

Abstract
The coordination of macrophage polarization is essential for the robust regenerative potential of skeletal muscle. Repair begins with an inflammatory monocyte/pro-inflammatory macrophage (M1)-mediated phase followed by polarization to a pro-regenerative (M2) phenotype. Recently, regulatory T cells (Tregs) were described as necessary for this M1 to M2 transition. Here, we report that chronic infection with the protozoan parasite Toxoplasma gondii causes a non-resolving Th1 myositis with prolonged tissue damage associated with persistent M1 accumulation. Surprisingly, Treg ablation during chronic infection rescues macrophage homeostasis and skeletal muscle fiber regeneration showing that Tregs can directly contribute to muscle damage. This study provides evidence that the tissue environment established by the parasite could lead to a paradoxical pathogenic role for Tregs. As such, these findings should be considered when tailoring therapies directed at Tregs in inflammatory settings.
Keywords: Tregs, Macrophages, Infectious Myositis
Introduction
The current paradigm for muscle repair is based on the sequential and discrete actions of inflammatory monocytes/pro-inflammatory macrophages (IM/M1) and pro-regenerative macrophages (M2) (1–3). IM/M1 mediate the acute inflammatory phase of repair by production of lytic factors to break down necrotic cells, phagocytosis of cellular debris, and satellite cell activation, a population of myofiber progenitor cells (2, 4, 5). M2 facilitate a regenerative phase through suppression of inflammatory cells and production of tissue remodeling products such as collagen matrix enzymes. Critically, the IM/M1 to M2 balance in the muscle environment drives both the phase and efficiency of repair (1, 2, 6). Prolonged IM/M1 presence during muscle injury may lead to delayed or aberrant repair (7–13), while exuberant M2 activity may cause muscle fibrosis (6). However, it remains unclear how muscles are repaired in the context of chronic inflammation and the factors preventing repair from proceeding. Recent studies implicate a tissue-specific role for resident regulatory T-cells (Tregs) in the transition from IM/M1 to M2 during tissue repair in acute sterile injury and dystrophy models (12, 13). Still, greater insight into how repair proceeds in the face of immune dysfunction and chronic inflammation is needed as these are hallmark features of a variety of debilitating inflammatory myopathies such as polymyositis, dermatomyositis, and idiopathic inflammatory myositis (6, 14–16).
Chronic infections of muscle can represent a source of chronic inflammation that may have detrimental consequences for muscle physiology. Here, we examine skeletal muscle injury and potential tissue repair in the context of infection with Toxoplasma gondii in mice. T. gondii persists as a life-long chronic infection characterized by parasite-containing tissue cysts in the brain and skeletal muscle (17, 18). Ingestion of tissue cysts or oocysts shed in feline feces results in a robust Th1 immune response to the parasite in the gastrointestinal tract that controls the fast growing tachyzoite life stage. Parasites that escape this response then disseminate throughout the host and IFNγ production by innate and adaptive cells is required for control of parasite growth (19). The mechanisms by which IFNγ operate to limit tachyzoite proliferation include induction of immunity-related GTPases and inducible nitric oxide synthase in a number of host cell types including M1 (20, 21). Immune pressure induces the transition of parasite to slow growing tissue cysts (bradyzoites) that resides in the skeletal muscle and central nervous system. As contaminated meats represent a major parasitic reservoir and source of food-borne transmission, tight regulation of both tissue integrity and immunity in skeletal muscle during chronic infection is important for survival of not only the host but also the parasite (22).
In this report, we ask how tissue integrity is maintained during of chronic infection. Specifically, we investigate the status of macrophage-mediated tissue repair mechanisms in the tissue environment established by chronic infection. Here, we find chronic infection in mice with T. gondii elaborates extensive muscle damage and prolonged disruption of macrophage homeostasis. In the setting of a highly Th1-polarized environment established during chronic skeletal muscle infection, Tregs are altered and strikingly, hinder skeletal muscle repair by promoting accumulation of M1. Our results provide the first evidence for detrimental effects of chronic inflammation on muscle Treg function and their role in skeletal muscle tissue repair.
Materials and Methods
Mice
Female C57BL/6 and B6.SJL (CD45.1) mice were obtained from Taconic Farms (Germantown, NY). C57BL/6-Tg(Foxp3-DTR/EGFP)23.2Spar/Mmjax DEREG mice were obtained from Jackson Laboratories (Bar Harbor, ME). Foxp3eGFP knock-in reporter mice were obtained from M. Oukka (Seattle Children's Research Institute). All procedures involving mice were reviewed and approved by the Institutional Animal Care and Use Committee at the University at Buffalo. Mice used were aged and sex matched and 8 to 10 weeks of age.
Oral infection with Toxoplasma gondii
Brains from chronically infected mice (30 to 60 days post-infection) were isolated and homogenized in phosphate-buffered saline pH 7.2 (PBS) for oral infection (Oldenhove et al., 2009). The total number of RFP-expressing ME49 cysts (graciously provided by Michael Grigg) within the brain homogenate was counted using a fluorescence microscope on the basis of 3 or more aliquots of 20 ul. Mice were orally gavaged with five ME49 cysts and monitored by weight and posture daily for the first 14 days of infection, and weekly thereafter.
Muscle histopathology
Skeletal muscle hindlimb muscles were fixed in 10% paraformaldehyde (Sigma-Aldrich, St. Louis, MO). Fixed tissue samples were embedded in paraffin and sectioned at 5μM for hematoxylin and eosin (H&E) staining. H&E stained skeletal muscle sections were blindly scored by an independent pathologist for muscle damage by degree of immune infiltration by location, necrotic fibers, and centralized nuclei (0: normal – 3: severe). Images were taken with an AperioScanScope slide scanner. Centralized nuclei per mm2 was calculated by counting the number of centrally nucleated myofibers on skeletal muscle sections. Muscle tissue cross-sectional area was calculated using ImageScope software (Leica, Wetzlar, Germany).
Muscle functional strength testing
Mice were placed on a screen consisting of 1 × 1 cm squares. The screen was slowly inverted and held two feet above a padded container. Muscle strength was scored based on time-elapsed between full inversion and maximum duration of hanging (maximum 60 seconds).
Screening of musculoskeletal disease pathways
RNA from naïve (n=5) and chronically infected skeletal muscle tissue (n=5) was isolated by TRIzol extraction (Thermo Fisher Scientific, Waltham, MA). Reverse transcriptase (iScript, Bio-Rad, Hercules, CA) was used to convert extracted RNA to cDNA. Pooled samples of cDNA from either naïve mice or chronically infected mice were used as a template (100 ng/sample) for a SYBR green (SsoAdvaced Universal SYBR green supermix, Bio-rad) qPCR-based screen of 379 unique targets implicated in musculoskeletal disease pathways (Prime PCR musculoskeletal disease pathway tier 1-4, Bio-rad). 384-well plates were run on a Bio-rad CFX384 Touch™ real-time PCR detection system and analyzed using Bio-rad CFX manager.
Isolation of tissue lymphocytes from organ tissues
Mice were euthanized and immediately perfused with PBS pH 7.2. Skeletal muscle was harvested and minced into digestion media (RPMI, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 0.1% β-mercatoethaol, 25 mM HEPES, 150μg/ml Dnase I [Sigma-Aldrich], 50 μg/ml liberase TL [Roche]). Tissues were digested at 37°C for 55 minutes and subsequently passed through a 70μM filter. Mononuclear cells were purified by a percoll gradient (37.5% Percoll [GE healthcare, Chicago, IL]/62.5% Hank's buffered saline solution [HBSS]) and resuspended in 10% media (RPMI with 10% FBS, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 0.1% β-mercatoethaol, 25 mM HEPES) for a single-cell suspension.
