

Summary
ETAA1 (Ewing tumor-associated antigen 1), also known as ETAA16, was identified as a tumor-specific antigen in the Ewing family of tumors. However, the biological function of this protein remains unknown. Here, we report the identification of ETAA1 as a DNA replication stress response protein. ETAA1 specifically interacts with RPA via two conserved RPA-binding domains and is therefore recruited to stalled replication forks. Interestingly, further analysis of ETAA1 function revealed that ETAA1 participates in the activation of ATR signaling pathway via a conserved ATR-activating domain (AAD) located near its N-terminus. Importantly, we demonstrate that both RPA binding and ATR activation are required for ETAA1 function at stalled replication forks to maintain genome stability. Therefore, our data suggest that ETAA1 is a new ATR activator involved in replication checkpoint control.
Keywords: ETAA1, ATR, TOPBP1, AAD, DNA replication stress
eTOC Blurb
Lee et al. report the identification of a DNA damage sensor protein ETAA1 which is recruited to the sites of DNA damage via specific interaction with single stranded DNA-binding protein RPA. They further demonstrate that ETAA1 is a new ATR kinase activator involved in replication and DNA damage checkpoint control.
Introduction
The human genome is continuously challenged by genotoxic pressure from both endogenous and exogenous sources. A variety of DNA lesions resulting from these insults must be properly repaired for the maintenance of genomic integrity. Cells have evolved a coordinated network of DNA damage response (DDR) involving multiple cellular processes, such as cell-cycle checkpoint control, DNA replication, DNA repair and chromosome segregation, to maintain genome stability.
DNA damage response is orchestrated by two major damage-induced protein kinases, the ataxia-telangiectasia mutated (ATM) and the ataxia telangiectasia mutated and Rad3-related (ATR). ATM and ATR share a lot of biochemical and functional similarities. These two kinases target overlapping sets of substrates involved in numerous cellular processes. However, while ATM-depleted cells are viable, ATR is essential for the viability of replicating cells [1–3]. ATM functions predominantly in response to double-strand breaks (DSBs), and ATR, in a complex with its functional partner ATRIP, is activated by a broad spectrum of DNA damage and replication stress that involves the exposure of RPA-coated single stranded DNA (ssDNA) together with adjacent stretch of double-stranded DNA (dsDNA) that presents a 5′ junction [4–6]. Once activated, ATR phosphorylates a variety of substrates including CHK1 in order to promote cell cycle arrest, DNA repair, and recovery from replication stress [6]. ATR activation depends on the temporal and spatial interactions between ATR/ATRIP complex and its associated proteins containing ATR-activating domains (AAD). In budding yeast, three proteins, Dpb11TopBP1, Ddc1Rad9 and Dna2, can interact with and activate Mec1ATR[7–9]. Each of these proteins contains an AAD that binds directly to Mec1ATR:Ddc2ATRIP complex and any of these AADs is sufficient to activate Mec1ATR in vitro [8–11]. However, TOPBP1 is so far the only protein reported containing an AAD that interacts with and activates ATR/ATRIP complex in Xenopus and humans [12].
In the ATR-dependent replication checkpoint pathway, the key sensor protein is RPA, which efficiently binds to and protects ssDNA generated during replication fork stalling and recruits factors such as ATRIP, RAD17, and RAD9 to promote ATR activation. Human RPA is a stable heterotrimer composed of three subunits, RPA70, RPA32 and RPA14 (also named as RPA1, 2 and 3) that are conserved among eukaryotes. RPA is essential in eukaryotic cells and is involved in a number of key cellular activities including DNA replication, repair, recombination and DNA damage signaling pathways. RPA is a ssDNA-binding and scaffold protein complex that interacts with multiple proteins and facilitates various biochemical reactions that occur at or involve ssDNA. Recent studies have revealed a number of novel RPA-binding proteins that play important roles in DNA replication and/or replication checkpoint control. For example, PRP19/PSO4 directly binds RPA and localizes to DNA damage sites via RPA, where it acts as a ubiquitin ligase for RPA and facilitates the accumulation of ATR/ATRIP at DNA damage sites [13, 14]. Another E3 ligase RFWD3 is also reported to bind to and ubiquitinate RPA in response to replication fork stalling, therefore promoting replication fork restart and homologous recombination at stalled forks [15–17]. Schlafen 11 (SLFN11) was shown to interact directly with RPA1 and is recruited to sites of DNA damage in an RPA1-dependent manner [18]. SLFN11 inhibits checkpoint maintenance and homologous recombination repair by promoting the destabilization of the RPA-ssDNA complex [18]. Another RPA-binding protein helicase B (HELB) was recently published by two different groups [19–21]. While HELB was proposed in one study to play an inhibitory role for DNA end resection in G1 phase and is exported to the cytoplasm to allow efficient DNA end resection in S/G2 phase [21], in another study, HELB was reported to promote homologous recombination [20].
