

ABSTRACT
MTOR (mechanistic target of rapamycin [serine/threonine kinase]) plays a crucial role in many major cellular processes including metabolism, proliferation and macroautophagy/autophagy induction, and is also implicated in a growing number of proliferative and metabolic diseases. Both MTOR and autophagy have been suggested to be involved in lung disorders, however, little is known about the role of MTOR and autophagy in pulmonary epithelium in the context of acute lung injury (ALI). In the present study, we observed that lipopolysaccharide (LPS) stimulation induced MTOR phosphorylation and decreased the expression of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β)-II, a hallmark of autophagy, in mouse lung epithelium and in human bronchial epithelial (HBE) cells. The activation of MTOR in HBE cells was mediated by TLR4 (toll-like receptor 4) signaling. Genetic knockdown of MTOR or overexpression of autophagy-related proteins significantly attenuated, whereas inhibition of autophagy further augmented, LPS-induced expression of IL6 (interleukin 6) and IL8, through NFKB signaling in HBE cells. Mice with specific knockdown of Mtor in bronchial or alveolar epithelial cells exhibited significantly attenuated airway inflammation, barrier disruption, and lung edema, and displayed prolonged survival in response to LPS exposure. Taken together, our results demonstrate that activation of MTOR in the epithelium promotes LPS-induced ALI, likely through downregulation of autophagy and the subsequent activation of NFKB. Thus, inhibition of MTOR in pulmonary epithelial cells may represent a novel therapeutic strategy for preventing ALI induced by certain bacteria.
KEYWORDS: acute lung injury, autophagy, epithelium, inflammation, MTOR
Introduction
Acute lung injury (ALI) is a severe clinical condition characterized by respiratory distress, refractory hypoxemia, and noncardiogenic pulmonary edema, and pathologically by widespread lung inflammation and loss of epithelial and endothelial integrity.1 Cellular characteristics include damage of alveolar-capillary membrane, excessive transepithelial neutrophil migration, and release of pro-inflammatory, cytotoxic mediators in ALI.1 The epithelium, considered as the primary host defense of the alveolus, forms a robust barrier in response to infectious and noninfectious stimulation in both the pathogenesis and resolution of ALI.2,3
Severe pneumonia or sepsis is the primary cause of ALI.4 As a major constituent of the outer membrane of Gram-negative bacteria, lipopolysaccharide (LPS) causes a diverse spectrum of infections including life-threatening pneumonia and septicemia, and subsequent signaling leads to the downstream activation of NFKB (nuclear factor of kappa light polypeptide gene enhancer in B cells) and upregulation of inflammatory mediators, such as IL6 (interleukin 6), IL8, TNF (tumor necrosis factor), and IL1B/IL1β.5 Hence, LPS has been widely used as a clinically relevant model of ALI.6 Existing therapeutic strategies for ALI include reducing of bacterial load and reducing organ damage brought about by excessive inflammation. However, despite the improvements in critical care and mechanical ventilation protocols, the mortality of pneumonia or sepsis with ALI is still high, 37% or 43%, respectively,1 and thus novel approaches for ALI prevention are necessitated.
The protein kinase MTOR (mechanistic target of rapamycin [serine/threonine kinase]) and one of its major downstream processes, autophagy, are central regulators of cellular responses to organism growth and homeostasis.7 MTOR is well-defined as belonging to 2 distinct complexes named MTOR complex 1 (MTORC1) and MTORC2.7 Accumulating evidence suggests MTORC1 activation is associated with various signaling pathways including the downstream target RPS6 (ribosomal protein S6), and inflammatory responses.8 In addition, MTORC1 is the major negative regulator of autophagy.7 Autophagy is an evolutionarily conserved cellular process that influences many aspects of innate and adaptive immunity in almost all human cell types,9,10 and this process controls inflammation through regulatory interactions with innate immune signaling pathways.11 However, as recognized by many scientists,12,13 the role of MTOR and autophagy in disease pathogenesis including ALI is extremely cell and pathogen dependent. Thus, the functions and the underlying mechanisms by which MTOR and autophagy interrelate in ALI pathogenesis, especially in the pulmonary epithelial cells, have yet to be fully understood.
In this study, we demonstrate that activation of MTOR in lung epithelial cells is essential for LPS-induced ALI in vivo. LPS activates MTOR and decreased autophagic marker MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β)-II in pulmonary epithelium via the TLR4 (toll-like receptor 4) pathway, and the MTOR-autophagy axis eventually orchestrates LPS-induced NFKB activation and subsequent induction of inflammatory cytokines, thereby playing pivotal roles in ALI pathogenesis.
Results
LPS activates MTOR and decreases the autophagic protein LC3B in mouse airway epithelium and in human bronchial epithelial cells
To determine if MTOR and autophagy were biologically modulated in airway epithelial cells in LPS-induced ALI, we examined the expression of MTOR and related molecules in vivo and in vitro. As shown in Figure 1A, the expression of phosphorylated (p)-MTOR and p-RPS6 were significantly increased in lung homogenates after LPS exposure, while the autophagy-related hallmark LC3B was markedly downregulated (Fig. 1A). Histological analyses further revealed that the activation of MTOR, as indicated by the expression of p-RPS6, was predominant in both bronchial and alveolar epithelial cells upon LPS treatment (Fig. 1B, upper panels). In contrast, the expression of the autophagy-related protein LC3B was significantly decreased in those epithelial cells (Fig. 1B, lower panels). In human bronchial epithelial (HBE) cells, we also observed that the levels of p-MTOR and p-RPS6 were gradually increased, while LC3B expression was again suppressed in a time-dependent manner upon LPS exposure (Fig. 1C). Interestingly, in the presence of bafilomycin A1 (Baf A1), a widely used lysosome inhibitor that blocks the fusion of autophagosomes with lysosomes, LPS could still decrease the levels of LC3B (Fig. 1D), suggesting that LPS treatment indeed decreased the basal levels of autophagy rather than induced a high degradation capacity in HBE cells. These findings imply that LPS activates MTOR and may suppress autophagy in both airway and alveolar epithelial cells.
Figure 1.
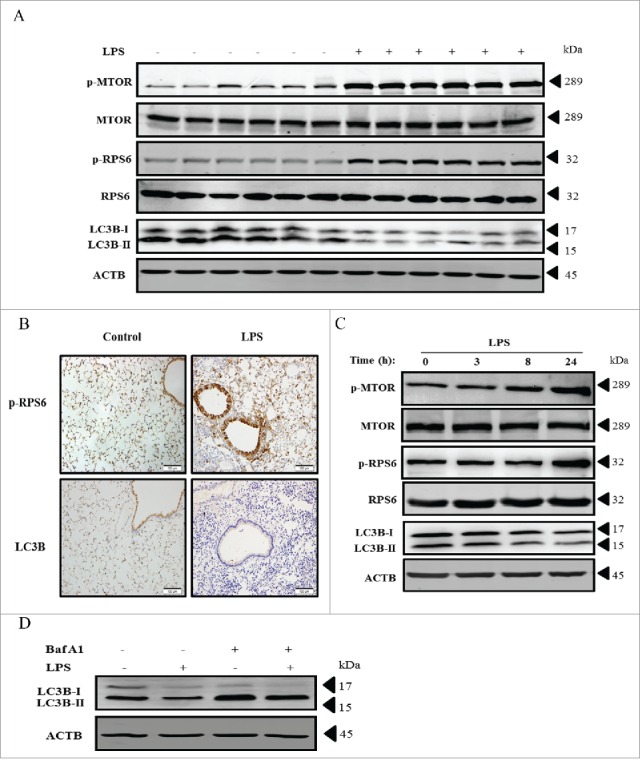
LPS activates MTOR and decreases the autophagic protein LC3B in mouse airway epithelium and in HBE cells. (A) Protein levels of p-MTOR, MTOR, p-RPS6, RPS6 and LC3B in lung tissues of C57BL/6 mice at 24 h after LPS injection. (B) Representative immunohistochemistry images of p-RPS6 and LC3B in mouse lungs injected with or without LPS. Scale bars: 100 μm. (C) Time-dependent expression of p-MTOR, MTOR, p-RPS6, RPS6 and LC3B in HBE cells stimulated with LPS. (D) Levels of LC3B in HBE cells after LPS stimulation with or without Baf A1. Data are representative of 3 independent experiments. Baf A1, bafilomycin A1.
