

Abstract
The Gal repressosome is a higher-order nucleoprotein complex that represses transcription of the gal operon in Escherichia coli. During the repressosome assembly, a DNA loop is formed by the interaction of two GalR dimers, bound to two spatially separated operators, OE and OI, flanking the gal promoters. Structure-based genetic analysis indicated that GalR homodimers interact directly and form a V-shaped stacked tetramer in repressosome, further stabilized by HU binding to an architecturally critical position on the DNA. In this scheme of GalR tetramerization, the alignment of the operators in the DNA loop could be in either parallel (PL) or antiparallel (AL) mode. As each mode can have two alternative geometries differing in the mutual stacking of the OE- and OI-bound GalR dimers, it is possible to have four different DNA trajectories in the repressosome. Feasibilities of these trajectories were tested by in vitro transcription repression assays, first by isolating GalR mutants with altered operator specificity and then by constructing four different potential loops with mutant GalR heterodimers bound to specifically designed hybrid operators in such a way as to give rise to only one of the four putative trajectories. Results show that OE and OI adopt a mutual antiparallel orientation in an under-twisted DNA loop, consistent with the energetically optimal structural model. In this structure the center of the HU-binding site is located at the apex of the DNA loop. The approach reported here can be used to distinguish between otherwise indistinguishable DNA trajectories in complex nucleoprotein machines.
Keywords: Gal repressor, transcription regulation, DNA loop, oriented heterodimers, operators, HU
Complex nucleoprotein machines carry out various DNA transaction reactions, transcription, replication, and recombination (Echols 1990). In these complexes, proteins bound to spatially separated sites on the DNA interact with each other, resulting in a DNA loop in the intervening region. In many cases, architectural nucleoid proteins stabilize the DNA loops (Santero et al. 1992; Aki et al. 1996; Nash 1996; Wassem et al. 2000; Thomas and Travers 2001; de Beer et al. 2002). One such higher-order nucleoprotein complex, the Gal repressosome, represses transcription of the gal operon of Escherichia coli. Assembly of the Gal repressosome requires (1) binding of two dimeric GalR proteins to two spatially separated operator elements, OE and OI, (2) negatively supercoiled DNA, (3) optimal angular orientation of the two operator sites, and (4) specific binding of the architectural protein HU to a DNA site (hbs) in the interoperator region (Aki and Adhya 1997; Lewis and Adhya 2002). Protein–protein interactions in the repressosome structure have been extensively studied. Isolation and characterization of GalR mutants, which bind to DNA but do not repress transcription, indicated that DNA-bound GalR dimers form tetramers in loop formation (Geanacopoulos and Adhya 2002). The intrinsic ability of GalR to tetramerize was also shown by isolation of GalR mutants which function without HU, as well as by chemical cross-linking (Semsey et al. 2002). Results of homology-based site-directed mutagenesis of the GalR dimer suggested that DNA-bound GalR dimers form a V-shaped, stacked tetramer in the repressosome (Geanacopoulos et al. 1999, 2001; Geanacopoulos and Adhya 2002).
The structure of the GalR tetramer allows the operators in the loop to be either parallel (wrapping mode; Friedman et al. 1995; Tsodikov et al. 1999) or antiparallel (looping mode; Geanacopoulos et al. 2001). In each mode the DNA loop can have two alternative trajectories, which differ in the relative stacking arrangements of the two operator-bound GalR dimers (see Fig. 3, below). Evaluation of DNA elastic energies in stereochemical models of the different loop conformations indicated that antiparallel looping is energetically less expensive in the Gal repressosome structure (Geanacopoulos et al. 2001). Visualization of the structures by atomic force microscopy (AFM) also supported the antiparallel model (Virnik et al. 2003). Although a specific interaction of GalR and HU was shown to be essential for loop formation, the HU-GalR contact is unlikely to be present in the repressosome; it appears that GalR piggybacks HU to its architecturally critical binding site on DNA (Kar and Adhya 2001; S. Roy, E. Dimitriadis, S. Kar, M. Geanacopoulos, M.S. Lewis, and S. Adhya, in prep.).
Figure 3.