Spleens were harvested and passed through a 70μm filter to obtain a single cell suspension. Red blood cells were lysed in ACK lysing buffer (Lonza, Basel, Switzerland) and resuspended in 10% media. Blood was collected by cardiac puncture and immediately suspended in RPMI with 10% FBS, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 0.1% β-mercatoethaol, 25 mM HEPES, and 5 mM EDTA on ice. Red blood cells were lysed in ACK lysing buffer for 3 minutes on ice, twice. For single-cell suspensions of bone marrow cells, femurs were collected, cleared of muscle and connective tissue, flushed with PBS, and filtered through a 70μm filter.
Flow cytometric analysis
Single-cell suspensions were stained with HBSS containing extracellular antibody stains and Live/Dead Fixable Aqua dead cell stain (Thermo Fischer Scientific). After extracelluar staining, cells were fixed and permeabilized (Intracellular Fixation and Permeabilization Buffer Set, eBioscience, Santa Clara, CA). Cells were then rinsed and stained with eBioscience Permeabilization Buffer containing intracellular antibody stains. For intracellular stains containing biotinylated antibodies, streptavidin staining was performed separately in permeabilization buffer (eBioscience) after intracellular staining. Samples were washed and resuspended in flow cytometry buffer/FACS Buffer (PBS, 1% bovine serum albumin [Sigma-Aldrich], 2 mM EDTA [Thermo Fischer Scientific]) for acquisition. Absolute numbers were calculated using Count Bright absolute counting beads (Life Technologies).
In vitro re-stimulation
Lymphocyte single-cell suspensions from tissue were prepared as above and stimulated with 50 ng/mL phorbolmyristate acetate (PMA) (Sigma-Aldrich), 5 μg/mL ionomycin (Sigma-Aldrich) in the presence of brefeld in A (GolgiPlug, BD Biosciences, Franklin Lakes, NJ) for 3 hours at 37°C in 5% CO2. Extracellular staining was performed as above, fixed in 2% paraformaldehyde, and permeabilized with 0.5% Saponin in FACS buffer. Cells were then stained with intracellular antibodies and FACS with 0.5% Saponin, washed, and resuspended in FACS buffer for flow cytometric acquisition.
Toxoplasma gondii-specific tetramer staining
Lymphocyte single-cell suspensions from tissue were prepared as above. The following tetramer was obtained through the NIH Tetramer Facility: (APC-conjugated MHC class II tetramers bound to T. gondii ME49 hypothetical antigenic peptide I-A(b) (AVEIHRPVPGTAPPS) (Atlanta, GA). Cells were incubated with the T. gondii-specific tetramer or CLIP tetramer for 1 hour followed by extracellular and intracellular staining described above.
Antibodies
Antibodies used were: anti-TCRβ-APC-Cy7 (BD Pharmigen, clone H57-597), anti-CD44-BV605 (BD Horizon, clone IM7), anti-CD4-PE-Cy7 (BD Pharmigen, clone RM4-5), anti-CD8β-PE (BD Pharmigen, clone H35-17.2), anti-CD8β-PerCP-Cy5.5 (BioLegend, clone YTS156.7.7, San Diego, CA), anti-Foxp3-FITC (eBioscience, clone FJK-16s), anti-CD25-PerCP-Cy5.5 (BD Pharmigen, clone PC61), anti-CD278-PE-Cy5 (eBioscience, clone 7E.17G9), (anti-Tbet-ef660 (eBioscience, clone eBio4B10), anti-Ki67-AF700 (BD Pharmigen, clone B56), anti-IL-10-APC (BD Pharmigen), anti-TNFα (BD Pharmigen), anti-IFNγ BV650 (BD Horizon, clone, XMG1.2), anti-mAmphiregulin-Biotin (R&D systems, Minneapolis, MN), anti-Ly6C-PerCp-Cy5.5 (eBioscience, clone HK1.4), anti-CD11b-BV605 (BD Horizon, clone M1/70), anti-Ly6G-PECF94 (BD Horizon, clone 1A8), anti-CD45-V450 (BD Horizon, clone 30-F11), anti-Nos2-AF488 (eBioscience, clone CXNFT), anti-CD68-PE-Cy7 (eBioscience, clone FA-11), anti-CD206-APC (BioLegend, clone C068C2), anti-TCRβ-FITC (ebioscience, clone H57-597), anti-Ly6G-FITC (BioLegend, clone 1A8), anti-NK1.1-FITC (eBioscience, clone PK136), Anti-SiglecF-BB515 (BD Horizon, clone E50-2440), anti-TER119-FITC (BD Pharmigen, clone TER-119), anti-CD45-APC-eFluor 710 (eBioscience, clone 30-F11), and anti-CD115-BV421 (BioLegend, clone AFS98). Flow cytometry data was acquired using a BD LSRFortessa Cell Analyzer and analyzed using FlowJo version 10.0.8 (Tree Star, Ashland, OR).
Treg Adoptive Transfer
CD4+CD44-Foxp3- cells were FACS sorted from Foxp3-eGFP knock-in reporter mice. Isolated cells were stimulated with 1 μg/mL plate bound anti-CD3/anti-CD28 (ebioscience) for 48 hours in conditioning media. After the initial 24 hours, 100 U/mL recombinant IL-2 (Peprotech, Rocky Hill, NJ) was added to the wells/conditioning media. To generate non-polarized Tregs, conditioning media contained 10 ng/mL TGFβ, 5 μg/mL anti-IL-4 and anti-IFNγ blocking antibodies (BD Pharmigen). To generate Th1-Tregs, conditioning media contained 10 ng/mL TGFβ (Peprotech, Rocky Hill, NJ), 5 μg/mL anti-IL-4 blocking antibodies and 50 ng/mL IFNγ (Peprotech, Rocky Hill, NJ). Cells were expanded in vitro for a total of 8 days. HBSS, 7.5×105 non-polarized Tregs, or Th1-Tregs were adoptively transfered intravenously into CD45+ B6.SJL 23 days post-infection. Organs were harvested for analysis six days following adoptive transfer.
Treg-specific depletion in DEREG mice
DEREG mice (previously described in (Lahl et al., 2007)) were orally infected with ME49 cysts. Starting 23 days post-infection, both DTR+ and DTR- mice were treated with 1 μg diphtheria toxin (DT) i.p. for a total of three injections every other day. Organs were harvested for analysis one day following the last injection.
qRT-PCR of muscle differentiation factors
Tissues from experimental mice were isolated and preserved in RNAlater (Qiagen, Venlo, Netherlands) for RNA isolation by Trizol extraction. Isolated RNA was converted to cDNA (iScript, BD Biosciences) assayed for muscle differentiation factors MyoD and myogenin by real-time PCR (iTaq Universal SYBR Green supermix, BD Biosciences) using primers purchased from BioRad (PrimePCR assay, BioRad laboratories). Ct values were normalized the housekeeping gene 18s rRNA (Forward: 5′-GCAATTATTCCCCATGAACG-3′, Reverse: 5′-GGGACTTAATCAACGCAAGC-3′). 100 ng of skeletal muscle RNA was used for qRT-PCR analysis.