In this study, we report the identification of a previously uncharacterized protein ETAA1 (Ewing Tumor-Associated Antigen 1) as a novel DNA damage sensor. We showed that ETAA1 is recruited to stalled replication forks in an RPA-dependent manner. We further demonstrate that ETAA1 is the second identified ATR activator in humans. In addition, we were able to identify a conserved ATR-activating domain in ETAA1, which, together with its RPA-binding domains, is critically important for ETAA1 function at stalled replication forks.
Results
ETAA1 is an RPA-interacting protein involved in cellular response to DNA damage
Our tandem affinity purification (TAP) of RPA protein complex repeatedly revealed Ewing Tumor-Associated Antigen 1 (ETAA1) as a candidate RPA-binding protein (Figure S1A). When our manuscript was under preparation, ETAA1 was also found in a list of potential RPA-associated proteins from a proteomics study [21]. To ensure that ETAA1 indeed associates with RPA, we performed reverse TAP using whole cell lysate prepared from 293T cells stably expressing triple-epitope (S-protein, FLAG and streptavidin binding peptide) tagged ETAA1 (SFB-ETAA1). Mass spectrometry analysis revealed RPA1/2/3 as major ETAA1-associated proteins (Figure 1A). We also found several known RPA-binding proteins in this list of ETAA1-associated proteins (Figure 1A), which include HARP/SMARCAL1 previously identified by us and others [22–26], PRPF19/PSO4 [13, 14], and BLM/TOP3A/RMI1 [27]. These data strongly suggest that ETAA1 is a bona fide RPA-binding protein. We confirmed the in vivo interaction of SFB-tagged ETAA1 with myc-tagged RPA1 (Figure 1B). HARP and AH2 were included respectively as positive and negative controls in this experiment. While HARP associated with RPA complex, AH2 did not bind to RPA (Figure 1B), but instead AH2 predominantly binds to PCNA as we and others reported previously [28, 29]. To further confirm the endogenous interaction between ETAA1 and RPA, we raised two ETAA1 antibodies against residues 1–300 and residues 626–926 of ETAA1 respectively. Using these ETAA1 antibodies, we verified the in vivo interaction between endogenous ETAA1 and RPA complex (Figure 1C).
Figure 1. ETAA1 is an RPA-interacting protein involved in cellular response to DNA damage.

(A) Tandem affinity purification was performed using 293T cells stably expressing tagged ETAA1. The results from the mass spectrometry analysis are shown in the table. Unique: number of unique peptides; Total: number of total peptides. (B) ETAA1 specifically interacts with RPA. 293T cells were transfected with plasmids encoding SFB-tagged HARP, AH2 or ETAA1 together with plasmids encoding myc-tagged RPA1. Co-precipitation was carried out using S-protein beads and immunoblotting was performed using antibodies as indicated. (C) Association of endogenous ETAA1 with RPA in 293T cells was analyzed by co-immunoprecipitation using two anti-ETAA1 antibodies and immunoblotting using antibodies as indicated. Anti-ETAA1 antibodies were raised by immunizing rabbits with GST-ETAA1 fusion proteins containing residues 1–300 and residues 626–926 of human ETAA1 proteins. (D) HeLa cells were transfected with the plasmid encoding SFB-tagged ETAA1. Immunostaining experiments were performed 6 hr after HU, CPT, or IR treatment using indicated antibodies. (E) ETAA1 depletion in ETAA1 knockout HeLa cells was confirmed by immunoblotting. (F and G) Survival curves in response to increasing doses of HU (F) and CPT (G) for indicated cell lines are presented. Cell survival assays were performed as described in the Materials and methods. Data are presented as mean ± s.d. from three different experiments. See also Figure S1 and S2.
Since quite a few RPA-binding proteins are recruited to stalled replication forks or resected DSB ends, we first tested whether ETAA1 would also participate in cellular response to replication stress and/or other types of DNA damage. As shown in Figure 1D and Figure S1B, discrete foci of FLAG-tagged ETAA1, which partially co-localized with γ H2AX, were readily detected in HeLa cells following hydroxyurea (HU), camptothecin (CPT), and ionizing radiation (IR) treatment, indicating that ETAA1 is involved in DNA damage response. Similarly, we detected the co-localization of ETAA1 and RPA foci in U2OS cells following HU, CPT, and IR treatment (Figure S1C), indicating that ETAA1 is recruited to RPA-bound ssDNA regions in response to DNA damage. Thus, we further examined the effect of ETAA1 depletion on cell survival following DNA damage. We generated two ETAA1 knockout HeLa cell lines using CRISPR-cas9 mediated gene editing (Figures 1E and S2A). HU treatment reduces the production of deoxyribonucleotides via inhibition of ribonucleotide reductase, therefore causing replication stress. Although cells with ETAA1 depletion had relatively normal cell cycle distribution (Figure S2B), ETAA1 knockout cells showed a marked hypersensitivity to HU treatment (Figure 1F). We also observed increased sensitivity of ETAA1 depleted cells to the topoisomerase I inhibitor CPT which also induces replication stress (Figure 1G).
