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci 2014; 127:3-9; PMID:24345374; http://dx.doi.org/ 10.1242/jcs.140426 [DOI] [PubMed] [Google Scholar]
- [16].Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to MTORC1. Science 2008; 320:1496-501; PMID:18497260; http://dx.doi.org/ 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. MTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011; 334:678-83; PMID:22053050; http://dx.doi.org/ 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10:935-45; PMID:18604198; http://dx.doi.org/ 10.1038/ncb1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al.. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015; 17:288-99; PMID:25720963; http://dx.doi.org/ 10.1038/ncb3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al.. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/ 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, et al.. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via MTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/ 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852-66; PMID:21752829; http://dx.doi.org/ 10.1093/hmg/ddr306 [DOI] [PubMed] [Google Scholar]
- [23].Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 2014; 39:159-69; PMID:24630600; http://dx.doi.org/ 10.1016/j.tibs.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease–locating the primary defect. Neurobiol Dis 2011; 43:38-45; PMID:21296668; http://dx.doi.org/ 10.1016/j.nbd.2011.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al.. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146-58; PMID:20541250; http://dx.doi.org/ 10.1016/j.cell.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005; 64:113-22; PMID:15751225; http://dx.doi.org/ 10.1093/jnen/64.2.113 [DOI] [PubMed] [Google Scholar]
- [27].Suzuki K, Terry RD. Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathol 1967; 8:276-84; PMID:6039977; http://dx.doi.org/ 10.1007/BF00688828 [DOI] [PubMed] [Google Scholar]
- [28].Kawai M, Cras P, Richey P, Tabaton M, Lowery DE, Gonzalez-DeWhitt PA, Greenberg BD, Gambetti P, Perry G. Subcellular localization of amyloid precursor protein in senile plaques of Alzheimer's disease. Am J Pathol 1992; 140:947-58; PMID:1562053 [PMC free article] [PubMed] [Google Scholar]
- [29].Anderton BH. Alzheimer's disease. Paired helical filaments and the cytoskeleton. Nature 1988; 335:497-8; PMID:3138546; http://dx.doi.org/ 10.1038/335497a0 [DOI] [PubMed] [Google Scholar]
- [30].Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120:885-90; PMID:6375662; http://dx.doi.org/ 10.1016/S0006-291X(84)80190-4 [DOI] [PubMed] [Google Scholar]
- [31].Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, et al.. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep 2015; 12:1430-44; PMID:26299959; http://dx.doi.org/ 10.1016/j.celrep.2015.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson DW, Bandyopadhyay U, Jiang Y, et al.. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011; 7:788-9; PMID:21464620; http://dx.doi.org/ 10.4161/auto.7.7.15596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 2008; 28:6926-37; PMID:18596167; http://dx.doi.org/ 10.1523/JNEUROSCI.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al.. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118:2190-9; PMID:18497889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA. Alteration of MTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 2015; 133:739-49; PMID:25645581; http://dx.doi.org/ 10.1111/jnc.13037 [DOI] [PubMed] [Google Scholar]
- [36].Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, et al.. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A 2010; 107:14164-9; PMID:20660724; http://dx.doi.org/ 10.1073/pnas.1009485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sun YX, Ji X, Mao X, Xie L, Jia J, Galvan V, Greenberg DA, Jin K. Differential activation of MTOR complex 1 signaling in human brain with mild to severe Alzheimer's disease. J Alzheimers Dis 2014; 38:437-44; PMID:23979023 [DOI] [PubMed] [Google Scholar]
- [38].Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, et al.. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry 2010; 68:885-93; PMID:20655510; http://dx.doi.org/ 10.1016/j.biopsych.2010.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Alldred MJ, Duff KE, Ginsberg SD. Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol Dis 2012; 45:751-62; PMID:22079237; http://dx.doi.org/ 10.1016/j.nbd.2011.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ginsberg SD, Alldred MJ, Che S. Gene expression levels assessed by CA1 pyramidal neuron and regional hippocampal dissections in Alzheimer's disease. Neurobiol Dis 2012; 45:99-107; PMID:21821124; http://dx.doi.org/ 10.1016/j.nbd.2011.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011; 1:a006189; PMID:22229116; http://dx.doi.org/ 10.1101/cshperspect.a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004; 117:4837-48; PMID:15340014; http://dx.doi.org/ 10.1242/jcs.01370 [DOI] [PubMed] [Google Scholar]