Quantification of parasite burden
Tissues from experimental mice were isolated and preserved in RNAlater (Qiagen) for DNA extraction with the QiagenDNeasy Blood and Tissue kit or RNA isolation with Trizol (Life Technologies). Total parasite burden was quantified by PCR-amplification of a T. gondii specific gene, B1 from DNA isolated from target tissues (Forward: 5′-TCCCCTCTGCTGGCGAAAAGT-3′, Reverse: 5′-AGCGTTCGTGGTCAACTA TCGATTG - 3′). Ct values were compared to a standard curve constructed from B1 amplification of known T. gondii genomic DNA concentrations. Tachyzoite and bradyzoite parasite burden was quantified by qRT-PCR of Enolase2 (Forward: 5′-CCGTTACTCAACTTCCAACA-3′, Reverse: 5′-CCATCGGTCAACAAGTCAA-3′) and Bag1 (Forward: 5′-GGGATGTACCAAGCATCCTG-3′, Reverse: AGGGTAGTACGCCAGAG CAA-3′), respectively. Ct values were compared to a standard curve constructed from Enolase2 or Bag1 amplification of known T. gondii RNA concentrations. 1 μg of skeletal muscle DNA or RNA was used for PCR analysis of parasite burden.
Statistics
All statistics were generated using Graphpad Prism v6.0c (GraphPad Software, La Jolla, CA).
Results
T. gondii infection causes myositis and loss of function in skeletal muscle
Following oral infection with five type II ME49 T. gondii cysts, C57BL/6 mice experience significant weight loss acutely 9-12 days post-infection and fail to recover weight similar to that of their naïve counter parts as chronic infection (day 30) is established (Figure 1A). We speculated that the inability to efficiently regain weight was due, in part, to ongoing skeletal muscle damage following acute infection. To this end, we compared inflammatory cell infiltration and muscle fiber damage in mock and parasite-infected mice by H&E staining (Figure 1B,C). Specifically, analysis of cellular damage elaborated during infection shows little inflammatory infiltrate in muscle at day 12 but by day 30 post infection, extensive inflammatory infiltrate in perivascular, perimysial, and endomysial locations was observed (Figure 1B,C). The presence of pre-necrotic hyaline and necrotic fibers is comparably increased at day 12 and 30 post infection (Figure 1B,C). Additionally, there is an increased number of myofibers with central nuclei and prominent nucleoli, an indicator of regenerating fibers at 30 days post infection (Figure 1B,C). To determine if this damage evidenced in histology resulted in physiological consequences and alterations in muscle function, we measured muscle strength in infected mice using an inverted screen test. Chronically, infected mice (>day 30) displayed significant muscle weakness as compared to their naïve counterparts (Figure 1D).
Figure 1. T. gondii causes myositis and skeletal muscle damage.

(A) C57BL/6 mice were orally infected with 5 ME49 T. gondii cysts. Body weight was monitored for up to 35 dpi. (B) Representative images of H&E stained skeletal muscle sections from naïve and infected (30 dpi.) mice. (C) Blinded histopathological scoring of muscle damage and inflammation naïve and infected (12 and 30 dpi.) skeletal muscle. (D) Functional muscle strength was quantified in naïve and infected (>30 dpi.) mice by measuring hang time on Kondziela's inverted screen test. ***P< 0.001, Student's t test (A-D) Results are representative of at least two experiments with n = 4 mice per group/experiment; error bars are the SD.
To investigate myositis elicited by T. gondii in skeletal muscle, we next characterized the immunological landscape of infected skeletal muscle. To this end, we analyzed whole tissue RNA from naïve and chronically infected skeletal muscle for differential expression of gene targets and pathways previously implicated in musculoskeletal diseases. 379 unique targets were screened by qRT-PCR using a predesigned musculoskeletal disease pathway panel (Biorad). A Th1 driven immune response is protective during both acute and chronic stages (19, 23–26). As expected we find a robust increase in whole tissue expression of Th1 cytokines including Il12a, il1a, il2, il12b and Ifng in day 30 infected muscle that were below the level of detection in naïve skeletal muscle (Figure 2A left panel). Of note, transcripts encoding proteins involved in immunomodulatory pathways, such as il1rn, il10 and il10ra, in d30 infected muscle are expressed at a several fold increase over its expression in naïve skeletal muscle (Figure 2A right panel). The data from this screen describe an environment in which Th1 inflammatory pathways are active despite a concurrent increase in immunomodulatory mechanism, ultimately propagating unresolved inflammation. Furthermore, at the cellular level total CD4+ and CD8+ T cells dramatically increase in infected muscle while muscle from naïve controls shows few infiltrating T cells (Figure 2B,C). Both muscle CD4+ and CD8+ effector T cells highly express the Th1 canonical transcription factor Tbet (Figure 2D). Upon restimulation, both CD4+ and CD8+ effectors from day 24 infected muscle produce IFNγ but not IL-17A (Figure 2E,F). Notably, these populations persist in the infected muscle as evidenced by the continued expression of high levels of Tbet and the production of IFNγ that was detectable directly ex vivo 60 days post infection (Figure 2G).
Figure 2. Inflammatory landscape of infected skeletal muscle is highly Th1-polarized.

(A) Inflammatory targets involved in known musculoskeletal disease pathways were explored by screening RNA isolated from the skeletal muscle of naïve mice and mice 30 dpi against 379 unique targets by qRT-PCR. Left: Heatmap was generated from CT values of targets normalized to housekeeping (GAPDH) from naïve and infected mice. Right: Relative gene expression of infected skeletal muscle targets normalized to housekeeping and naïve skeletal muscle. Statistics were calculated on log-transformed fold change values. (B,C) Quantification of (B) TCRβ+CD4+ T cells and (C) TCRβ+CD8+ T cells in spleen and skeletal muscle after infection (D) Representative FACS plots of Tbet expression by Tconv (TCRβ+CD4+Foxp3-CD44+) in naïve and infected muscle (30 dpi). (G) Representative FACS plots of Tbet and IFNγ by Tconv from naïve and infected muscle ex vivo (60 dpi.). (E,F) Representative FACS plots of IL-17 and IFNγ production by (E) Tconv and (F) CD44+CD8+ T cells from naïve and infected muscle following 3-hour PMA/Ionomycin stimulation (24 dpi.). (A-F) Results are representative of at least three experiments with n = 4 mice per group/experiment; error bars are the SD. **P < 0.01; ***P <0.001; ****P < 0.0001; (A) ANOVA with Tukey's multiple comparisons test (B,C) Student's t test.
Long lasting alterations in skeletal muscle macrophage populations during infection
Due to the fact we observed such striking muscle damage and loss of function, we next investigated macrophage polarization in infected muscle, as its coordination is essential for proper muscle fiber regeneration (4). Consistent with previous reports, naïve muscle consists mostly of M2-polarized macrophages (∼80%) ((27) and Figure 3A-C). By day 12 post infection, we observe an increase in IM/M1-polarized macrophages and a dramatic reduction in M2, which markedly shifts the IM/M1 to M2 ratio heavily in favor of IM/M1s (Figure 3A-C). By 24 days post infection, muscle macrophages express M2 surface markers, such as CD206 and the absolute number of M2 increases (Figure 3A-C). Notably, they may not be fully functionally polarized to the M2 phenotype as they continue to express iNOS, an IM/M1 feature (Figure 3D). Furthermore, this increase in M2 is not paralleled by a contraction of IM/M1 as typically seen during skeletal muscle repair from sterile injury. Rather IM/M1 accumulation is sustained as chronic infection is established (Figure 3C). We postulated this altered macrophage polarization and/or function results in impairment of muscle regeneration leading to on-going muscle damage.