Different modes of DNA looping in the Gal repressosome and transcription repression. Four possible arrangements of the GalR heterodimers on the operator sites are shown on the left. Each GalR dimer has two crescent-shaped tetramerization interfaces. Functional interfaces are colored green; the inactivated ones are red. GTGTAANC operator half sites are orange; GTGGTANC are light green. Arrows indicate the orientation of the operators. A V-shaped, stacked GalR tetramer (middle panel) forms upon interaction of the functional tetramerization interfaces (colored green on the left panel). The schematic DNA trajectory in each looping mode is shown in the middle panel. For detailed description of the antiparallel loops see Figure 6. Transcription of the gal promoters in different arrangements of GalR is shown on the right. Reactions contained 80 nM HU to allow repressosome formation. The transcript marked RNA1 served as an internal control (see text for details).
In this study, we used heterodimers of wild-type and nonlooping mutants of GalR specifically oriented on each operator to determine the feasibility of the four DNA trajectories. The oriented heterodimer formation followed the principle of Zhou et al. (1993, 1994). The results of transcription repression studies reported here support an antiparallel DNA loop in which the GalR dimer bound to OI stacks upon the one bound to OE, consistent with the previous theoretical calculations (Geanacopoulos et al. 2001). The approach reported here can be generally applicable to trace DNA trajectories in DNA loop-containing multiprotein complexes based on the “modular” principle (Antson et al. 2000; Dodd et al. 2001).
Results
HU independent repression of the P2 promoter in vivo
The HU protein was found to play a major role in DNA looping-dependent repression of the gal promoters (Aki et al. 1996). In the absence of HU, GalR binding to OE partially represses the P1 promoter and activates the P2 promoter (Choy and Adhya 1992). Although P2 repression is completely HU-dependent in vitro, P2 is not completely derepressed in vivo in the absence of HU. In the latter situation, addition of D-galactose, the inducer of the gal operon, derepressed P2 by an additional 10% (Lewis et al. 1999). However, the mechanism of this residual repression was unknown. In a similar setup, the homologous dimeric LacI repressor lacking the C-terminal domain also displayed residual repression, believed to be due to residual dimer–dimer interactions resulting in weak DNA looping (Muller et al. 1998). To investigate the mechanism and specificity of the weak HU-independent looping repression in the gal system, we investigated the effect of the GalR mutants GalRT322R and GalRH327R, defective in tetramerization, on the P2 activity in the absence of HU (Geanacopoulos et al. 2001). Neither of these mutations have any effect on the repression of the P1 promoter in vivo, suggesting that the two mutants have normal DNA-binding ability. In this study, the GalRT322R and GalRH327R mutations were created in multicopy plasmid pSEM1029 (Semsey et al. 2002) by PCR mutagenesis. Plasmids carrying the mutations were introduced into DM0022 ( ∼gusA) and DM0100 (
∼gusA) and DM0100 ( ∼gusA ΔhupAΔhupB; Lewis et al. 1999) cells. DNA looping was monitored by assaying β-glucuronidase activity in vivo. Both GalR mutants resulted in higher P2 promoter activity compared to that in the wild-type GalR in the absence of the HU protein, showing that the tetramerization interface of GalR dimers is involved in the residual repression of the P2 promoter (Fig. 1).
∼gusA ΔhupAΔhupB; Lewis et al. 1999) cells. DNA looping was monitored by assaying β-glucuronidase activity in vivo. Both GalR mutants resulted in higher P2 promoter activity compared to that in the wild-type GalR in the absence of the HU protein, showing that the tetramerization interface of GalR dimers is involved in the residual repression of the P2 promoter (Fig. 1).
Figure 1.

(A) Schematic structure of the P2∼gusA fusion used to study looping repression. In this construct the gal regulatory region contains the external (OE) and internal (OI) operator sites, the P2 promoter, and the HU-binding site hbs. The P1 promoter is inactivated by a point mutation. The transcription start site +1 of P1 is used as a reference in the numbering system. (B) Repression of the P2 promoter in vivo. Differential rates of β-glucuronidase synthesis from P2∼gusA in hup+ DM0022 (left panel) and in ΔhupAΔhupB strain DM0100 (right panel) cells in the presence of plasmid carrying galR+ (filled squares), galRH327R (open circles), and galRT322R (open squares).