Together, these data suggest that ETAA1 is an RPA-interacting protein located at stalled replication forks upon DNA replication stress to maintain cell viability.
Multiple binding surfaces on ETAA1 tether it to stalled replication forks
We were interested in how ETAA1 and RPA interact and how ETAA1 is recruited to sites of DNA damage. We already showed that ETAA1 foci co-localized with RPA foci following DNA damage (Figure S1C). To further confirm that RPA-bound ssDNA regions are required for ETAA1 recruitment to sites of DNA damage, we examined the foci formation of ETAA1 in CtIP-depleted cells following DNA damage. As shown in Figure S3A, two gRNAs were designed for a lentiviral CRISPR/Cas9 system and could be used to efficiently deplete CtIP in these cells. We found that RPA and ETAA1 foci were dramatically reduced in CtIP-depleted cells following CPT treatment (Figures S3B and S3C), suggesting that ETAA1 is recruited by RPA-bound ssDNA generated by DNA end resection mediated by CtIP.
In the initial experiments, we found that the C-terminal half of ETAA1 (500C) is sufficient to bind to RPA and localizes to stalled replication forks induced by HU (Figures 2A and 2B). When aligning human ETAA1 protein sequence with its orthologs from different species, we found that ETAA1 has four distinct and conserved regions (Figure S4). However, none of them is related to domains or motifs with known function. To further determine which region is required for the recruitment of ETAA1 to DNA damage sites, we generated a serious of truncation and internal deletion mutants of ETAA1 based on the conserved regions revealed by the alignment (Figure 2C). Interestingly, all the ETAA1 mutants formed clear foci after HU treatment (Figure 2D), suggesting that there are more than one RPA-binding surface on the C-terminal half of ETAA1, which have redundant roles in tethering ETAA1 to stalled replication forks.
Figure 2. Multiple binding surfaces on ETAA1 tether it to stalled replication forks.

(A) C-terminal half of ETAA1 binds to RPA. Co-precipitation was performed as described in Figure 1B. 500N: N-terminal half of ETAA1; 500C: C-terminal half of ETAA1. (B) C-terminal half of ETAA1 is sufficient to locate at stalled replication forks upon replication stress. HeLa cells were transfected with indicated plasmids. Immunostaining experiments were performed 6 hr after HU treatment using indicated antibodies. (C) Diagram of internal deletion mutants of ETAA1 used in this study. Conserved regions are highlighted in different colors. (D) A series of deletion mutations did not affect the foci formation of ETAA1 at DNA damage sites. Plasmids encoding ETAA1 deletion mutants were transfected into HeLa cells. Immunostaining experiments were performed 6 hr after HU treatment. See also Figure S3 and S4.
Two distinct RPA-binding domains contribute to the function of ETAA1 at stalled replication forks
Our alignment of different ETAA1 orthologs revealed two conserved regions within the C-terminal half of ETAA1 (Figure S4). To test whether these regions are required for the RPA binding and foci formation of ETAA1, we generated mutants lacking single or both regions (Figure 3A). Co-precipitation assays were performed to test the interaction between mutant ETAA1 and RPA1 or RPA2. Our results clearly showed that deletion of the first region (D572-700) disrupted the binding between ETAA1 and RPA1, while deletion of the second region (1–872, which is D873-926) abolished its interaction with RPA2 (Figures 3B and 3C). As expected, the ETAA1 mutant lacking both regions (DD) totally lost the ability to bind RPA complex (Figures 3B and 3C). We further introduced these mutants into HeLa cells and tested the foci formation of these mutants after HU treatment. Our results showed that either of the RPA-binding domains is sufficient to guide ETAA1 to stalled replication forks, and disruption of both RPA-binding domains from ETAA1 abolished its localization at stalled replication forks (Figures 3D). We further tested whether the binding of ETAA1 to RPA is required for its function in vivo. As shown in Figures 3E and 3F, the expression of wild-type ETAA1 rescued HU hypersensitivity in ETAA1 knockout cells. However, the mutant lacking both RPA1 and RPA2-binding domains (DD) failed to do so. These data indicate that the RPA-binding ability is required for ETAA1 function at stalled replication forks in vivo. Therefore, we concluded that ETAA1 has two distinct RPA-binding domains, RBD1 and RBD2 (Figure 3A), which are responsible for binding to RPA1 and RPA2 respectively, and either RBD1 or RBD2 is sufficient for tethering ETAA1 at stalled replication forks and contributes to ETAA1 function in response to replication stress.