Figure 3. T. gondii infection expands IM/M1 populations.
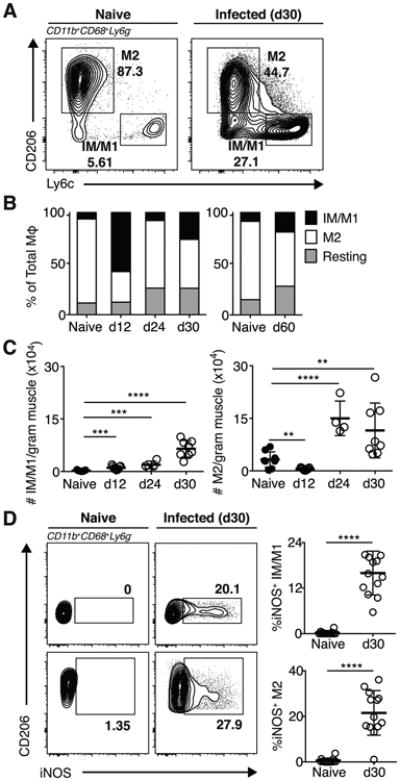
(A) Gating scheme for IM/M1 and M2 MΦ were gated on CD45+CD11b+Ly6G-CD68+ cells.(B) Frequencies and (C) absolute numbers of IM/M1 (Ly6ChiCD206lo), M2 (Ly6CloCD206hi), and resting (Ly6CloCD206lo) MΦ in naïve and infected muscle. (A-B) Results are representative of at least two experiments with n ≥ 4 mice per group/experiment; error bars are the SD. (D) Representative FACS plots and graphical representation of iNOS expression by muscle IM/M1 and M2 in naïve and infected mice. Results are representative of at least four experiments with n = 4 mice per group/experiment. **P < 0.01; ***P < 0.001; ****P < 0.0001; (C) ANOVA with Tukey's multiple comparisons test, (D) Mann-Whitney U test.
T. gondii infection results in sustained reduction of Tregs frequencies throughout infection
Recent studies of models of sterile muscle injury and muscular dystrophy (mdx) have shown during proper muscle repair a required presence of Tregs in promoting the shift of macrophage populations from pro-inflammatory IM/M1 to the pro-regenerative M2 (12, 13). Critically, the emergence of Tregs in muscle after sterile injury coincides with the transition from IM/M1 to M2-mediated phases of repair (12, 13). Prior works have described a collapse of the Treg compartment both locally in the GI tract, as well as systemically during acute lethal infection with T. gondii (28–30). Decreased Treg numbers are associated with limited Treg conversion and a lack of IL-2 availability in the gut (28). However, the long-term dynamics of the Treg compartment, especially the muscle, have not been investigated. During infection, we observe a steady increase in the number of Tregs during early-infection, accompanied by increased proliferative potential at day 24 of infection (Figure 4A). However, the substantial infiltration of CD4+Foxp3- Tconv and CD8+ T cells into the muscle results in significantly reduced proportions of Tregs to Tconv (< 6% Tregs) during chronic infection (Figure 4A). These findings are in striking contrast to the aforementioned sterile injury model of muscle damage where Tregs represent ∼40% of all CD4+ T cells (31). This is significant because relatively fewer Tregs are available to regulate the expanded effector populations.
Figure 4. Treg kinetics and phenotype during chronic infection.
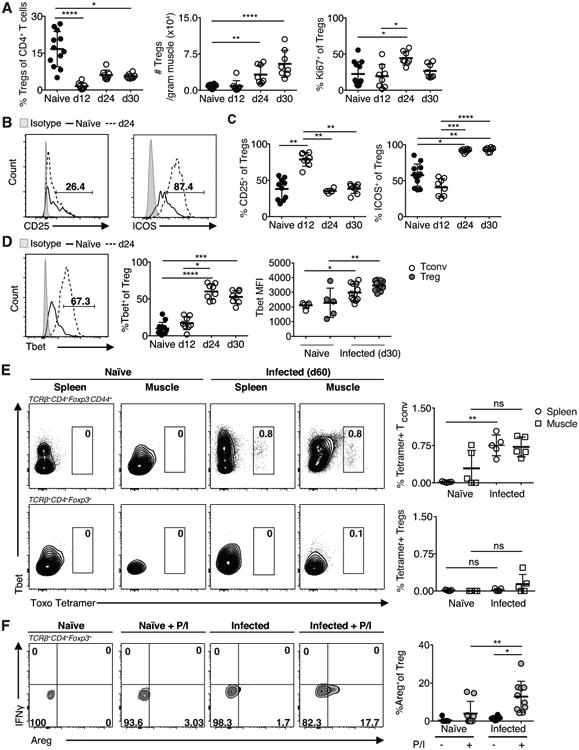
(A) Quantification of Treg (TCRβ+CD4+Foxp3+) frequency, absolute number and proliferation (KI67+) during infection. (B) Representative FACS plots (C) and graphical summary of CD25 and ICOS expression in Tregs from naïve and infected (24-days p.i.) muscle. (D) Left and middle: Representative FACS plots and graphical summary of Tbet expression in skeletal muscle Tregs during infection. Right: Tbet MFI in Tconv and Treg 30 dpi. (E) Representative FACS plots and graphical summaries of antigen specific CD44+CD4+Tconv and Tregs in the spleen and skeletal muscle during long-term infection as detected by staining with MHC class II tetramers loaded with T. gondii antigenic peptide (F) FACS plots and graphical representation of Areg expression by Tregs isolated from naïve and infected (24-days p.i.) muscle following 3-hour PMA/Ionomycin (P/I) restimulation. Results are representative of at least three experiments with n ≥ 4 mice per group/experiment; error bars are the SD. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Kruskal-Wallis test.
Treg populations can be subset by expression of a series of markers into two populations of central memory (CD25hi) and effector memory (ICOShi) as described in (32). We analyzed the expression of these proteins and found at day 12 there are high-levels of CD25 expression that are not sustained during later time points. Instead ICOS expression is favored by muscle Tregs (Figure 4B,C). Also, phenotypically, a large proportion of these Tregs express the Th1-associated transcription factor Tbet (Figure 4D). Of these Tbet+Tregs, the per cell expression of Tbet is comparable to that of Th1 (Figure 4D). Furthermore, in contrast to Tconv, the presence of a durable population of T. gondii antigen specific Tregs is not detected in the spleen or skeletal muscle during infection (Figure 4E).
As a readout of function, we assessed expression of various pro- and anti-inflammatory cytokines by muscle Tregs from day 24 infected mice by restimulating with the mitogen PMA and ionomycin. Stimulated muscle Tregs did not produce IL-10 (data not shown). Previously it was shown muscle Tregs express high levels of the epidermal growth factor family member amphiregulin (Areg); this expression is thought to promote muscle fiber regeneration by acting on stem cells in the skeletal muscle (12). We find infection increases Areg expression and restimulation increases that expression (Figure 4F). We next assessed pro-inflammatory cytokine IFNγ from muscle Tregs in response to restimulation and did not find skeletal muscle Tregs producing IFNγ (Figure 4F).