- [43].Talaber G, Miklossy G, Oaks Z, Liu Y, Tooze SA, Chudakov DM, Banki K, Perl A. HRES-1/Rab4 promotes the formation of LC3(+) autophagosomes and the accumulation of mitochondria during autophagy. PLoS One 2014; 9:e84392; PMID:24404161; http://dx.doi.org/ 10.1371/journal.pone.0084392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ 2014; 21:348-58; PMID:24440914; http://dx.doi.org/ 10.1038/cdd.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 2014; 15:65-74; PMID:24326622; http://dx.doi.org/ 10.1038/nrm3716 [DOI] [PubMed] [Google Scholar]
- [46].Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci 1996; 16:186-99; PMID:8613784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 1995; 14:671-80; PMID:7695914; http://dx.doi.org/ 10.1016/0896-6273(95)90324-0 [DOI] [PubMed] [Google Scholar]
- [48].Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol 2000; 48:77-87; PMID:10894219; http://dx.doi.org/ 10.1002/1531-8249(200007)48:1%3c77::AID-ANA12%3e3.0.CO;2-A [DOI] [PubMed] [Google Scholar]
- [49].Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett 2009; 583:3966-73; PMID:19850042; http://dx.doi.org/ 10.1016/j.febslet.2009.10.036 [DOI] [PubMed] [Google Scholar]
- [50].Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 2007; 120:4081-91; PMID:18032783; http://dx.doi.org/ 10.1242/jcs.019265 [DOI] [PubMed] [Google Scholar]
- [51].Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci 2010; 31:7817-30; PMID:NOT_FOUND; http://dx.doi.org/ 10.1523/JNEUROSCI.6412-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 1990; 296:1-22; PMID:2358525; http://dx.doi.org/ 10.1002/cne.902960102 [DOI] [PubMed] [Google Scholar]
- [53].West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet 1994; 344:769-72; PMID:7916070; http://dx.doi.org/ 10.1016/S0140-6736(94)92338-8 [DOI] [PubMed] [Google Scholar]
- [54].Cavallucci V, D'Amelio M, Cecconi F. Abeta toxicity in Alzheimer's disease. Mol Neurobiol 2012; 45:366-78; PMID:22415442; http://dx.doi.org/ 10.1007/s12035-012-8251-3 [DOI] [PubMed] [Google Scholar]
- [55].Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- [56].Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al.. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 2013; 15:647-58; PMID:23604321; http://dx.doi.org/ 10.1038/ncb2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 2014; 7:ra9; PMID:24448649; http://dx.doi.org/ 10.1126/scisignal.2004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol 2015; 210:435-50; PMID:26240184; http://dx.doi.org/ 10.1083/jcb.201501002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al.. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463 [DOI] [PubMed] [Google Scholar]
- [60].Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer's disease. Alzheimers Res Ther 2013; 5:53; PMID:24171818; http://dx.doi.org/ 10.1186/alzrt217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Piras A, Collin L, Gruninger F, Graff C, Ronnback A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 2016; 4:22; PMID:26936765; http://dx.doi.org/ 10.1186/s40478-016-0292-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/ 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, Pradier L, Hugon J. MTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem 2005; 94:215-25; PMID:15953364; http://dx.doi.org/ 10.1111/j.1471-4159.2005.03187.x [DOI] [PubMed] [Google Scholar]
- [64].Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (MTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 2010; 285:13107-20; PMID:20178983; http://dx.doi.org/ 10.1074/jbc.M110.100420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Xue X, Wang LR, Sato Y, Jiang Y, Berg M, Yang DS, Nixon RA, Liang XJ. Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer's disease. Nano Lett 2014; 14:5110-7; PMID:25115676; http://dx.doi.org/ 10.1021/nl501839q [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of MTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J 2005; 272:4211-20; PMID:16098202; http://dx.doi.org/ 10.1111/j.1742-4658.2005.04833.x [DOI] [PubMed] [Google Scholar]
- [67].Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-MTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener 2009; 4:14; PMID:19291319; http://dx.doi.org/ 10.1186/1750-1326-4-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci 1997; 17:6142-51; PMID:9236226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al.. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain 2011; 134:258-77; PMID:21186265; http://dx.doi.org/ 10.1093/brain/awq341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Coffey EE, Beckel JM, Laties AM, Mitchell CH. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer's disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014; 263:111-24; PMID:24418614; http://dx.doi.org/ 10.1016/j.neuroscience.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci 2013; 37:1949-61; PMID:23773064; http://dx.doi.org/ 10.1111/ejn.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer's disease: partners in crime. Biochem Soc Trans 2013; 41:1495-502; PMID:24256243; http://dx.doi.org/ 10.1042/BST20130201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev 2016; In press; PMID:27125852; http://dx.doi.org/ 10.1016/j.arr.2016.04.010 [DOI] [PubMed] [Google Scholar]