Knockdown of MTOR reduces LPS-induced inflammatory responses in HBE cells
We next sought to explore the function of MTOR in LPS-induced injury of airway epithelial cells. We utilized 3 different small interfering RNAs (siRNAs) of MTOR to rule out possible off-target effects, and all the MTOR-siRNAs worked effectively (Fig. S1A–C). Interestingly, in MTOR-siRNA treated HBE cells, the LPS-induced mRNA transcripts and secreted protein levels of IL6 and IL8 were remarkably downregulated (Fig. 2A–D). As expected, knockdown of MTOR eventually enhanced the basal and the LPS-decreased levels of LC3B (Fig. 2E). The results suggest that inhibition of MTOR attenuates the LPS-induced inflammatory response in airway epithelium, accompanying an induction of autophagy.
Figure 2.
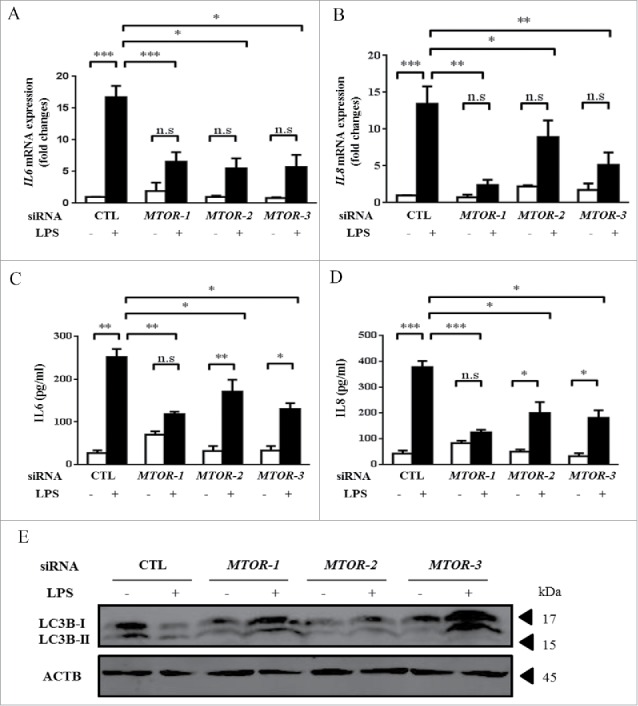
Knockdown of MTOR reduces LPS-induced inflammatory responses in HBE cells. Cells were transfected with the indicated siRNA for 24 h, and incubated with 100 μg/ml LPS for an additional 24 h. Cells were then harvested for analyzing the mRNA expression of IL6 (A) and IL8 (B) by quantitative real-time PCR. Cell culture supernatants were analyzed for IL6 (C) and IL8 (D) by ELISA. (E) The levels of LC3B were analyzed by western blot. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s, p > 0.05. Data are presented as mean ± SEM of 3 independent experiments. CTL, control.
Autophagic proteins suppress LPS-induced inflammation in HBE cells
As shown above, LPS activated MTOR and eventually decreased autophagy (Fig. 1), and MTOR-siRNA enhanced autophagy (Fig. 2). Also, autophagy plays an important role in infection, inflammation and immunity.9,10 We thus would like to examine whether autophagy exerts any effects on the LPS-induced inflammatory response in HBE cells. We utilized siRNAs or cDNAs of ATG5 (autophagy related 5) and LC3B, and the effects of knockdown or overexpression were demonstrated (Fig. S1D-G). Consistent with the role of MTOR, genetic knockdown of autophagy-related proteins ATG5 or LC3B, or pharmacological inhibition of autophagy by Baf A1 or chloroquine (CQ), further augmented the levels of IL6 and IL8 induced by LPS infection (Fig. 3A–D). Conversely, these pro-inflammatory cytokines were significantly reduced by overexpression of ATG5 or LC3B in LPS-treated HBE cells (Fig. 3E-F).
Figure 3.
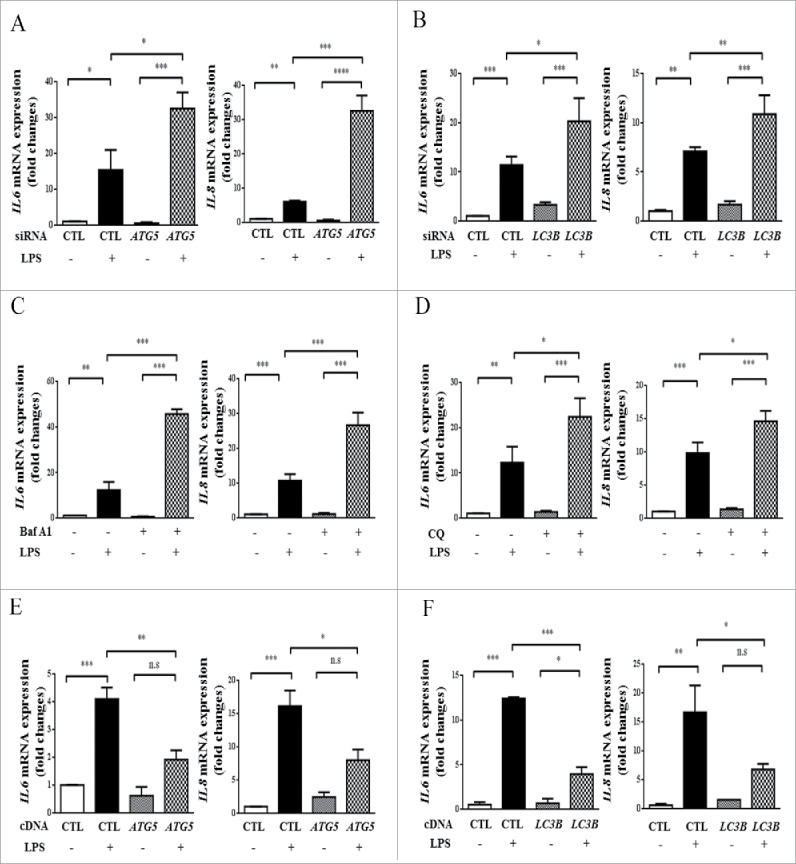
Autophagic proteins suppress LPS-induced inflammation in HBE cells. ((A)-B) Cells were transfected with the indicated siRNA for 24 h, and incubated with 100 μg/ml LPS for an additional 24 h. The mRNA levels of IL6 and IL8 were assessed by quantitative real-time PCR assays. (C-D) During LPS stimulation, HBE cells were co-cultured with or without Baf A1 (C) or CQ (D) and the mRNA levels of IL6 and IL8 were examined. (E-F) HBE cells were transfected with CTL-, ATG5- (E), or LC3B-expressing (F) plasmid, and then were treated with LPS for 24 h to detect the mRNA levels of IL6 or IL8. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s, p > 0.05. Data are presented as mean ± SEM of 3 independent experiments. CTL, control.
LPS activates MTOR via upstream TLR4 (toll-like receptor 4) in HBE cells
We next sought to examine the upstream signals that orchestrate LPS-induced MTOR activation in HBE cells. TLR4 is a well-recognized membrane receptor for LPS and it is also abundantly expressed in epithelial cells;14-16 we therefore attempted to investigate the role of TLR4 signals in regulation of LPS-induced MTOR activation. As expected, the treatment with LPS upregulated the levels of TLR4 and MYD88 in HBE cells (Fig. 4A). Moreover, genetic knockdown of TLR4 or MYD88 by siRNAs effectively decreased the LPS-induced MTOR activation and subsequently enhanced LC3B expression (Fig. 4B). More autophagic vacuoles (AVs) were observed in HBE cells knocked down for TLR4 or MYD88, especially in cells with LPS treatment (Fig. 4C-D). Not surprisingly, the siRNAs of TLR4 and MYD88 markedly reduced the LPS-induced production of IL6 and IL8 (Fig. 4E-F). The knockdown effects of TLR4 or MYD88 are shown in Fig. S1 (H-I).
Figure 4.