Figure 3. Two distinct RPA-binding domains contribute to the function of ETAA1 at stalled replication forks.

(A) Schematic representation of wild-type ETAA1 and the mutants used in the following study. (B and C) The interaction between wild-type or mutant ETAA1 and RPA was tested by co-precipitation assays as described in Figure 1B. (D) Disrupting both C-terminal domains in ETAA1 disables its localization at stalled replication forks. DD: double deletion mutant (deleted with amino acids 572–700 and 873–926). (E) The exogenous ETAA1 expression in ETAA1 KO1 cells was confirmed by immunoblotting. (F) The ETAA1 mutant deleted of both RPA-binding domains failed to rescue HU hypersensitivity in cells with ETAA1 depletion. The empty vector was included as a control. Survival curves are shown for indicated cell lines in response to increasing doses of HU. Data are presented as mean ± s.d. from three different experiments.
ETAA1 participates in the activation of ATR signaling pathway
To further understand how ETAA1 may participate in DNA replication stress response, we first checked the ETAA1 TAP results (Figure 1A). Besides some known RPA-binding proteins, we noticed that ATR, the master regulator of genome integrity, is a putative ETAA1-binding protein. ATR is activated in response to a variety of DNA lesions that induce the formation of ssDNA [6, 30]. One of the best characterized ATR substrates is the checkpoint kinase 1 (CHK1), an effector kinase of ATR in the ATR-mediated DNA damage checkpoint pathway [31]. CHK1 is not only important for the checkpoint response during S phase but is also crucial for maintaining the stability of DNA replication forks. Activation of CHK1 after DNA replication stress is known to require the phosphorylation of two C-terminal residues by ATR kinase at Ser317 and Ser345, which is a reliable indicator of CHK1 activation [32–34]. We speculated that ETAA1 might have a role in the activation of ATR signaling pathway. Therefore, we tested CHK1 phosphorylation at Ser317 in wild-type and ETAA1 knockout cells following replication stress. As shown in Figure 4A, CHK1 phosphorylation at Ser317 is impaired in ETAA1 knockout cells following HU treatment. We further tested other ATR phosphorylation sites: CHK1 Ser345 and RPA2 Ser33. As shown in Figure 4B, after CPT treatment, both CHK1 Ser317 and 345 phosphorylation as well as RPA2 Ser33 phosphorylation were diminished in ETAA1 depleted cells. However, we did not detect any difference in CHK2 Thr68 phosphorylation, an ATM-dependent event, in wild-type and ETAA1 depleted cells following CPT treatment. These results suggest that ETAA1 is specifically required for the activation of ATR signaling pathway following replication stress.
Figure 4. ETAA1 participates in the activation of ATR signaling pathway.

(A) ETAA1 depletion impairs CHK1 phosphorylation upon replication stress induced by HU treatment. Parental and ETAA1 KO cells were treated with HU (2 mM or 10 mM), and cell lysates were collected one hour after treatment. Immunoblotting was performed using antibodies as indicated. (B) ETAA1 depletion impairs CHK1 and RPA2 phosphorylation upon replication stress induced by CPT treatment. Parental and ETAA1 KO cells were treated with CPT (1 μM), and the cell lysates were collected one hour after treatment. (C) Exogenous expression of ETAA1 activates ATR-CHK1 signaling. HeLa cells were transfected with indicated plasmids. Immunostaining experiments were performed using indicated antibodies. NLS-AAD: the AAD from TOPBP1 with an N-terminal nuclear localization sequence (NLS).
We further explored mechanistically how ETAA1 may activate the ATR/CHK1 pathway. An early study demonstrated that TOPBP1 contains an ATR-activating domain (AAD), which interacts with and activates ATR/ATRIP complex in vitro [12]. Indeed, overexpression of this domain by itself could lead to ATR activation [12]. We cloned TOPBP1-AAD (residues 978–1286) and confirmed that overexpression of TOPBP1-AAD triggered robust CHK1 phosphorylation at Ser317 (Figure 4C). Interestingly, in ETAA1-overexpressing cells, we also observed clear CHK1 phosphorylation (Figure 4C), indicating that ETAA1 may activate ATR kinase. As a negative control, we showed that we could not detect CHK1 phosphorylation in HARP-overexpressing cells (Figure 4C). These observations provided the first evidence that ETAA1 participates in the activation of ATR signaling pathway.