Transfer of Th1-iTregs promote IM/M1-accumulation in infected skeletal muscle
We postulated the reduced proportion of Tregs in infected muscle was inadequate to promote efficient transition from IM/M1 to M2 during chronic infection, resulting in the observed accumulation of IM/M1. Furthermore, chronic exposure to IFNγ has been shown to sensitize Tregs to IL-12 signaling and potentially promote IFNγ production by Tregs (28, 33), which could hinder their capacity to promote M2 differentiation. To test if insufficient numbers of Tregs or their chronic exposure to IFNγ results in the accumulation of IM/MI during infection, we adoptively transferred inducible Tregs cultured in the absence (iTreg) or presence (Th1-iTreg) or recombinant IFNγ for 7-9 days. To generate iTregs for transfer, we FACS sorted CD4+Foxp3- T cells from Foxp3eGFP reporter mice, converted them to iTregs in the presence of TGFβ, and expanded/conditioned them in vitro for 7-9 days. iTregs and Th1-iTregs were resorted on the basis of GFP+ (Foxp3+) cells prior to transfer at day 23 of infection to ensure transfer of a pure population of iTregs (> 98%, data not shown). Day 23 of infection was chosen as the transfer date because M2 populations re-emerge and Tregs are actively proliferating at day 24 post-infection (Figure 3C, 4A). Skeletal muscle was harvested 6 days post-transfer to monitor IM/M1 and M2. Transfer of iTregs did not significantly alter IM/M1 and M2 numbers, proportions, and proliferative capacity remained similar to transfer controls (Figure 5A-D). Surprisingly, Th1-iTregs adoptively transferred into mice 23 days post infection increases IM/M1 in the skeletal muscle (Figure 5A-C). This increase was not due to the action of transferred Th1-iTregs on in situ IM/M1 proliferation (Figure 5D). Transfer of 7.5×105 iTregs or Th1-iTregs did not appreciably alter the absolute number of skeletal muscle Treg (Supplemental 2A). Furthermore, these changes occurred in the absence of enhanced immunopathology assessed by elevated liver enzyme (AST/ALT) levels (Supplemental 2B). To better understand how transfer of iTregs and Th1-iTregs result in differential effects on the macrophage population, we assessed their ability to produce a variety of cytokines following stimulation with PMA/Ionomycin after culture. Notably, no differences were observed in their capacity to produce IL-4, IL-13, IL-17, or IL-10 (Supplemental 1A,B). Both iTregs and Th1-iTregs produced Areg (Supplemental 1B). While there was a small but significantly increased ability of Th1-iTregs to produce IFNγ statistically, IFNγ-producing Th1-iTregs still only represent ∼2% of total Th1-iTregs (Supplemental 1C). Our data suggests that Th1-iTreg IFNγ production is likely not the distinguishing factor promoting the IM/M1 accumulation in the infected skeletal muscle. Together, our data suggests long-term exposure of Tregs to IFNγ, may alter their canonical function and lead to the accumulation of IM/M1 instead of M2.
Figure 5. Th1-iTreg Transfer increases M1 populations.
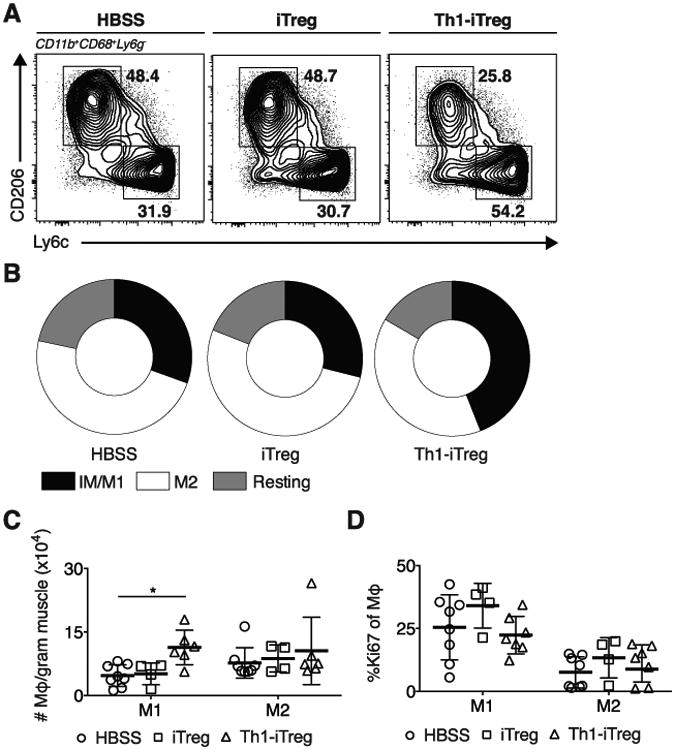
HBSS, iTregs, or Th1-iTreg were adoptively transferred into infected mice 23 dpi. (A) Representative FACS plots, (B) Distribution and (C) absolute number of IM/M1, M2 and resting MΦ six days after cell-transfer. (D) Effect of Treg transfer on skeletal muscle M1 and M2 proliferation (Ki67+) six days following transfer. Results are representative of n ≥ 5 per group from two experiments, error bars are the SD. *P < 0.05; Student's t test.
Treg ablation shifts macrophage populations in favor of M2 and rescues muscle regeneration
Given the remarkable finding that Th1-iTregs enhances IM/M1 numbers in infected muscle, we asked if skeletal muscle Tregs were directly contributing to the pathology observed during chronic infection. To this end, we used transgenic mice expressing the simian Diphtheria Toxin Receptor driven by Foxp3 (DTR) to systemically deplete Tregs 23 days post infection. DTR- and DTR+ mice were infected and treated with diphtheria toxin (DT) starting on day 23 and depletion of skeletal muscle Tregs was confirmed (Supplemental 2C). Given that adoptive transfer of Th1-iTregs increased IM/M1 (Figure 5C), we asked whether Treg depletion restores M2 proportions, leading to improved repair. Indeed, Treg ablation (DTR+) increases muscle M2 proportions while DT treatment in DTR- mice had no effect (Figure 6A,B). The shift towards a statistically significant increase in the M2 to IM/M1 ratio was due to the combinatory effect of both a decrease in IM/M1 and increase in M2 absolute numbers (Figure 6C). Furthermore, the effects of Treg depletion on IM/M1 and M2 numbers occurred in the absence of increased immunopathology as assayed by serum AST/ALT levels and total T cell/IFNγ-producing T cells in the skeletal muscle (Supplemental 2D,E). The frequency and number of total iNOS-producing macrophages remained unaltered following Treg depletion (Supplemental 2F). However, reactive nitrogen species may not be the key agent inciting damage, and the re-emergence of macrophages expressing M2 phenotypic markers such as the scavenger receptor CD206, indicates an increased proportion of macrophages that may have enhanced regenerative functions despite also producing iNOS.
Figure 6. Treg depletion is accompanied by shift toward increased M2.

Systemic depletion of Tregs following DT treatment of DTR transgenic mice. (A) Representative FACS plots, (B) distribution and (C) absolute number and fold change of IM/M1 and M2 one day following final DT treatment. (D) Effect of Treg depletion on skeletal muscle M1 and M2 proliferation (Ki67+) one day following final DT treatment. (A-D) Results are representative of n = 10 per group from four experiments. (E) Gating scheme for Ly6chi and Ly6clow monocytes. Frequencies and absolute numbers of (F) bone marrow and (G) blood Ly6chi and Ly6clow monocytes (CD45+TCRβ-Ly6G-SiglecF-NK1.1-TER119-CD11b+CD115+) one day following final DT treatment. Results are representative of n = 13 per group from three experiments; error bars are the SD. *P < 0.05; **P < 0.01; (C, E, F) Kruskal-Wallis test, (D-F) Student's t test.