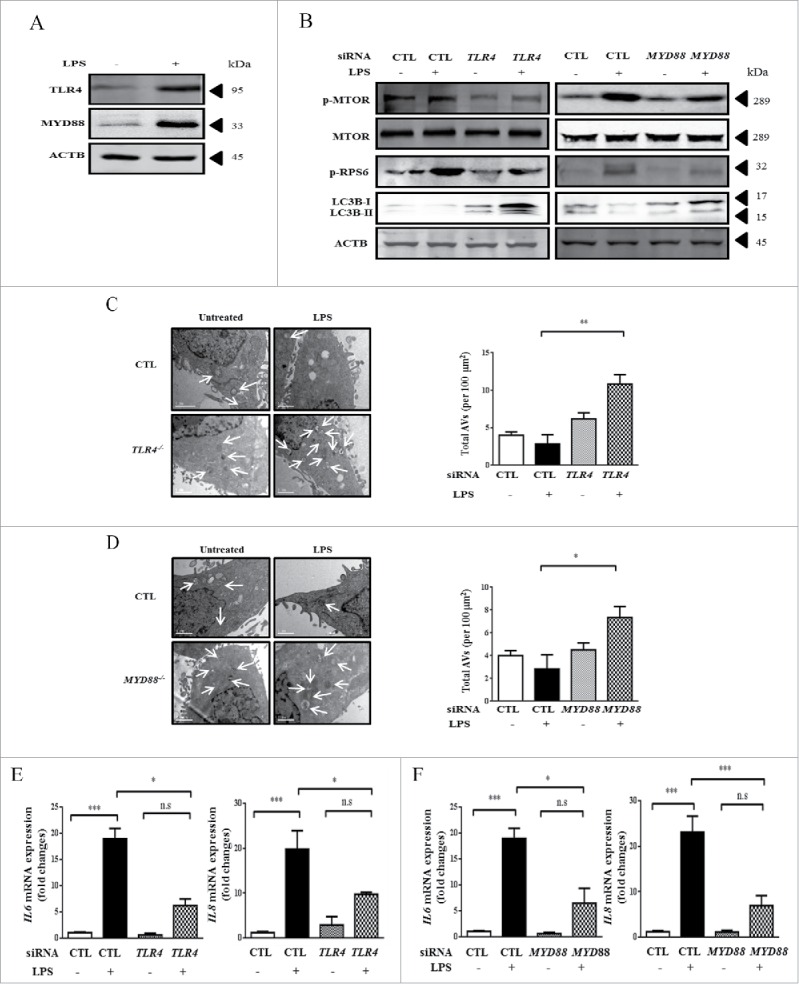
LPS activates MTOR via upstream TLR4-MYD88 signaling in HBE cells. (A) The expression of TLR4 and MYD88 in HBE cells after LPS stimulation for 24 h. (B-F) Cells were transfected with indicated the siRNA for 24 h, and incubated with 100 μg/ml LPS for an additional 24 h. The levels of MTOR and autophagy-related proteins were assessed by western blot (B). TEM images and semiquantified levels of AVs in HBE cells with knockdown of TLR4 (C) or MYD88 (D) and stimulated with LPS at 100 μg/ml for 24 h. AVs were marked by arrows. Scale bars: 1 μm. The mRNA expression of IL6 and IL8 (E-F) were analyzed by quantitative real-time PCR. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s, p > 0.05. Data are presented as mean ± SEM of 3 independent experiments. CTL, control.
MTOR promotes LPS-induced inflammatory cytokines through the NFKB pathway in HBE cells
We then explored the downstream pathways that mediate the role of MTOR in regulation of LPS-induced inflammatory cytokines. Not surprisingly, LPS activated NFKB signaling (Fig. 5A), and genetic knockdown of RELA (RELA proto-oncogene, NF-kB subunit) decreased the LPS-induced pro-inflammatory cytokines (Fig. 5B). The knockdown effects of RELA are shown in Fig. S1J. We next investigated the effect of MTOR inhibition on LPS-induced activation of NFKB. As expected, the levels of p-RELA were significantly reduced by MTOR siRNA in pulmonary epithelial cells treated with LPS (Fig. 5C). Conversely, the basal expression and phosphorylation of RELA in LPS-stimulated epithelial cells were notably increased by ATG5- or LC3B-siRNAs (Fig. 5D-E). These data suggest that MTOR positively, and autophagy negatively, regulates NFKB activation in HBE cells.
Figure 5.
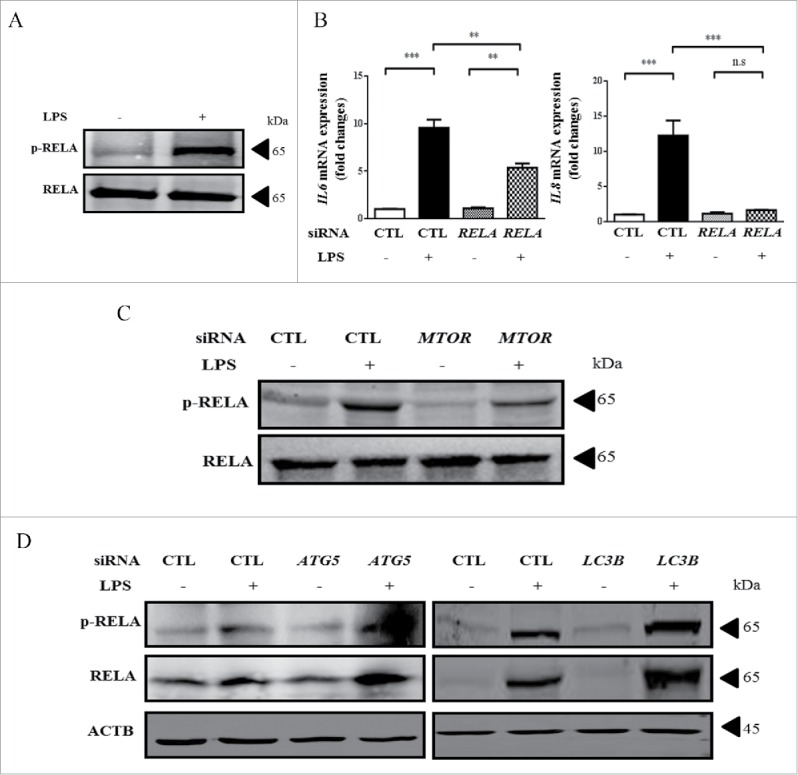
MTOR promotes LPS-induced inflammatory cytokines through the downstream NFKB pathway in HBE cells. (A) LPS induced an increase of p-RELA in HBE cells. (B) HBE cells were transfected with control (CTL-) or RELA-siRNAs for 24 h, and infected with LPS for an additional 24 h. The mRNA levels of IL6 and IL8 were quantified by quantitative real-time PCR assays. (C-D) Cells were transfected with the indicated siRNA for 24 h, and incubated with 100 μg/ml LPS for an additional 24 h. Levels of p-RELA and RELA were examined by western blot. **, p < 0.01; ***, p < 0.001; n.s, p > 0.05. Data are presented as mean ± SEM of 3 independent experiments. CTL, control.
Selective disruption of Mtor in pulmonary epithelium attenuated LPS-induced acute lung injury
To further confirm the function of MTOR activation in lung epithelium in the regulation of LPS-induced ALI pathogenesis in vivo, we attempted to conditionally deplete Mtor in alveolar or airway epithelial cells. Sftpc/SP-C-rtTA/(tetO)7-Cre/Mtorflox/flox and Scgb1a1/CC10-rtTA/(tetO)7-Cre/Mtorflox/flox mice were generated as described in Materials and Methods (Fig. S2A–B).17,18 After doxycycline treatment, the levels of MTOR were significantly decreased in lung tissues as detected by western blot (Fig. S2C). Moreover, the immune-staining of p-RPS6 revealed that MTOR activity was markedly decreased in both alveolar and airway epithelial cells (Fig. S2D-E).