Defining a conserved ATR-activating domain in ETAA1
In budding yeast, three proteins, Dpb11TopBP1, Ddc1Rad9 and Dna2, interact with and independently activate Mec1ATR. Each contains an ADD that interacts directly with the Mec1ATR:Ddc2ATRIP complex [8–11]. In human, only TOPBP1 has been identified as a direct ATR activator, and TOPBP1-AAD is both sufficient and necessary for ATR activation. Since we observed that ETAA1 could participate in the activation of ATR signaling pathway, we asked whether there is a potential AAD in ETAA1. We found that while the N-terminal half of ETAA1 (500N) is sufficient to stimulate CHK1 phosphorylation at Ser317, the C-terminal half of ETAA1 (500C) failed to do so (Figure 5A). We further narrowed down this potential AAD to a conserved region (residues 77–238) of ETAA1 (Figure S4). As shown in Figure 5B, similar to TOPBP1-AAD, ETAA1-AAD (1-238/D10-76) overexpression triggered robust CHK1 phosphorylation at Ser317. Further deletion of ETAA1-AAD (D77-238) disrupted the ability of ETAA1 to induce CHK1 phosphorylation (Figures 5C and 5D). To test whether ETAA1 can directly activate ATR in vitro, we performed ATR kinase assays. As shown in Figure S5A, like TOPBP1-AAD, ETAA1-AAD stimulated ATR kinase activity. We then tested whether this ETAA1-AAD is required for its function in vivo. As shown in Figures 5E and 5F, the expression of wild-type ETAA1 rescued HU hypersensitivity in ETAA1 knockout cells. However, the mutant lacking ETAA1-AAD failed to do so. Therefore, we identified an AAD in ETAA1, which is required for activating ATR signaling in human cells.
Figure 5. Defining a conserved ATR-activating domain in ETAA1.

(A) N-terminal half of ETAA1 is sufficient to activate ATR-CHK1 signaling. HeLa cells were transfected with indicated plasmids. Immunostaining was performed using indicated antibodies. (B) Narrowing down the potential ETAA1 AAD to an N-terminal conserved domain (amino acids 77–238). We retained the native NLS (within amino acids 1–10) in these ETAA1 deletion mutants. (C and D) A deletion mutant lacking amino acids 77–238 (D77-238) failed to activate ATR-CHK1 signaling and lost the ability to rescue HU hypersensitivity in cells with ETAA1 depletion (E and F). The empty vector was included as a control. Survival curves are shown for indicated cell lines in response to increasing doses of HU. Data are presented as mean ± s.d. from three different experiments. See also Figure S5.
Identification of a point mutation in the ATR-activating domain of ETAA1
It has been established that the ATR activation by TOPBP1 can be disrupted via introducing a point mutation that removes a key aromatic residue in AAD (W1138R in Xenopus, W1147R in Mouse and W1145R in Human) [12, 35]. Since we located an AAD in ETAA1, we are wondering whether we can identify a point mutation, which would similarly disrupt the ATR activation by ETAA1. We first aligned the residues 77–238 from human ETAA1 with corresponding segments from different species (Figure 6A, see also Figure S4). We were able to locate a conserved tryptophan (W107 in human ETAA1, Figure 6A). Further alignment of TOPBP1-AAD and ETAA1-AAD, and the residues surrounding W1145 from TOPBP1-AAD and W107 from ETAA1-AAD revealed a conserved sequence (IF/I/VWD) (Figure 6B, see also Figure S5B). To test whether W107 is the key residue required for ATR activation, we generated W107R mutation in both full-length ETAA1 and ETAA1-AAD (Figure 6C). Overexpression of those mutants in HeLa cell clearly revealed that W107R mutation abolished the CHK1 phosphorylation at Ser317 (Figure 6D) and the induction of γ H2AX (Figure S5C) by full-length ETAA1 and ETAA1-AAD. Taken together, these results indicate that, similar to TOPBP1, ETAA1 must have an intact AAD in order to support the ATR-dependent phosphorylation of CHK1 or other ATR substrates including H2AX that normally occurs in the presence of stalled replication forks.
Figure 6. Identification of a point mutation in the ATR-activating domain of ETAA1.

(A) Alignment of residues 77–238 from the ATR-activating domain of human ETAA1 with corresponding segments from different species. A conserved sequence (IF/I/VWD) is marked with a red box, and a conserved tryptophan (W107 in human ETAA1) is denoted with an asterisk. (B) Alignment of residues surrounding the conserved sequence IF/I/VWD from ETAA1-AAD and TOPBP1-AAD. Two essential tryptophans (W1145 in TOPBP1 and W107 in ETAA1) are denoted with asterisks. (C) Schematic representation of wild-type ETAA1 and the W107R mutants used in the following study. (D) W107R mutation disrupts the ability of ETAA1 in activation of ATR-CHK1 signaling. HeLa cells were transfected with indicated plasmids. Immunostaining experiments were performed using indicated antibodies. (E) A proposed model for ETAA1 in DNA replication stress response. Please refer to the text for details. See also Figure S5 and S6.