To understand how Treg ablation influences the macrophage compartment, we first investigated the in situ proliferation of IM/M1 and M2 macrophages following depletion. Ki67-expression in both IM/M1 and M2 remained unaltered (Figure 6D). We next asked whether Treg ablation acted on a systemic level, influencing M1 and M2 precursor monocytes from the bone marrow. Specifically, it has previously been reported that Ly6chi and Ly6clow monocytes preferentially give rise to M1 and M2, respectively (4, 34–37, gating scheme Figure 6E). Following Treg depletion during infection, the frequency of Ly6chi and Ly6clow monocytes is not notably altered (Figure 6F). Interestingly, the absolute number of Ly6clow monocytes is significantly increased whereas the number of Ly6chi monocytes trends upward (Figure 6F). We next asked whether this change was also reflected in the blood. However, no detectable changes were observed in either frequency or number of monocyte subsets in the blood (Figure 6G).
Strikingly, the depletion of Tregs is associated with an increased incidence of centrally nucleated muscle fibers demonstrating enhanced muscle regeneration (Figure 7A,B), suggesting that Tregs actively inhibit muscle regeneration during chronic infection. We did not observe alterations in the weights of mice during DT treatment (Supplemental 2G). Previously, it was demonstrated that M2 promote the early, proliferative stage of myogenesis following sterile injury (38). To assess whether increased proportions of M2 influence the myogenic processes, we measured levels of the transcription factors MyoD and myogenin, which regulate distinct phases of the myogenic differentiation program in skeletal muscle stem cell populations. MyoD is highly expressed in the mid-G1 phase and between the S and M phases of the cell cycle (39). Myogenin is expressed upon differentiation of myoblasts into multinucleated myotubes (39). While tissue level expression of MyoD is not significantly altered in infected DTR+ mice compared to DTR- mice, myogenin expression decreases following Treg depletion (Figure 7C).
Figure 7. Treg ablation rescues skeletal muscle fiber regeneration.

(A) Representative H&E stains of hindlimb muscles one day following final DT treatment in DTR- and DTR+ mice 28 dpi. Arrows indicate centrally nucleated myofibers. (B) Quantification of regenerating fibers (centrally nucleated myofibers) in muscle sections per mm2 (C) Fold expression of myogenic targets MyoD and myogenin in infected DTR+ skeletal muscle normalized to DTR- skeletal muscle following DT treatment Statistics were calculated on log-transformed fold change values. (D) qRT-PCR quantification of total parasite burden by B1 and (E) parasite stage specific transcripts Bag1 bradyzoite (left) and Eno2 tachyzoite (right) following DT treatment. Results are representative of n = 10 per group from four experiments; error bars are the SD. *P < 0.05; Student's t test
Further, it was previously demonstrated that M2 have anti-cyst activity (40). We assessed whether a shift in proportions to favor M2 improved regeneration in part through decreasing parasite burden. Total parasite burden was assessed by the T. gondii tandem-arrayed 35-fold repetitive gene, B1. Treg depletion resulted in a non-significant trend downward in global parasite burden (Figure 7D). Transcripts Eno2 (tachyzoite) and Bag1 (bradyzoite) were also measured to determine if changes in parasite life-stage following Treg depletion occur. While detection of Eno2 does not significantly change, expression of Bag1 decreases (Figure 7E). Collectively, our data shows that chronic T. gondii infection causes ongoing inflammation and muscle damage. Furthermore, skeletal muscle Tregs in T. gondii infected mice directly contribute to muscle pathology by hindering muscle regeneration through promoting IM/M1 accumulation, and consequently influencing the myogenic program.
Discussion
In recent years, great progress has been made in examining the role of inflammation in sterile muscle injury and genetic diseases. These studies have shown that muscle repair is highly dependent on the sequential and discrete actions of pro-inflammatory macrophages (M1) and pro-regenerative (M2) macrophages (1–3). Importantly, dysregulation of macrophage populations and the inability to repair tissue damage is central to the pathogenesis of many inflammatory myopathies. In many myopathies including polymyositis and dermatomyositis, inappropriate macrophage accumulation and activation lead to invasion and destruction of non-necrotic muscle fibers (7, 8, 16) Notably, prolonged M1 presence during muscle injury may lead to delayed or aberrant repair (7–13), while exuberant M2 activity may cause muscle fibrosis (6).
Previous reports have demonstrated sensorimotor deficits during T. gondii infection, however these findings were attributed solely to neurological abnormalities (41–43). Our data compellingly show muscle damage, ongoing myositis and loss of muscle function within infected muscle. This previously under appreciated pathology is associated with prolonged accumulation of IM/M1 in skeletal muscle during infection. Furthermore, it appears that M2 polarization is aberrant as we observe expression of iNOS from these cells. A previous report demonstrated that macrophages exposed to T. gondii express features of both M1 and M2 macrophages (44). Recent studies show a tissue-specific role for resident Tregs in the critical transition of pro-inflammatory IM/M1 to pro-regenerative M2 during tissue repair process (12, 13). Surprisingly, we find that during chronic toxoplasma infection Tregs acquire a pathogenic capacity to promote skeletal muscle damage.
The ability to adapt to local inflammatory environments is important for Treg homeostasis and function (30, 45). It is known that exposure to Th1 inflammation endows Tregs with an enhanced capacity to traffic to and survive within local Th1 inflamed environments through acquiring Th1 effector traits, such as Tbet and CXCR3 expression (30, 46). Furthermore, transcriptomic analysis of Tregs cultured in Th1 environments identifies Tregs with hallmark features like Tbet and CXCR3 as phenotypically distinct from Tregs cultured in neutral conditions (30). Consistent with this, we show that in the Th1 environment of chronically infected skeletal muscle, Tregs highly express Tbet. However, little is known about the long-term consequences of extended exposure to a pro-inflammatory milieu on Treg physiology such as that experienced during chronic inflammatory disease states. We show here that there can be detrimental consequences on the ability of Tregs to continue to perform homeostatic functions in the face of long-term inflammation. However it remains to be determined whether Tbet directly plays a role in the pathogenic capacity of these Th1-Tregs. Taken together with our current data, the paradigm of repair must be expanded to account for how pre-existing inflammation may modulate Treg physiology and ultimately alter progression of the repair process.
The role of Tregs during chronic infection with T. gondii was previously suggested to be limited during late-stage infection (47, 48). Studies showed that depletion of CD25-expressing cells with the anti-CD25 antibody administered after 2 months of infection showed no alterations in the course of disease. Interestingly, previous studies and our data here highlight that Tregs found in chronic infection express low levels of CD25 compared to Tregs during acute infection ((28, 47, 49), Figure 4). Our study provides compelling evidence that during chronic infection Tregs do indeed participate in the immune response, albeit in a pro-inflammatory role instead of a suppressive role. Our results show that these Tregs modulate the IM/M1 and M2 balance by promoting the accumulation of IM/M1 in the skeletal muscle. This role is independent of IFNγ production by Tregs as we do not find Tregs produce IFNγ ex vivo or with mitogenic stimulation. This is in contrast to previous studies that showed Foxp3-expressing cells producing the pro-inflammatory cytokine IFNγ during acute lethal infection with T. gondii (28).
We show that ablation of pathogenic Tregs induced during infection coincides with increases in the proportion of pro-regenerative M2 accompanied by increases in the number of regenerating fibers. We did not observe an appreciable decrease in the production of iNOS by M2 suggesting that reactive nitrogen species may not be the key agent inciting damage since we observe a dramatic increase in muscle fiber regeneration. Treg depletion does not alter in situ proliferation of either IM/M1 or M2. However, we do observe an increased numbers of Ly6clow monocytes in the bone marrow. While this increase is not reflected in the blood, increased extravasation into the tissue may counter the increased input of Ly6clow monocytes into the blood. Alternatively, the increase in Ly6clow monocytes may represent an accumulation in the bone marrow due to hindered egress and the effect of Treg depletion on the balance of IM/M1 and M2 occurs at the tissue-level. In either circumstance, our results indicate a correlative relationship between the development of pathogenic Tregs during chronic infection, altered tissue macrophage homeostasis, and impaired muscle regeneration.