LPS-induced lung inflammation was limited by selective disruption of Mtor in the alveolar epithelium, as demonstrated by a dramatic reduction in pro-inflammatory mediators IL6, CXCL1 (chemokine [C-X-C motif] ligand 1), and CXCL2 (chemokine [C-X-C motif] ligand 2) in bronchoalvoelar lavage fluid (BALF) (Fig. 6A). This protective effect was further confirmed by the reduced BALF protein, lung edema and pulmonary microvascular permeability (Fig. 6B-D). As expected, mice with impaired Mtor in the alveolar epithelium exhibited significantly higher survival rates relative to the control group after LPS administration (Fig. 6E). Moreover, histological analyses further revealed that LPS-induced pulmonary inflammation was significantly reduced in alveolar epithelium-specific mtorΔ/Δ mice (Fig. 6F–H). Consistent with the in vitro data, LPS also led to a significant increase in LC3B in lung homogenates of mice with selective disruption of Mtor in alveolar epithelium (Fig. 6I).
Figure 6.

Selective disruption of Mtor in alveolar epithelium attenuates LPS-induced ALI in mice. AT-II-mtorΔ/Δ mice (defined as mice with specific knockdown of Mtor in alveolar type-II epithelial cells) were generated as described in Materials and Methods. (A) The expression of IL6, CXCL1 and CXCL2 in BALF. (B) BALF protein, lung edema (C), alveolar-capillary permeability (D), and the survival rate (E) of mice after intratracheal administration of LPS for 24 h. (F) Representative images of H&E staining of lung tissue. Scale bars: 100 μm. Numbers of alveoli (G) and crowded area (H) of mouse lung sections 24 h following LPS challenge. (I) The levels of p-RPS6, RPS6 and LC3B were examined by western blot.*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s, p > 0.05. Data are presented as mean ± SEM of 3 independent experiments.
A similar protective effect was observed in airway epithelium-specific mtorΔ/Δ mice (Fig. 7). LPS-induced production of pro-inflammatory cytokines was significantly decreased in these mice (Fig. 7A). In addition, BALF protein, wet:dry ratio, and alveolar-capillary permeability were also reduced in these airway epithelium-specific mtorΔ/Δ mice at 24 h after LPS administration (Fig. 7B-D), although survival rate of the mice was only slightly improved (Fig. 7E). Not surprisingly, lung pathological changes caused by LPS were also notably recovered by the loss of Mtor in the airway epithelium (Fig. 7F-H). Furthermore, the expression of LC3B was again markedly upregulated in these specific mtorΔ/Δ mice when exposed to LPS (Fig. 7I).
Figure 7.

Conditional knockdown of Mtor in airway epithelium alleviates LPS-induced ALI in mice. AE-mtorΔ/Δ mice (defined as mice with specific knockdown of Mtor in airway epithelial cells) were generated as described in Materials and Methods. (A) The expression of IL6, CXCL1 and CXCL2 in BALF. (B) BALF protein, lung edema (C), alveolar-capillary permeability (D), and the survival rate (E) of mice after intratracheal administration of LPS for 24 h. (F) Representative images of H&E staining of lung tissue. Scale bars: 100 μm. Numbers of alveoli (G) and crowded area (H) of mouse lung sections 24 h following LPS challenge. (I) The levels of p-RPS6, RPS6 and LC3B were assessed by western blot.*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s, p > 0.05. Data are presented as mean ± SEM of 3 independent experiments.
Rapamycin ameliorated LPS-induced ALI in vivo
To further suggest a possible pharmaceutical approach for ALI, we investigated the effect of rapamycin (Rapa) on LPS-induced ALI in mice. As expected, Rapa treatment decreased the LPS-induced MTOR activity in the lungs, as revealed by the levels of p-RPS6 (Fig. S3A). Consistent with the genetic approaches, Rapa treatment also notably ameliorated LPS-induced inflammation, lung edema, and pulmonary microvascular permeability, although it only slightly improved the survival rate (Fig. S3B-H), suggesting a possible application of this compound for ALI.
Discussion
The major findings of this study can be summarized as follows: 1) LPS activated MTOR and suppressed autophagy, which orchestrated the LPS-induced inflammatory responses in the pulmonary epithelium; 2) the mechanisms of the autophagy axis in regulation of inflammatory responses in pulmonary epithelial cells were via the upstream TLR4-MYD88 pathway and downstream NFKB signaling; 3) selective disruption of Mtor in lung epithelium significantly attenuated LPS-induced ALI in vivo.
A growing body of literature has explored the function of autophagy in diverse animal models of ALI, showing that autophagy plays different roles upon various stimuli and in different cell types. The role of autophagy in macrophages in the context of ALI appears to be protective, as demonstrated by a number of studies.11,19-21 Mice lacking Atg7 or Atg5 in myeloid cells spontaneously develop pulmonary inflammation.22,23 However, the functions of autophagy in lung epithelial cells in ALI pathogenesis remain controversial. For instance, several studies indicated a deleterious role of autophagy in ALI. Li et al. have observed that nanoparticles trigger autophagic cell death by deregulating the AKT-TSC2-MTOR signaling pathway in airway epithelial cells, and the inhibition of autophagy rescues the nanoparticle-induced epithelial cell death and ameliorates ALI in mice.24 Several studies25-27 have also reported that H5N1 infection induces autophagic cell death or inflammatory response in lung epithelial cells, and blocking autophagy not only reduces the epithelial cell death, but also ameliorates H5N1-induced ALI and mortality. In the models of lung injury induced by cigarette smoke28,29 or environmental particulate matter,30 autophagy also appears to mediate the epithelial cell death, inflammatory response, and mucus hyperproduction. In contrast, 2 recent studies have demonstrated that the autophagic protein LC3B exerts a protective role against hyperoxia-induced lung epithelial injury and cell death.31,32 Our current results are consistent with these latter studies, demonstrating that autophagy suppresses LPS-induced inflammatory response in HBE cells. The detailed mechanisms as to why autophagy can be either cytoprotective or deleterious in epithelial injury remain unclear and need further investigation.
It should be noted that in a few studies LPS has been shown to induce autophagy in airway epithelial cells.33,34 Currently it is not clear why such a difference in findings exists. One plausible explanation is that the induction of autophagy in lung disease pathogenesis is exactly cell type and pathogen dependent. Given the facts that these studies used different types of cells and likely different sources of LPS, such inconsistencies might be rational. Nevertheless, even in the work showing that LPS triggers autophagy, mice lacking Atg4b exhibit increased inflammation and mortality after endotoxemia,33 which is in complete agreement with our conclusion that MTOR inactivation and subsequent autophagy induction protect against LPS-induced ALI.
MTOR is considered as a major negative regulator of autophagy although detailed mechanisms underlying this effect are only partially understood.35-37 However, accumulating evidence indicates that MTOR might not be the sole regulator of autophagy. For example, autophagy activation can be attributable to an increase in glutamine-derived ammonia, but this occurs via in an MTOR-independent pathway.38 Our data showed that following LPS exposure, MTOR was activated while the autophagy protein LC3B was downregulated both in mouse lung tissues and in HBE cells. Upon knockdown of TLR4 and MYD88, the reduction of MTOR accompanied an upregulation of LC3B. Moreover, the functions of MTOR and those dependent on ATG5 and LC3B were exactly opposite in regulation of LPS-induced inflammation in HBE cells. Thus, these results together suggested that in airway epithelial cells LPS-induced autophagy degradation was most likely regulated by MTOR.
The eventual functions of MTOR in ALI remain largely unknown. Most of the available studies only investigate the role of MTOR in the context of ALI in vitro, or just use MTOR inhibitors in vivo.24,27,39-45 Actually, most MTOR-related studies in ALI models per se focus on autophagy, taking MTOR as a pivotal upstream signal in the regulation of autophagy. For example, nanoparticles induce autophagic cell death by deregulating the AKT-TSC2-MTOR signaling pathway,24 and H5N1 infection also causes autophagy-mediated cell death in lung epithelial cells through suppression of MTOR.27 To the best of our knowledge, our study was the first to use an Mtor-specific knockout approach to investigate its functions in ALI, or even in pulmonary diseases. Our in vivo data demonstrated that epithelial MTOR activation was essential for LPS-induced ALI, and suggested that targeting MTOR in lung epithelial cells could potentially be an effective therapeutic strategy for ALI induced by bacteria infections. Also, Rapa notably inhibited inflammation and alleviated lung injury after LPS infection in vivo (Fig S3), and such a beneficial effect is likely through downregulation of MTOR in the lung. However, in some other lung disorders or disease models, Rapa can cause airway inflammation and lymphocytic pneumonitis.46-48 This inconsistency again further supports the conclusion that functions of MTOR and autophagy in disease pathogenesis are cell specific and pathogen dependent.