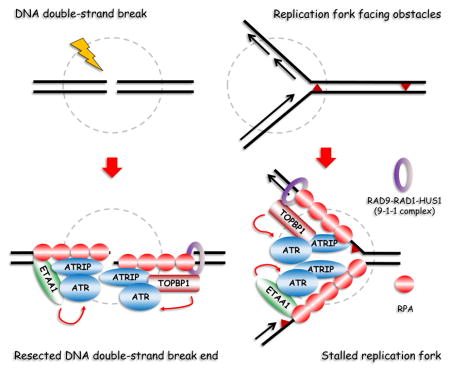
The current model of ATR activation indicates that ATR activation occurs in a manner that depends on RAD9-RAD1-HUS1 (the 9-1-1 complex) and TOPBP1. Therefore, we decided to further evaluate the relationship between ETAA1 and 9-1-1 complex/TOPBP1. First, we designed two gRNAs targeting RAD9, a subunit of the 9-1-1 complex. As shown in Figures S6A, S6B, and S6C, while depletion of RAD9 dramatically reduced TOPBP1 recruitment to stalled replication forks following CPT treatment, the ETAA1 recruitment was not affected and we observed clear foci formation of ETAA1 in RAD9-depleted cells which failed to induce TOPBP1 recruitment. Second, to test whether ETAA1 and TOPBP1 would compete for loading onto stalled replication forks, we overexpressed ETAA1 in cells, but did not observe its competition with TOPBP1 loading (Figure S6D). Finally, as shown in Figures S6E and S6F, cells co-depleted with TOPBP1 and ETAA1 showed more severe defects in checkpoint signaling and cell survival upon replication stress. These data suggest that ETAA1 is located in a parallel pathway and plays a role in ATR activation independent of 9-1-1 complex and TOPBP1 (Figure 6E).
Discussion
In this study, we demonstrate that 1) ETAA1 is an RPA-interacting protein that participates in cellular response to replication stress; 2) ETAA1 is recruited to DNA damage sites upon replication stress via two RPA-binding domains, RBD1 and RBD2; 3) In humans, ETAA1 is a novel ATR activator in addition to and independent of TOPBP1; 4) ETAA1 activates ATR via a conserved AAD located at its N-terminus, and a single mutation at W107 in this AAD can abolish ATR activation by ETAA1. Thus, ETAA1 is a new ATR activator and plays a role in replication checkpoint control (Figure 6E).
As a master kinase in the DDR pathway, ATR responds to a broad spectrum of DNA damage that leads to replication stress. ATR is essential for cell survival even in the absence of extrinsic DNA damage, underscoring the critical function of ATR in coping with intrinsic genomic stress. Although the mechanism underlying ATR activation is well studied and widely discussed, how ATR is activated by different types of DNA damage and replication stress in human is largely unknown. A single DNA structure containing ssDNA is proposed to be required for ATR activation in response to many different types of DNA damage, including DSBs, base adducts, crosslinks and replication stress [36, 37]. RPA coats most forms of ssDNA in the cell, including the ssDNA that is formed during DNA replication and DNA repair [38]. ATRIP, the regulatory partner of ATR, binds directly to RPA-coated ssDNA (RPA-ssDNA) and thereby enables the ATR/ATRIP complex to localize to sites of DNA damage [37, 39, 40]. The localization of ATR/ATRIP to sites of DNA damage, however, is not sufficient to fully activate ATR at the sites of DNA damage. Instead, it requires the functions of additional ATR regulators, including RAD17 and RAD9-RAD1-HUS1 (the 9-1-1 complex) which recognize DNA damage independently [6] (see also Figure 6E). ATR activation also requires TOPBP1, which is recruited to sites of DNA damage via RAD9 C-terminal phosphorylation at Ser387 [41, 42], or by MRE11-RAD50-NBS1 complex as published recently [43].
ATR-activating domains (AAD) play a key role in activating ATR kinase activity. Depleting wild type TopBP1 from Xenopus and replacing it with recombinant protein with a single aromatic residue mutation in its AAD (W1138R) totally abolished ATR activation in response to replication stress [12, 35]. The W1145R mutant of human TOPBP1 (analogous to the W1138R mutant of TopBP1 from Xenopus) also lost its ability to promote the phosphorylation of downstream targets of ATR. Similarly, analyzing a TopBP1-W1147R knock-in mouse model revealed the essential function for TopBP1 AAD in mouse development and cellular senescence [12, 35]. However, in the model organisms such as S. cerevisiae and S. pombe, while the AAD domain of the TopBP1 homologs was similarly sufficient to activate ATR, TopBP1 itself plays a relatively minor role in ATR signaling activation in response to DNA damage or replication stress. Recent studies from S. cerevisiae identified two further AADs which play partially redundant roles in ATR checkpoint activation: one is contained within the C-terminus of Ddc1Rad9, a 9-1-1 clamp subunit [8], while the other is found in the Dna2 replication protein [9]. Interestingly, disrupting the function of all three AAD domains from Dpb11TopBP1, Ddc1Rad9 and Dna2 in the same cells largely abolished the activation of ATR pathway in S. cerevisiae. These results indicate that multiple AADs promote ATR activation above basal level and that ATR activation is mediated by one or more AAD domains in yeasts [11].