The results presented are consistent with previous in vitro co-culture studies in which M2 stimulated myoblast proliferation (38). Earlier reports note that this effect of M2 is not reliant on myoblast MyoD or myogenin expression (38). We show, enhanced proportions of M2 following Treg depletion correlates with increases in regenerating fibers concurrent with a decrease in myogenin expression. While myogenin is important for myotube terminal differentiation and fusion, its expression has been show to inhibit myoblast responsiveness to epidermal growth factors (50). Additionally, decreased myogenin expression may represent a myogenic checkpoint to ensure genetic stability in differentiated cells and proper regeneration (51). Taken together, these data suggest that pathogenic Tregs may cause a defective myogenic differentiation program through modulating macrophage subsets.
Alternatively, it is possible that Treg interactions with satellite cells are involved (12, 52). During sterile injury it was shown that Treg recruitment only occurred during the time when satellite cells expand. Furthermore, in vitro studies showed iTregs enhanced proliferation of satellite cells while preventing their further differentiation (12, 52). It is unclear if this prevention of differentiation occurs in vivo. Furthermore, Tregs in injured muscle show marked increases in the production of epidermal growth factor Areg that is concurrent with the M2 accumulation and proliferation of satellite cells (12). We find that Tregs from infected skeletal muscle express higher levels of Areg than naïve skeletal muscle Tregs. Albeit they do not express as much as Tregs in a sterile injured muscle where greater than half the population produced Areg (12). Given that Treg ablation leads to dramatic increases in regenerating fibers our data suggests the regenerative program during T. gondii is not reliant on Areg production by Tregs.
Our studies highlight the complexity of the potential interactions between the parasite, skeletal muscle fiber and immune response. While the depletion of Tregs may affect total parasitic activity, we do not observe significant overall alterations in the parasite burden following depletion as measured by the B1 gene. Previous studies have shown that M2 have anti-cyst properties during T. gondii infection in the brain (40). Interestingly, we show that following Treg depletion, increases in M2 proportions are associated with decreased expression of the bradyzoite gene bag1. Conversely, we do not detect alterations in tachyzoite specific gene expression as evidenced by non-significant changes in the eno2 transcript levels between Treg replete and depleted mice. Host cell differentiation status can also directly influence parasite growth. Notably terminally differentiated syncytial myotubes promote stage conversion to bradyzoites (22). Taken together, these findings raise intriguing questions about whether the parasite may be influencing the local environment to induce pathogenic Tregs as a means to remain encysted, evade immune detection, and increase the likelihood of transmission via the infectious cyst stage as ingestion of undercooked contaminated meats is the primary route of transmission. In addition, it is tempting to speculate lasting loss of muscle function may have an impact on an organismal level such that the host cannot escape predators and thus the parasite is more likely to be transmitted.
While the importance of innate cells to muscle fiber regeneration has been well characterized, the role of the adaptive immune response is still being explored. Moreover, the regulatory mechanisms that allow for the progression of the wound repair response have just recently begun to be elucidated. The critical role that Tregs play in promoting tissue repair mechanisms in response to damage of the skeletal muscle has been recently recognized (12, 13, 52). The interplay between these cell populations during chronic infection of Toxoplasma gondii has not been examined until now. Previous work has showed that in the gut and other tissues, Tregs have a protective role during T. gondii infection (28, 30). Our data shows the unexpected finding that Tregs promote muscle damage during chronic infection. As Treg-directed therapies gain traction, a deeper understanding of the consequences of chronic inflammation on Treg physiology and tissue-specific reparative programing will be a powerful asset in producing safe and efficacious therapies. Our results show that Tregs residing in infected skeletal muscle play a decidedly different role than what is normally the suppressive function associated with Foxp3-expressing cells. Identifying the determinants of immunoregulation during skeletal muscle infection will provide targets for novel immune-modulatory therapeutics for a range of inflammatory myopathy etiologies, not limited to infection.
Supplementary Material
Acknowledgments
We thank the University at Buffalo Histology core and the Confocal Microscopy and Flow Cytometry Core Facility at the University at Buffalo for their technical assistance. We thank the NIH Tetramer Core Facility for the T. gondii ME49 hypothetical protein tetramers. We thank Drs. Yasmine Belkaid, Joanne Konkel and John Grainger for discussion and their critical reading of the manuscript.
This work was supported by the Jacobs School of Medicine State University of New York at Buffalo (E.W.), and National Institute of Health grants AI124677 (I.J.B.) and AI007614 (R.M.J.).
Footnotes
Disclosures: The authors declare no competing financial interests.
References
- 1.Bosurgi L, Manfredi AA, Rovere-Querini P. Macrophages in injured skeletal muscle: a perpetuum mobile causing and limiting fibrosis, prompting or restricting resolution and regeneration. Front Immunol. 2011;2:62. doi: 10.3389/fimmu.2011.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marino M, Scuderi F, Provenzano C, Bartoccioni E. Skeletal muscle cells: from local inflammatory response to active immunity. Gene Ther. 2011;18:109–16. doi: 10.1038/gt.2010.124. [DOI] [PubMed] [Google Scholar]
- 3.Pillon NJ, Bilan PJ, Fink LN, Klip A. Cross-talk between skeletal muscle and immune cells: muscle-derived mediators and metabolic implications. Am J Physiol Endocrinol Metab. 2013;304:E453–65. doi: 10.1152/ajpendo.00553.2012. [DOI] [PubMed] [Google Scholar]
- 4.Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–69. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiendl H, Hohlfeld R, Kieseier BC. Immunobiology of muscle: advances in understanding an immunological microenvironment. Trends Immunol. 2005;26:373–80. doi: 10.1016/j.it.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Muñoz-Cánoves P. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rostasy KM, Schmidt J, Bahn E, Pfander T, Piepkorn M, Wilichowski E, Schulz-Schaeffer J. Distinct inflammatory properties of late-activated macrophages in inflammatory myopathies. Acta Myol. 2008;27:49–53. [PMC free article] [PubMed] [Google Scholar]
- 8.Rostasy KM, Piepkorn M, Goebel HH, Menck S, Hanefeld F, Schulz-Schaeffer WJ. Monocyte/macrophage differentiation in dermatomyositis and polymyositis. Muscle Nerve. 2004;30:225–30. doi: 10.1002/mus.20088. [DOI] [PubMed] [Google Scholar]
- 9.Watson NB, Schneider KM, Massa PT. SHP-1-dependent macrophage differentiation exacerbates virus-induced myositis. J Immunol. 2015;194:2796–809. doi: 10.4049/jimmunol.1402210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rayavarapu S, Coley W, Kinder TB, Nagaraju K. Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness. Skelet Muscle. 2013;3:13. doi: 10.1186/2044-5040-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Melton DW, Porter L, Sarwar ZU, McManus LM, Shireman PK. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am J Pathol. 2014;184:1167–84. doi: 10.1016/j.ajpath.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–95. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, Margeta M, Spencer MJ, Bluestone JA. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med. 2014;6(258):258ra142. doi: 10.1126/scitranslmed.3009925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T, Bourdon J, Rafei M, Galipeau J, Divangahi M, Petrof BJ. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med. 2014;6:1476–92. doi: 10.15252/emmm.201403967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt J, Rakocevic G, Raju R, Dalakas MC. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain. 2004;127:1182–90. doi: 10.1093/brain/awh148. [DOI] [PubMed] [Google Scholar]