TLR4 regulates innate immunity or inflammatory processes, which can be expressed in endothelial cells, epithelial cells, monocytes and macrophages.49-52 TLR4 plays a crucial role in the recognition of Gram-negative bacteria following LPS stimulation.53 The MYD88-dependent and -independent signaling intermediates are downstream transduction pathways of TLR4,54 which lead to the activation of the transcription factor NFKB and upregulation of inflammatory mediators. The regulative relationship between TLR4 and the MTOR pathway has been investigated in a few studies. For example, MTOR activation has been essential for TLR4-mediated neutrophil41 and macrophage activation.55 Choi and colleagues56 have also observed that the TLR4 activity participates in autophagic degradation after LPS stimulation, which is in accordance with our current findings. However, some studies also demonstrated that LPS can induce cytokine expression in TLR4-independent pathways. For instance, IL1RN expression is induced via EIF2AK2 (eukaryotic translation initiation factor 2-α kinase 2)-dependent signaling,57 and secretion of proinflammatory cytokines partially results from phosphoinositide 3-kinase/PI3K or TRAF2-MAP3K5/ASK1-MAPK/JNK signaling pathways by LPS stimulation.58,59 In our study, MTOR phosphorylation was downregulated when TLR4- or MYD88-siRNA was given in HBE cells, suggesting that MTOR activation and subsequent inflammation were dependent on the TLR4 signaling pathway following LPS exposure.
MTOR and autophagy have been implicated in the processes of acute lung injury, while the detailed mechanisms for how MTOR and autophagy regulate inflammatory responses remain largely unknown. The effects of autophagic flux in the regulation of NFKB activities are complex, and the molecular mechanisms are extremely complicated, and likely vary depending on cell types and stimuli. NFKBIA, RELA, as well as other NFKB-related molecules, can all be targets for degradation by autophagy, resulting in completely different outcomes. Of note, some mechanistic studies are consistent with our current results showing MTOR acts positively, while autophagy acts negatively, in modulating RELA phosphorylation. For example, recent studies by 2 independent groups have shown that Rapa inhibits phosphorylation of RELA in LPS-stimulated neutrophils.41,60 Conversely, López-Alonso and colleagues have demonstrated that impairment of autophagy results in the accumulation of SQSTM1 (sequestosome 1) and ubiquitinated NFKBIA, and less NFKB activation, eventually ameliorating ventilator-induced inflammatory response and lung injury.61 Moreover, NFKB signaling can in turn modulate autophagy. For instance, it has been reported that activation of NFKB promotes autophagosome formation and autophagy-mediated inflammatory responses in H5N1 pseudotyped particle-induced lung inflammation.25 Nevertheless, further investigations are needed to fully understand the correlation between the MTOR-autophagy pathway and NFKB pathway in pulmonary epithelial cells and other cell types in ALI pathogenesis.
In summary, our present study demonstrates that activation of MTOR and subsequent inactivation of autophagy in lung epithelium are essential for LPS-induced inflammatory responses in vitro and ALI pathogenesis in vivo. Classical TLR4-MYD88 and NFKB pathways serve in critical upstream and downstream signaling, respectively, in mediating the functions of the MTOR-autophagy axis in ALI pathogenesis. Thus, inhibition of MTOR or activation of autophagy in pulmonary epithelial cells may represent novel therapeutic strategies for preventing ALI induced by bacteria.
Materials and methods
Chemicals and reagents
Antibodies against p-MTOR (Cell Signaling Technology, 5536), MTOR (Cell Signaling Technology, 2972), RPS6 (Cell Signaling Technology, 2217), p-RPS6 (Cell Signaling Technology, 4858), LC3B (Abcam, ab168831), ATG5 (Santa Cruz Biotechnology, sc-515347), RELA (Cell Signaling Technology, 8242), p-RELA (Cell Signaling Technology, 4812), SFTPC/SP-C (Santa Cruz Biotechnology, sc-7705), SCGB1A1/CC10 (Santa Cruz Biotechnology, sc-9772), and ACTB (Cell Signaling Technology, 3700) were used. Trizol reagents (9109) were purchased from Takara. Phosphate-buffered saline (PBS) was ordered from Biotopped (150814). Lipopolysaccharide (L2630) and doxycycline (D9891) were obtained from Sigma-Aldrich. Rapa was from Selleck (s1039). The small interfering RNAs (siRNAs) of control (sc-37007), TLR4 (sc-40260), MYD88 (sc-35986), LC3B (sc-43390), ATG5 (sc-41445), and RELA (sc-29410) were purchased from Santa Cruz Biotechnology. Different MTOR siRNAs (SR301656) were purchased from Origene. Baf A1 (B1793) and CQ (C6628) were from Sigma-Aldrich, and the final concentrations in the culture medium were 10 nM and 15 nM, respectively.
Mice
Sftpc-rtTAtg/− or (tetO)7CMV-Cretg/− transgenic mice (C57BL/6 background) were provided by Yuehai Ke (Zhejiang University School of Medicine). Mtorflox/flox mice (C57BL/6 background) were purchased from the Jackson laboratory (011009). Sftpc-rtTAtg/− or (tetO)7CMV-Cretg/− transgenic mice were mated with Mtorflox/flox mice to generate double-transgenic Sftpc-rtTA/Mtorflox/flox or (tetO)7-Cre/Mtorflox/flox mice. Then, these mice were mated to generate Sftpc-rtTA/(tetO)7-Cre/Mtorflox/flox triple-transgenic mice. Triple transgenic mice and littermate control mice (Sftpc-rtTA/Mtorflox/flox, (tetO)7-Cre/Mtorflox/flox and Mtorflox/flox) were used for the experiments. The Scgb1a1-rtTA/(tetO)7 -Cre/Mtorflox/flox transgenic mice were generated following the same method as above. We extracted genomic DNA from tails or lungs for genotyping. PCR primers are shown in Table 1.
Table 1.
Primers used for PCR experiments.
| Gene | Primer sequence (5′–3′) |
|---|---|
| Sftpc-rtTA | Forward primers: GACACATATAAGACCCTGGTCA Reverse primers: AAAATCTTGCCAGCTTTCCCC |
| Scgb1a1-rtTA | Forward primers: AAAATCTTGCCAGCTTTCCCC Reverse primers: ACTGCCCATTGCCCAAACAC |
| (tetO)7CMV-Cre | Forward primers: TGCCACGACCAAGTGACAGCAATG Reverse primers: AGAGACGGAAATCCATCGCTCG |
| Mtorflox/flox | Forward primers: TTATGTTTGATAATTGCAGTTTTGGCTAGCAGT Reverse primers: TCCTTTCCAGGTCAGTTA |
To induce the activation of Cre recombinase in transgenic mice, 6- to 8-wk-old mice were fed with doxycycline in their drinking water (2 mg/ml) for 20 d. After confirming the knockdown effect, the Sftpc-rtTA/(tetO)7-Cre/Mtorflox/flox and Scgb1a1-rtTA/(tetO)7-Cre/Mtorflox/flox mice treated with DOX were then designated as AT-II-mtorΔ/Δ or AE-mtorΔ/Δ mice, respectively.
C57BL/6 mice (18–20 g) were purchased from the Animal Center of Slaccas (Shanghai, China) and maintained in the animal facility of the laboratory animal center of Zhejiang University. All animal experiments were in accordance with the “Guide for the care and use of laboratory animals,” and were approved by the Animal Care and Use Committee at Zhejiang University.
ALI mouse model
Mice were injected intraperitoneally with 1.5% pentobarbital in PBS (90 mg/kg), and then the LPS (2 mg/50 μl) or PBS was injected into the trachea with a microsyringe. After intratracheal instillation, the mice were kept vertical for 1 min to ensure the distribution of the LPS or PBS in the lungs. Mice were sacrificed 24 h after LPS injection.