Whether there are other ATR activators in humans other than TOPBP1 has been a long-standing question in the field. Our study uncovered ETAA1 as another ATR activator in humans. ETAA1 is not a very conserved protein, and we can only trace ETAA1 homologs to Xenopus (Figure S4), but not to yeast, suggesting that in higher eukaryotes, ETAA1 has evolved as an alternative ATR activator, similar to Ddc1Rad9 or Dna2 in yeast. Interestingly, TOPBP1-AAD, and now ETAA1-AAD, can trigger ATR activation in the absence of DNA damage [8–11]. Therefore, it is assumed that ATR activation by TOPBP1 should be tightly regulated, but the detailed mechanism remains to be determined. In humans, the AAD of TOPBP1 interacts with the ATR/ATRIP complex via an internal region of ATRIP and the PIKK regulatory domain (PRD) near the C-terminus of ATR [44]. Further studies are needed to characterize the detailed interaction between ETAA1-AAD and ATR/ATRIP complex and how this interaction may be regulated following DNA damage.
ETAA1 was first noticed and named in a study working on Ewing’s family of tumors [45]. By immunoscreening of an Ewing’s family of tumor (EFT)-derived cDNA library using 16 EFT-specific antibodies, Borowski and colleagues isolated a 3.5 kb cDNA, named Ewing’s tumour-associated antigen 16 (ETAA16, also known as ETAA1) [45]. As demonstrated by flow cytometry, the cell surface expression of ETAA1 antigen is restricted to Ewing tumor cell lines, suggesting that ETAA1 may function as a tumor-specific cell surface antigen in EFTs. However, our results showed that overexpressed FLAG-tagged ETAA1 is predominantly located in the nucleus, and we did not observe cell surface expression or even cytoplasmic staining of FLAG-tagged ETAA1 in HeLa cells (Figure 1D). Further studies should be conducted to detect the endogenous expression of ETAA1 and its cellular localization in both EFT and non-EFT cells.
More recently, a genome-wide association study (GWAS) of pancreatic cancer revealed significant association at 2p13.3 (ETAA1, rs1486134) with susceptibility to pancreatic cancer [46]. As a matter of fact, this ETAA1 locus has also been implicated in another GWAS study of pancreatic cancer in Han Chinese [47]. While it remains to be determined whether or not ETAA1 expression would be altered in pancreatic cancers, it is worthwhile to point out that inherited high-penetrance mutations in a number of DDR genes, including BRCA2, ATM, PALB2, and BRCA1, have been reported to be associated with increased risk for pancreatic cancer. As a novel ATR activator in humans, ETAA1 deregulation may also contribute to cancer development, which warrants further experimentation.
Experimental Procedures
Antibodies
Anti-ETAA1 antibodies were raised by immunizing rabbits with GST-ETAA1 fusion proteins containing residues 1–300 and residues 626–926 of human ETAA1 proteins respectively. Antisera were affinity-purified using AminoLink plus Immobilization and purification kit (Pierce). Antibody against γ H2AX was previously described [28, 48]. The anti-myc and anti-CHK1 were obtained from Santa Cruz Biotechnology. Anti-p-CHK1 (Ser317 and Ser345) and anti-p-CHK2 (Thr68) antibodies were purchased from Cell Signaling. Anti-p-RPA2 (Ser33) was obtained from Bethyl Laboratories. Anti-RPA2 antibody was obtained from Abcam. Anti-β-actin and anti-FLAG were obtained from Sigma.
Other Methods
See the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
ETAA1 participates in cellular response to replication stress
ETAA1 is recruited to the sites of DNA damage via two RPA-binding domains
In humans, ETAA1 is a novel ATR activator in addition to and independent of TOPBP1
ETAA1 activates ATR kinase via a conserved ATR-activating domain (AAD)
Acknowledgments
We would like to thank all members of the DNA Dynamics Club at Columbia University Medical Center, Dr. Lee Zou, Dr. Tom Hei, and Dr. David Brenner for insightful discussion. We would like to thank Dr. Zihua Gong for providing the anti-TOPBP1 antibody, Dr. Howard Lieberman for the anti-RAD9 antibody, and Dr. Zhenkun Lou for the TOPBP1 shRNA. Images were collected in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grant #P30 CA013696 (National Cancer Institute). Cell sorting was performed in the CCTI Flow Cytometry Core at Columbia University, supported in part by the Office of the Director, National Institutes of Health under awards S10OD020056. This work was supported in part by grants from the National Institutes of Health to J.C. (CA089239, CA092312, and CA100109), a Susan G. Komen CCR Grant to J.Y. and an NCI Transition Career Development Award (K22) to J.Y. J.C. is a member of MD Anderson Cancer Center (CA016672).