- 16.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–82. doi: 10.1016/S0140-6736(03)14368-1. [DOI] [PubMed] [Google Scholar]
- 17.Frenkel JK. Pathophysiology of toxoplasmosis. Parasitol Today. 1988;4:273–278. doi: 10.1016/0169-4758(88)90018-x. [DOI] [PubMed] [Google Scholar]
- 18.Sullivan WJ, Jeffers V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol Rev. 2012;36:717–33. doi: 10.1111/j.1574-6976.2011.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki Y, Conley FK, Remington JS. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J Immunol. 1989;143:2045–50. [PubMed] [Google Scholar]
- 20.Takács AC, Swierzy IJ, Lüder CGK. Interferon-γ restricts Toxoplasma gondii development in murine skeletal muscle cells via nitric oxide production and immunity-related GTPases. PLoS One. 2012;7:e45440. doi: 10.1371/journal.pone.0045440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunter Ca, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol. 2012;10:766–778. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swierzy IJ, Muhammad M, Kroll J, Abelmann A, Tenter AM, Lüder CGK. Toxoplasma gondii within skeletal muscle cells: a critical interplay for food-borne parasite transmission. Int J Parasitol. 2014;44:91–8. doi: 10.1016/j.ijpara.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Yap G, Pesin M, Sher A. Cutting Edge: IL-12 Is Required for the Maintenance of IFN- Production in T Cells Mediating Chronic Resistance to the Intracellular Pathogen, Toxoplasma gondii. J Immunol. 2000;165:628–631. doi: 10.4049/jimmunol.165.2.628. [DOI] [PubMed] [Google Scholar]
- 24.Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A. Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-gamma production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J Immunol. 1991;146:286–92. [PubMed] [Google Scholar]
- 25.Sturge CR, Yarovinsky F. Complex immune cell interplay in the gamma interferon response during Toxoplasma gondii infection. Infect Immun. 2014;82:3090–7. doi: 10.1128/IAI.01722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munoz M, Liesenfeld O, Heimesaat MM. Immunology of Toxoplasma gondii. Immunol Rev. 2011;240:269–85. doi: 10.1111/j.1600-065X.2010.00992.x. [DOI] [PubMed] [Google Scholar]
- 27.Honda H, Kimura H, Rostami A. Demonstration and phenotypic characterization of resident macrophages in rat skeletal muscle. Immunology. 1990;70:272–7. [PMC free article] [PubMed] [Google Scholar]
- 28.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–86. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benson A, Murray S, Divakar P, Burnaevskiy N, Pifer R, Forman J, Yarovinsky F. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J Immunol. 2012;188:800–10. doi: 10.4049/jimmunol.1100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno EDT, Wagage S, Roos DS, Scott P, Turka La, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter Ca. The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity. 2012;37:511–23. doi: 10.1016/j.immuni.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–95. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, Campbell DJ. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. 2014;211:121–36. doi: 10.1084/jem.20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch Ma, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor β2. Immunity. 2012;37:501–10. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geissmann F, Jung S, Littman DR. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 35.Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M, Randolph GJ. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J Exp Med. 2006;203:583–97. doi: 10.1084/jem.20052119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of Blood Vessels and Tissues by a Population of Monocytes with Patrolling Behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 37.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–74. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol. 2012;189:3669–80. doi: 10.4049/jimmunol.1103180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee EJ, Malik A, Pokharel S, Ahmad S, Mir BA, Cho KH, Kim J, Kong JC, Lee DM, Chung KY, Kim SH, Choi I. Identification of genes differentially expressed in myogenin knock-down bovine muscle satellite cells during differentiation through RNA sequencing analysis. PLoS One. 2014;9:e92447. doi: 10.1371/journal.pone.0092447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nance JP, Vannella KM, Worth D, David C, Carter D, Noor S, Hubeau C, Fitz L, Lane TE, Wynn TA, Wilson EH. Chitinase dependent control of protozoan cyst burden in the brain. PLoS Pathog. 2012;8:e1002990. doi: 10.1371/journal.ppat.1002990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vyas A, Kim SK, Giacomini N, Boothroyd JC, Sapolsky RM. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc Natl Acad Sci USA. 2007;104:6442–7. doi: 10.1073/pnas.0608310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hermes G, Ajioka JW, Kelly KA, Mui E, Roberts F, Kasza K, Mayr T, Kirisits MJ, Wollmann R, Ferguson DJP, Roberts CW, Hwang JH, Trendler T, Kennan RP, Suzuki Y, Reardon C, Hickey WF, Chen L, McLeod R. Neurological and behavioral abnormalities, ventricular dilatation, altered cellular functions, inflammation, and neuronal injury in brains of mice due to common, persistent, parasitic infection. J Neuroinflammation. 2008;5:48. doi: 10.1186/1742-2094-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gulinell M, Acquarone M, Kim JH, Spray DC, Barbosa HS, Sellers R, Tanowitz HB, Weiss LM. Acquired infection with Toxoplasma gondii in adult mice results in sensorimotor deficits but normal cognitive behavior despite widespread brain pathology. Microbes Infect. 2011;12:528–537. doi: 10.1016/j.micinf.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patil V, Zhao Y, Shah S, Fox BA, Rommereim LM, Bzik DJ, Yap GS. Co-existence of classical and alternative activation programs in macrophages responding to Toxoplasma gondii. Int J Parasitol. 2014;44:161–4. doi: 10.1016/j.ijpara.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell DJ, Koch Ma. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11:119–30. doi: 10.1038/nri2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Couper KN, Lanthier PA, Perona-Wright G, Kummer LW, Chen W, Smiley ST, Mohrs M, Johnson LL. Anti-CD25 antibody-mediated depletion of effector T cell populations enhances susceptibility of mice to acute but not chronic Toxoplasma gondii infection. J Immunol. 2009;182:3985–94. doi: 10.4049/jimmunol.0803053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, Wynn TA, Kamanaka M, Flavell RA, Sher A. Conventional T-bet(+)Foxp3(-) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–83. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wohlfert E, Grainger J, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, Paul WE, Bosselut R, Wei G, Zhao K, Oukka M, Zhu J, Belkaid Y. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121:4503–4515. doi: 10.1172/JCI57456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf JR, Hirschhorn RR, Steiner SM. Growth factor responsiveness: role of MyoD and myogenin. Exp Cell Res. 1992;202:105–12. doi: 10.1016/0014-4827(92)90409-2. [DOI] [PubMed] [Google Scholar]
- 51.Yang ZJP, Broz DK, Noderer WL, Ferreira JP, Overton KW, Spencer SL, Meyer T, Tapscott SJ, Attardi LD, Wang CL. p53 suppresses muscle differentiation at the myogenin step in response to genotoxic stress. Cell Death Differ. 2015;22:560–73. doi: 10.1038/cdd.2014.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, Almada AE, Mondino A, Wagers AJ, Manfredi AA, Rovere-Querini P. FOXP3+ T Cells Recruited to Sites of Sterile Skeletal Muscle Injury Regulate the Fate of Satellite Cells and Guide Effective Tissue Regeneration. PLoS One. 2015;10:e0128094. doi: 10.1371/journal.pone.0128094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.