Rapamycin experiments
For in vivo Rapa experiments, we used a protocol based on intraperitoneal instillation of Rapa (1 mg/kg) or vehicle (1.8% DMSO) to wild-type mice for 7 d before LPS was administered.47
Cell culture
HBE cells were purchased from American Type Culture Collection (CRL-2741), and were cultured in RPMI 1640 (Gibco, C11875500BT) with 10% fetal bovine serum (Gibco, 10082147) in 5% CO2 at 37˚C. After LPS (100 μg/ml) stimulation for 24 h, cells were subjected to further analysis.
siRNA preparation and transfection
The cells were seeded in a 6-well plate for 24 h. Then the siRNA was transfected into HBE cells using the siRNA transfection reagent (SignaGen Laboratories, SL100568) following the manufacturer's instructions.
cDNA constructs and transfection
The recombinant pCMV6-LC3B (Origene, SC108102) and EGFP-ATG5 (Addgene, 22952, deposited by Noboru Mizushima) plasmids were used, and were prepared for later experiments using the EndoFree Plasmid Maxi Kit (Biomiga, PD1514-01). The HBE cells were transfected with plasmid using the PolyJet in vitro DNA Transfection reagent (SignaGen Laboratories, SL100688) following the manufacturer's protocol.
Transmission electron microscopy
After LPS stimulation for 24 h, cells were fixed in 2.5% glutaradehyde. The samples were prepared following standard methods and using a TECNA1 10 transmission electron microscope (FEI, Hillsboro, OR, USA) for the image analysis. To quantify the alteration of the number of AVs, the area of the cell cytoplasm was measured by using Image-Pro Plus 6.0. The data were represented as AVs per 100 μm2.
RNA isolation and RT-PCR
RNA of lungs and cells was extracted with Trizol reagent. After isolation, the quality of the RNA was assessed with the NanoDrop2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA) according to the manufacturer's instructions. The 260/280 absorbance ratios of 1.8–2.0 indicated a pure RNA sample.
Real-time RT-PCR was performed using the StepOnePlus PCR system and gene expression assays (Applied Biosystems, Foster City, CA, USA). A ΔCt method was used to quantify mRNA levels for IL6 and IL8, etc. Gene expression was quantified using ABI and MS Excel software. Each sample was assayed in triplicate. Real-time RT-PCR-specific primers as shown in Table 2 were used to evaluate gene expression.
Table 2.
Primers used for QRT-PCR experiments.
| Species | Gene | Primer sequence (5′–3′) |
|---|---|---|
| Human | ACTB | Forward: AGAGGGAAATCGTGCGTGAC Reverse: CAATAGTGATGACCTGGCCGT |
| Human | IL6 | Forward: ACTCACCTCTTCAGAACGAATTG Reverse: CCATCTTTGGAAGGTTCAGGTTG |
| Human | IL8 | Forward: CACCATGACTTCCAAGCTGGC Reverse: TTATGAATTCTCAGCCCTCTTC |
ELISA
BALF and cell culture medium samples were centrifuged at 400 g for 10 min, and the supernatant fractions were collected and stored at −80°C. Inflammatory protein CXCL1 (MKC00B), CXCL2 (MM200), IL6 (D6050, M6000B), or IL8 (D8000C) in the supernatants were measured by ELISA (R&D Systems), according to the manufacturer's instructions. BALF protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, 23227).
Western blot analysis
Protein was extracted from cells or lung tissues using RIPA buffer (Beyotime, P0013C) with protease and phosphatase inhibitors (Roche Diagnostics GmbH, 04906845001). Samples were electrophoresed through 6–15% polyacrylamide gels and immunoblotted with the relevant antibodies using standard methods.
Histological assessment
Twenty-four h after LPS infection, lungs of mice were inflated, gravity fixed (20 cm) with 4% paraformaldehyde, and paraffin embedded. Sections (5 μm) were sliced for haematoxylin and eosin (H&E) staining, immunohistochemistry (IHC) and immunofluorescence (IF). The sections were visualized using an Olympus BX53 inverted microscope (Olympus, Melville, NY) and Zeiss LSM confocal laser scanning microscope (Carl Zeiss, GÖttingen, Germany).
The average values were taken as a modified semi-quantitative histological index of lung injury using the following criteria: complete alveolar numbers of alveolar sacs were analyzed and 3 randomly selected high-power fields (HPFs) (100×) were examined in each section. The pulmonary crowded area was defined as the area of lung parenchyma and stroma possession, crowded area/per high-power field (100×): 0, no detectable crowded area; +1, crowded area less than 15%; +2, crowded area involving 16%–25%; +3, crowded area involving 26%–50%; +4, crowded area involving 51%–75% of lung; +5, crowded area involving more than 76% of the lung.62
Mouse lung wet/dry ratio assay
Twenty-four h after intratracheal instillation of LPS, mice were killed and the lungs after removal of excess blood were weighed for the wet weight measurement. The lungs then dried in an oven at 60°C for 72 h for dry weight measurement.
Mouse alveolar-capillary leakage assay
Twenty-two h after LPS intratracheal injection, the mice were injected with 20 mg/kg Evans blue solution (Sigma-Aldrich, E2129) by the tail vein. Two h later, the mice were exsanguinated through the heart with saline. Then the lungs were removed and placed in 100 mg/ml formamide (BIOSHARP, BS335A). The tissues were incubated at 60°C for 24 h and the absorbance of formamide was measured at 620 nm.
Survival rate
Mice were treated intratracheally with LPS. The survival percentages and body weights in each group were monitored daily for 96 h. Body weight with a decrease resulting in 20% or less of the initial weight was considered equivalent to death.
Statistical analysis
All results were expressed as mean ± SEM. Comparisons between 2 groups were done with an unpaired t test. A one-way ANOVA followed by Bonferroni correction for multiple-comparison testing was implemented. For analysis of survival rate, Log-rank (Mantel-Cox) Test was performed. Data were considered statistically significant if p < 0.05. Statistical analysis was performed using the Prism software program for Windows (GraphPad software, San Diego, CA, USA).
Supplementary Material
Abbreviations
- Ab
antibody
- ALI
acute lung injury
- AT-II
alveolar type-II
- AE
airway epithelial
- ATG
autophagy-related
- ACTB
actin, β
- AVs
autophagic vacuoles
- BALF
bronchoalveolar lavage fluid
- Baf A1
bafilomycin A1
- BCA
bicinchoninic acid
- CTL
control
- CQ
chloroquine
- CXCL1
chemokine (C-X-C motif) ligand 1
- CXCL2
chemokine (C-X-C motif) ligand 2
- DOX
doxycycline
- HBE
human bronchial epithelial
- H&E
hematoxylin and eosin
- IF
immunofluorescence
- i.t.
intratracheal instillation
- IHC
immunohistochemistry
- IL
interleukin
- LH
lung homogenate
- LPS
lipopolysaccharide
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- MTORC
MTOR complex
- MYD88
myeloid differentiation primary response gene 88
- NFKB
nuclear factor of kappa light polypeptide gene enhancer in B cells
- NFKBIA
nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, α
- Rapa
rapamycin
- RELA
RELA proto-oncogene, NF-kB subunit
- RPS6
ribosomal protein S6
- siRNA
small interfering RNA
- TLR4
toll-like receptor 4
- TNF
tumor necrosis factor
Disclosure of potential conflicts of interest
There were no potential conflicts of interest to be disclosed.
Funding
This work was supported in part by Major, Key, and General Projects of the NSFC (81490532 and 81130001 to H.H.S., and 81370142 to Z.H.C.), the National Key Technologies R&D Program for the 12th Five-year Plan (2012BAI05B01), the Key Science-Technology Innovation Team of Zhejiang Province (2011R50016), and the program for Key Site of National Clinical Research Center for Respiratory Disease.