Footnotes
Author Contributions
Y.-C.L., Q.Z. and J.Y. performed the experiments and analyzed the data; Writing-Original Draft, J.Y.; Writing-Review & Editing, J.C. and J.Y.; Supervision, J.C. and J.Y.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 2.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 3.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 4.MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navadgi-Patil VM, Burgers PM. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J Biol Chem. 2008;283:35853–35859. doi: 10.1074/jbc.M807435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navadgi-Patil VM, Burgers PM. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell. 2009;36:743–753. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S, Burgers PM. Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev. 2013;27:313–321. doi: 10.1101/gad.204750.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mordes DA, Nam EA, Cortez D. Dpb11 activates the Mec1-Ddc2 complex. Proc Natl Acad Sci U S A. 2008;105:18730–18734. doi: 10.1073/pnas.0806621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou L. Four pillars of the S-phase checkpoint. Genes Dev. 2013;27:227–233. doi: 10.1101/gad.213306.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 13.Marechal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jimenez AE, Jin J, Zou L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol Cell. 2014;53:235–246. doi: 10.1016/j.molcel.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wan L, Huang J. The PSO4 protein complex associates with replication protein A (RPA) and modulates the activation of ataxia telangiectasia-mutated and Rad3-related (ATR) J Biol Chem. 2014;289:6619–6626. doi: 10.1074/jbc.M113.543439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong Z, Chen J. E3 ligase RFWD3 participates in replication checkpoint control. J Biol Chem. 2011;286:22308–22313. doi: 10.1074/jbc.M111.222869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R, Elledge SJ. RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol Cell. 2015;60:280–293. doi: 10.1016/j.molcel.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Chu J, Yucer N, Leng M, Wang SY, Chen BP, Hittelman WN, Wang Y. RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J Biol Chem. 2011;286:22314–22322. doi: 10.1074/jbc.M111.222802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, Chen J, Huang J. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep. 2016;17:94–109. doi: 10.15252/embr.201540964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guler GD, Liu H, Vaithiyalingam S, Arnett DR, Kremmer E, Chazin WJ, Fanning E. Human DNA helicase B (HDHB) binds to replication protein A and facilitates cellular recovery from replication stress. J Biol Chem. 2012;287:6469–6481. doi: 10.1074/jbc.M111.324582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Yan P, Fanning E. Human DNA helicase B functions in cellular homologous recombination and stimulates Rad51-mediated 5′-3′ heteroduplex extension in vitro. PLoS One. 2015;10:e0116852. doi: 10.1371/journal.pone.0116852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tkac J, Xu G, Adhikary H, Young JT, Gallo D, Escribano-Diaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol Cell. 2016;61:405–418. doi: 10.1016/j.molcel.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brosh RM, Jr, Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem. 2000;275:23500–23508. doi: 10.1074/jbc.M001557200. [DOI] [PubMed] [Google Scholar]
- 28.Yuan J, Ghosal G, Chen J. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol Cell. 2012;47:410–421. doi: 10.1016/j.molcel.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou L. Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007;21:879–885. doi: 10.1101/gad.1550307. [DOI] [PubMed] [Google Scholar]
- 31.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 32.Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- 33.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci U S A. 2001;98:11289–11294. doi: 10.1073/pnas.191557598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou ZW, Liu C, Li TL, Bruhn C, Krueger A, Min W, Wang ZQ, Carr AM. An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet. 2013;9:e1003702. doi: 10.1371/journal.pgen.1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 37.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 38.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Namiki Y, Zou L. ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc Natl Acad Sci U S A. 2006;103:580–585. doi: 10.1073/pnas.0510223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- 42.Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol Cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008;22:1478–1489. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borowski A, Dirksen U, Lixin L, Shi RL, Gobel U, Schneider EM. Structure and function of ETAA16: a novel cell surface antigen in Ewing’s tumours. Cancer Immunol Immunother. 2006;55:363–374. doi: 10.1007/s00262-005-0017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, Li D, Neale RE, Olson SH, Scelo G, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911–916. doi: 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu C, Miao X, Huang L, Che X, Jiang G, Yu D, Yang X, Cao G, Hu Z, Zhou Y, et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2012;44:62–66. doi: 10.1038/ng.1020. [DOI] [PubMed] [Google Scholar]
- 48.Yuan J, Chen J. FIGNL1-containing protein complex is required for efficient homologous recombination repair. Proc Natl Acad Sci U S A. 2013;110:10640–10645. doi: 10.1073/pnas.1220662110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.