- [74].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, Jiang Y, Nixon RA. Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol Psychiatry 2015; 21:707-16; PMID:2619481.20080541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, Nixon RA. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 2010; 107:1630-5; PMID:20080541; http://dx.doi.org/ 10.1073/pnas.0908953107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nixon RA. Niemann-Pick Type C disease and Alzheimer's disease: the APP-endosome connection fattens up. Am J Pathol 2004; 164:757-61; PMID:14982829; http://dx.doi.org/ 10.1016/S0002-9440(10)63163-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet 2012; 21:4876-87; PMID:22872701; http://dx.doi.org/ 10.1093/hmg/dds324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991; 82:239-59; PMID:1759558; http://dx.doi.org/ 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- [80].Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997; 56:1095-7; PMID:9329452; http://dx.doi.org/ 10.1097/00005072-199710000-00002 [DOI] [PubMed] [Google Scholar]
- [81].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41:479-86; PMID:2011243; http://dx.doi.org/ 10.1212/WNL.41.4.479 [DOI] [PubMed] [Google Scholar]
- [82].Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology 2002; 59:198-205; PMID:12136057; http://dx.doi.org/ 10.1212/WNL.59.2.198 [DOI] [PubMed] [Google Scholar]
- [83].Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, Saragovi HU, Kordower JH. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol 2000; 427:19-30; PMID:11042589; http://dx.doi.org/ 10.1002/1096-9861(20001106)427:1%3c19::AID-CNE2%3e3.0.CO;2-A [DOI] [PubMed] [Google Scholar]
- [84].Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, Wuu J, Bennett DA, Jaffar S, Gilmor ML, Levey AI, Kordower JH. Loss of basal forebrain P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer's disease. J Comp Neurol 2002; 443:136-53; PMID:11793352; http://dx.doi.org/ 10.1002/cne.10122 [DOI] [PubMed] [Google Scholar]
- [85].Arvanitakis Z, Grodstein F, Bienias JL, Schneider JA, Wilson RS, Kelly JF, Evans DA, Bennett DA. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 2008; 70:2219-25; PMID:18519870; http://dx.doi.org/ 10.1212/01.wnl.0000313813.48505.86 [DOI] [PubMed] [Google Scholar]
- [86].Alvarez-Buylla A, Ling CY, Kirn JR. Cresyl violet: a red fluorescent Nissl stain. J Neurosci Methods 1990; 33:129-33; PMID:2232864; http://dx.doi.org/ 10.1016/0165-0270(90)90016-9 [DOI] [PubMed] [Google Scholar]
- [87].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al.. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676-82; PMID:22743772; http://dx.doi.org/ 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Ginsberg SD, Che S. Expression profile analysis within the human hippocampus: comparison of CA1 and CA3 pyramidal neurons. J Comp Neurol 2005; 487:107-18; PMID:15861457; http://dx.doi.org/ 10.1002/cne.20535 [DOI] [PubMed] [Google Scholar]
- [89].Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, Che S. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Chem Neuroanat 2011; 42:102-10; PMID:21669283; http://dx.doi.org/ 10.1016/j.jchemneu.2011.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ginsberg SD. Transcriptional profiling of small samples in the central nervous system. Methods Mol Biol 2008; 439:147-58; PMID:18370101; http://dx.doi.org/ 10.1007/978-1-59745-188-8_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Alldred MJ, Lee SH, Petkova E, Ginsberg SD. Expression profile analysis of vulnerable CA1 pyramidal neurons in young-Middle-Aged Ts65Dn mice. J Comp Neurol 2015; 523:61-74; PMID:25131634; http://dx.doi.org/ 10.1002/cne.23663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Alldred MJ, Lee SH, Petkova E, Ginsberg SD. Expression profile analysis of hippocampal CA1 pyramidal neurons in aged Ts65Dn mice, a model of Down syndrome (DS) and Alzheimer's disease (AD). Brain Struct Funct 2015; 220:2983-96; PMID:25031177; http://dx.doi.org/ 10.1007/s00429-014-0839-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3:RESEARCH0034; PMID:12184808; http://dx.doi.org/ 10.1186/gb-2002-3-7-research0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lipska BK, Peters T, Hyde TM, Halim N, Horowitz C, Mitkus S, Weickert CS, Matsumoto M, Sawa A, Straub RE, et al.. Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum Mol Genet 2006; 15:1245-58; PMID:16510495; http://dx.doi.org/ 10.1093/hmg/ddl040 [DOI] [PubMed] [Google Scholar]
- [95].Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-beta quantitation. Methods Mol Biol 2005; 299:267-78; PMID:15980611 [DOI] [PubMed] [Google Scholar]
- [96].Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem 1985; 150:76-85; PMID:3843705; http://dx.doi.org/ 10.1016/0003-2697(85)90442-7 [DOI] [PubMed] [Google Scholar]
- [97].Mohan PS, Nixon RA. Purification and properties of high molecular weight calpastatin from bovine brain. J Neurochem 1995; 64:859-66; PMID:7830080; http://dx.doi.org/ 10.1046/j.1471-4159.1995.64020859.x [DOI] [PubMed] [Google Scholar]
- [98].Cataldo AM, Thayer CY, Bird ED, Wheelock TR, Nixon RA. Lysosomal proteinase antigens are prominently localized within senile plaques of Alzheimer's disease: evidence for a neuronal origin. Brain Res 1990; 513:181-92; PMID:2350688; http://dx.doi.org/ 10.1016/0006-8993(90)90456-L [DOI] [PubMed] [Google Scholar]