References
- [1].Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 2010; 23:243-52; PMID:20073554; http://dx.doi.org/ 10.1089/jamp.2009.0775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 2010; 298:L715-31; PMID:20363851; http://dx.doi.org/ 10.1152/ajplung.00361.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gropper MA, Wiener-Kronish J. The epithelium in acute lung injury acute respiratory. Curr Opin Crit Care 2008; 14:11-5; PMID:18195620; http://dx.doi.org/ 10.1097/MCC.0b013e3282f417a0 [DOI] [PubMed] [Google Scholar]
- [4].Tsushima K, King LS, Aggarwal NR, De Gorordo A, D'Alessio FR, Kubo K. Acute lung injury review. Intern Med 2009; 48:621-30; PMID:19420806; http://dx.doi.org/ 10.2169/internalmedicine.48.1741 [DOI] [PubMed] [Google Scholar]
- [5].Chaudhuri NWM, Sabroe I. Reducing the toll of inflammatory lung disease. Chest 2007; 131:1550; PMID:17494804; http://dx.doi.org/ 10.1378/chest.06-2869 [DOI] [PubMed] [Google Scholar]
- [6].Wang HM, Bodenstein M, Markstaller K. Overview of the pathology of three widely used animal models of acute lung injury. Eur Surg Res 2008; 40:305-16; PMID:18349543; http://dx.doi.org/ 10.1159/000121471 [DOI] [PubMed] [Google Scholar]
- [7].Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006; 124:471-84; PMID:16469695; http://dx.doi.org/ 10.1016/j.cell.2006.01.016 [DOI] [PubMed] [Google Scholar]
- [9].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual Rev Genetics 2009; 43:67-93; PMID:19653858; http://dx.doi.org/ 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/ 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al.. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222-30; PMID:21151103; http://dx.doi.org/ 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hu Y, Liu J, Wu YF, Lou J, Mao YY, Shen HH, Chen ZH. mTOR and autophagy in regulation of acute lung injury: a review and perspective. Microbes Infect 2014; 16:727-34; PMID:25084494; http://dx.doi.org/ 10.1016/j.micinf.2014.07.005 [DOI] [PubMed] [Google Scholar]
- [13].Mizumura K, Cloonan S, Choi ME, Hashimoto S, Nakahira K, Ryter SW, Choi AM. Autophagy: Friend or foe in lung disease? Ann Am Thorac Soc 2016; 13 Suppl 1:S40-7; PMID:27027951; http://dx.doi.org/ 10.1513/AnnalsATS.201508-529OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim Y, Lee EJ, Jang HK, Kim CH, Kim DG, Han JH, Park SM. Statin pretreatment inhibits the LPS-induced EMT via the downregulation of TLR4 and NF-kappaB in human biliary epithelial cells. J Gastroenterol Hepatol 2016; 31:1220-8; PMID:26574150; http://dx.doi.org/26507165 10.1111/jgh.13230 [DOI] [PubMed] [Google Scholar]
- [15].Jiang Q, Yi M, Guo Q, Wang C, Wang H, Meng S, Liu C, Fu Y, Ji H, Chen T. Protective effects of polydatin on lipopolysaccharide-induced acute lung injury through TLR4-MyD88-NF-kappaB pathway. Int Immunopharmacol 2015; 29:370-6; PMID:26507165; http://dx.doi.org/ 10.1016/j.intimp.2015.10.027 [DOI] [PubMed] [Google Scholar]
- [16].Ojo OO, Ryu MH, Jha A, Unruh H, Halayko AJ. High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 2015; 309:L1354-1366; PMID:26432865 [DOI] [PubMed] [Google Scholar]
- [17].Zhang X, Zhang Y, Tao B, Teng L, Li Y, Cao R, Gui Q, Ye M, Mou X, Cheng H, et al.. Loss of Shp2 in alveoli epithelia induces deregulated surfactant homeostasis, resulting in spontaneous pulmonary fibrosis. FASEB J 2012; 26:2338-50; PMID:22362894; http://dx.doi.org/ 10.1096/fj.11-200139 [DOI] [PubMed] [Google Scholar]
- [18].Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, et al.. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest 2012; 122:2032-45; PMID:22546858; http://dx.doi.org/ 10.1172/JCI60132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, Nakahira K, Ryter SW, Choi AM. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal 2014; 20:432-42; PMID:23971531; http://dx.doi.org/ 10.1089/ars.2013.5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chuang SY, Yang CH, Chou CC, Chiang YP, Chuang TH, Hsu LC. TLR-induced PAI-2 expression suppresses IL-1beta processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci U S A 2013; 110:16079-84; PMID:24043792; http://dx.doi.org/ 10.1073/pnas.1306556110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/ 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- [22].Kanayama M, He YW, Shinohara ML. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J Immunol 2015; 194:5465-71; PMID:25911758; http://dx.doi.org/ 10.4049/jimmunol.1403249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol 2015; 194:5407-16; PMID:25888640; http://dx.doi.org/ 10.4049/jimmunol.1402277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li C, Liu H, Sun Y, Wang H, Guo F, Rao S, Deng J, Zhang Y, Miao Y, Guo C, et al.. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol 2009; 1:37-45; PMID:19516051; http://dx.doi.org/ 10.1093/jmcb/mjp002 [DOI] [PubMed] [Google Scholar]
- [25].Pan H, Zhang Y, Luo Z, Li P, Liu L, Wang C, Wang H, Li H, Ma Y. Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-kappaB and p38 MAPK signaling pathways. Am J Physiol Lung Cell Mol Physiol 2013; 306:L183-195; PMID:24242010; http://dx.doi.org/ 10.1152/ajplung.00147.2013 [DOI] [PubMed] [Google Scholar]
- [26].Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, et al.. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 2012; 5:ra16; PMID:22355189; http://dx.doi.org/ 10.1126/scisignal.2001931 [DOI] [PubMed] [Google Scholar]
- [27].Ma J, Sun Q, Mi R, Zhang H. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J Genet Genomics 2011; 38:533e7; http://dx.doi.org/ 10.1016/j.jgg.2011.10.002 [DOI] [PubMed] [Google Scholar]
- [28].Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A 2010; 107:18880-5; PMID:20956295; http://dx.doi.org/ 10.1073/pnas.1005574107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou JS, Zhao Y, Zhou HB, Wang Y, Wu YF, Li ZY, Xuan NX, Zhang C, Hua W, Ying SM, et al.. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol 2016; 310(11):L1042-52; PMID:27036871; http://dx.doi.org/ 10.1152/ajplung.00418.2015 [DOI] [PubMed] [Google Scholar]
- [30].Chen ZH, Wu YF, Wang PL, Wu YP, Li ZY, Zhao Y, Zhou JS, Zhu C, Cao C, Mao YY, et al.. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy 2016; 12:297-311; PMID:26671423; http://dx.doi.org/ 10.1080/15548627.2015.1124224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia-induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol 2012; 46:507-14; PMID:22095627; http://dx.doi.org/ 10.1165/rcmb.2009-0415OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AM, Kim YS. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol 2011; 45:867-73; PMID:21441382; http://dx.doi.org/ 10.1165/rcmb.2010-0352OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Aguirre A, Lopez-Alonso I, Gonzalez-Lopez A, Amado-Rodriguez L, Batalla-Solis E, Astudillo A, Blázquez-Prieto J, Fernández AF, Galván JA, dos Santos CC, et al.. Defective autophagy impairs ATF3 activity and worsens lung injury during endotoxemia. J Mol Med 2014; 92:665-76; PMID:24535031; http://dx.doi.org/ 10.1007/s00109-014-1132-7 [DOI] [PubMed] [Google Scholar]
- [34].Li S, Guo L, Qian P, Zhao Y, Liu A, Ji F, Chen L, Wu X, Qian G. Lipopolysaccharide induces autophagic cell death through the PERK-dependent branch of the unfolded protein response in human alveolar epithelial A549 cells. Cell Physiol Biochem 2015; 36:2403-17; PMID:26279443; http://dx.doi.org/ 10.1159/000430202 [DOI] [PubMed] [Google Scholar]
- [35].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/ 10.1091/mbc.E08-12-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1 ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/ 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973-9; PMID:19597335; http://dx.doi.org/ 10.4161/auto.5.7.9296 [DOI] [PubMed] [Google Scholar]
- [38].Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal 2010; 3:ra31; PMID:20424262 [DOI] [PubMed] [Google Scholar]
- [39].Chen D, Jiao G, Ma T, Liu X, Yang C, Liu Z. The mechanism of rapamycin in the intervention of paraquat-induced acute lung injury in rats. Xenobiotica 2015; 45:538-46; PMID:25523308; http://dx.doi.org/ 10.3109/00498254.2014.995149 [DOI] [PubMed] [Google Scholar]
- [40].Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, Zhao Y, Wang YG, Zhang GG, Yang P, et al.. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis 2011; 2:e159; PMID:21593791; http://dx.doi.org/ 10.1038/cddis.2011.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lorne E, Zhao X, Zmijewski JW, Liu G, Park YJ, Tsuruta Y, Abraham E. Participation of mammalian target of rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol 2009; 41:237-45; PMID:19131641; http://dx.doi.org/ 10.1165/rcmb.2008-0290OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ma C, Zhu L, Wang J, He H, Chang X, Gao J, Shumin W, Yan T. Anti-inflammatory effects of water extract of Taraxacum mongolicum hand.-Mazz on lipopolysaccharide-induced inflammation in acute lung injury by suppressing PI3K/Akt/mTOR signaling pathway. J Ethnopharmacol 2015; 168:349-55; PMID:25861954; http://dx.doi.org/ 10.1016/j.jep.2015.03.068 [DOI] [PubMed] [Google Scholar]
- [43].Nadon AM, Perez MJ, Hernandez-Saavedra D, Smith LP, Yang Y, Sanders LA, Gandjeva A, Chabon J, Koyanagi DE, Graham BB, et al.. Rtp801 suppression of epithelial mTORC1 augments endotoxin-induced lung inflammation. Am J Pathol 2014; 184:2382-9; PMID:25016184; http://dx.doi.org/ 10.1016/j.ajpath.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ustun S, Lassnig C, Preitschopf A, Mikula M, Muller M, Hengstschlager M, Weichhart T. Effects of the mTOR inhibitor everolimus and the PI3K/mTOR inhibitor NVP-BEZ235 in murine acute lung injury models. Transpl Immunol 2015; 33:45-50; PMID:26073719; http://dx.doi.org/ 10.1016/j.trim.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang L, Gui YS, Tian XL, Cai BQ, Wang DT, Zhang D, Zhao H, Xu KF. Inactivation of mammalian target of rapamycin (mTOR) by rapamycin in a murine model of lipopolysaccharide-induced acute lung injury. Chin Med J (Engl) 2011; 124:3112-7; PMID:22040565 [PubMed] [Google Scholar]
- [46].Fujitani Y, Trifilieff A. In vivo and in vitro effects of SAR 943, a rapamycin analogue, on airway inflammation and remodeling. Am J Respir Crit Care Med 2003; 167:193-8; PMID:12406821; http://dx.doi.org/ 10.1164/rccm.200205-455OC [DOI] [PubMed] [Google Scholar]
- [47].Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, Chukwueke U, Mao T, Richter A, Brown E, et al.. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke – induced pulmonary injury and emphysema. Nat Med 2010; 16:767-73; PMID:20473305; http://dx.doi.org/ 10.1038/nm.2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Singer SJ, Tiernan R, Sullivan EJ. Interstitial pneumonitis associated with sirolimus therapy in renal-transplant recipients. N Engl J Med 2000; 343:1815-6; PMID:11185606 [DOI] [PubMed] [Google Scholar]
- [49].Sheu JJ, Chang LT, Chiang CH, Youssef AA, Wu CJ, Lee FY, Yip HK. Prognostic value of activated toll-like receptor-4 in monocytes following acute myocardial infarction. Int Heart J 2008; 49:1-11; PMID:18360060; http://dx.doi.org/ 10.1536/ihj.49.1 [DOI] [PubMed] [Google Scholar]
- [50].Jagavelu K, Routray C, Shergill U, O'Hara SP, Faubion W, Shah VH. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 2010; 52:590-601; PMID:20564354; http://dx.doi.org/ 10.1002/hep.23739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wei W, Xiao HT, Bao WR, Ma DL, Leung CH, Han XQ, Ko CH, Lau CB, Wong CK, Fung KP, et al.. TLR-4 may mediate signaling pathways of Astragalus polysaccharide RAP induced cytokine expression of RAW264.7 cells. J Ethnopharmacol 2016; 179:243-52.; PMID:26743224; http://dx.doi.org/ 10.1016/j.jep.2015.12.060 [DOI] [PubMed] [Google Scholar]
- [52].Go H, Koh J, Kim HS, Jeon YK, Chung DH. Expression of toll-like receptor 2 and 4 is increased in the respiratory epithelial cells of chronic idiopathic interstitial pneumonia patients. Respir Med 2014; 108:783-92; PMID:24613046; http://dx.doi.org/ 10.1016/j.rmed.2013.12.007 [DOI] [PubMed] [Google Scholar]
- [53].He X, Wei Z, Zhou E, Chen L, Kou J, Wang J, Yang Z. Baicalein attenuates inflammatory responses by suppressing TLR4 mediated NF-kappaB and MAPK signaling pathways in LPS-induced mastitis in mice. Int Immunopharmacol 2015; 28:470-6; PMID:26202808; http://dx.doi.org/ 10.1016/j.intimp.2015.07.012 [DOI] [PubMed] [Google Scholar]
- [54].Liu Y, Ma D, Zhang L, Cui Y, Guo M. TN-2 modulates LPS-induced inflammatory response in human renal tubular epithelial cells by blocking TLR4-mediated NF-kappaB activation via MyD88- and TRIF-dependent mechanism. Inflamm Res 2015; 64:937; http://dx.doi.org/ 10.1007/s00011-015-0853-6 [DOI] [Google Scholar]
- [55].Yu M, Kang X, Xue H, Yin H. Toll-like receptor 4 is up-regulated by mTOR activation during THP-1 macrophage foam cells formation. Acta Biochim Biophys Sin (Shanghai) 2011; 43:940-7; PMID:22015781; http://dx.doi.org/ 10.1093/abbs/gmr093 [DOI] [PubMed] [Google Scholar]
- [56].An CH, Wang XM, Lam HC, Ifedigbo E, Washko GR, Ryter SW, Choi AM. TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol 2012; 303:L748-5; PMID:22983353; http://dx.doi.org/ 10.1152/ajplung.00102.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Na HY, Mazumdar K, Moon HJ, Chang S, Seong SY. TLR4-independent and PKR-dependent interleukin 1 receptor antagonist expression upon LPS stimulation. Cell Immunol 2009; 259:33-40; PMID:19559408; http://dx.doi.org/ 10.1016/j.cellimm.2009.05.010 [DOI] [PubMed] [Google Scholar]
- [58].Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol 2005; 25:1213-9; PMID:15718493; http://dx.doi.org/ 10.1161/01.ATV.0000159891.73193.31 [DOI] [PubMed] [Google Scholar]
- [59].Chassin C, Goujon JM, Darche S, du Merle L, Bens M, Cluzeaud F, Werts C, Ogier-Denis E, Le Bouguénec C, Buzoni-Gatel D, et al.. Renal collecting duct epithelial cells react to pyelonephritis-associated escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J Immunol 2006; 177:4773-84; PMID:16982918; http://dx.doi.org/ 10.4049/jimmunol.177.7.4773 [DOI] [PubMed] [Google Scholar]
- [60].Fielhaber JA, Carroll SF, Dydensborg AB, Shourian M, Triantafillopoulos A, Harel S, Hussain SN, Bouchard M, Qureshi ST, Kristof AS. Inhibition of mammalian target of rapamycin augments lipopolysaccharide-induced lung injury and apoptosis. J Immunol 2012; 188:4535-42; PMID:22450807; http://dx.doi.org/ 10.4049/jimmunol.1003655 [DOI] [PubMed] [Google Scholar]
- [61].Lopez-Alonso I, Aguirre A, Gonzalez-Lopez A, Fernandez AF, Amado-Rodriguez L, Astudillo A, Batalla-Solís E, Albaiceta GM. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-kappaB pathway. Am J Physiol Lung Cell Mol Physiol 2013; 304:L844-852; PMID:23585228; http://dx.doi.org/ 10.1152/ajplung.00422.2012 [DOI] [PubMed] [Google Scholar]
- [62].Li J, Li D, Liu X, Tang S, Wei F. Human umbilical cord mesenchymal stem cells reduce systemic inflammation and attenuate LPS-induced acute lung injury in rats. J Inflamm 2012; 9:33; http://dx.doi.org/ 10.1186/1476-9255-9-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.