Regulation of P2 in vitro
Previously, cross-linking experiments indicated an intrinsic ability of GalR to tetramerize at high concentrations (Semsey et al. 2002). We tested the effect of GalR concentrations on the regulation of the gal promoters in vitro. In a purified system, P2 transcription was enhanced by 80 nM GalR in the absence of HU, as observed previously, and D-galactose eliminated the activation. Hexahistidine-tagged wild-type GalR and GalRT322R mutant proteins were purified to >95% homogeneity as described in Materials and Methods. In vitro transcription assays were performed on supercoiled pSA850 plasmid DNA template carrying the wild-type gal regulatory region ( ; Lewis and Adhya 2002) at different GalR concentrations. The maximum enhancement of P2 activity, 1.5-fold, was obtained at ∼60 nM wild-type GalR concentration. The enhancement gradually diminished at a higher level of GalR (Fig. 2A,F). In contrast, the P2 enhancement by GalRT322R reached its saturation point at about 100 nM protein concentration, resulting in an 1.85-fold higher level (Fig. 2B,F). Both proteins showed the full enhancement when the OI operator was inactivated (Fig. 2C,D,F). Stronger repression of the P1 promoter in the presence of wild-type protein (Fig. 2E) can also be an indication of cooperativity between the OE- and OI-bound GalR dimers. These data demonstrate that the residual P2 repression is the result of specific interaction of operator-bound GalR dimers. Because the GalRT322R dimers did not show any repression, we propose that the nonassisted DNA loop formed in the absence of HU contains the same V-shaped stacked GalR tetramer that was described in the repressosome.
; Lewis and Adhya 2002) at different GalR concentrations. The maximum enhancement of P2 activity, 1.5-fold, was obtained at ∼60 nM wild-type GalR concentration. The enhancement gradually diminished at a higher level of GalR (Fig. 2A,F). In contrast, the P2 enhancement by GalRT322R reached its saturation point at about 100 nM protein concentration, resulting in an 1.85-fold higher level (Fig. 2B,F). Both proteins showed the full enhancement when the OI operator was inactivated (Fig. 2C,D,F). Stronger repression of the P1 promoter in the presence of wild-type protein (Fig. 2E) can also be an indication of cooperativity between the OE- and OI-bound GalR dimers. These data demonstrate that the residual P2 repression is the result of specific interaction of operator-bound GalR dimers. Because the GalRT322R dimers did not show any repression, we propose that the nonassisted DNA loop formed in the absence of HU contains the same V-shaped stacked GalR tetramer that was described in the repressosome.
Figure 2.

Effect of GalR on gal promoters in vitro. Results of in vitro transcription at varying concentrations of GalR (A–D). Plasmid DNA template  (pSA850) with GalR+ (A), and GalRT322R (B). Plasmid DNA template
(pSA850) with GalR+ (A), and GalRT322R (B). Plasmid DNA template 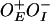 (pSA886) with GalR+ (C) and GalRT322R (D). Quantification of activity of P1 (E) and P2 (F) at varying concentrations of GalR. RNA1 transcript, which does not vary with GalR concentration, was used as an internal control between lanes. Promoter activities in the absence of GalR were used as a reference (1.00) in each experiment. Plasmid DNA template
(pSA886) with GalR+ (C) and GalRT322R (D). Quantification of activity of P1 (E) and P2 (F) at varying concentrations of GalR. RNA1 transcript, which does not vary with GalR concentration, was used as an internal control between lanes. Promoter activities in the absence of GalR were used as a reference (1.00) in each experiment. Plasmid DNA template 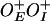 (pSA850) with wild-type GalR (black squares), and GalRT322R (red dots). Plasmid DNA template
(pSA850) with wild-type GalR (black squares), and GalRT322R (red dots). Plasmid DNA template 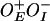 (pSA886) with wild-type GalR (green triangles) and GalRT322R (blue triangles).
(pSA886) with wild-type GalR (green triangles) and GalRT322R (blue triangles).
DNA trajectory in the repressosome
Two identical, symmetrically positioned tetramerization interfaces can be distinguished, one interface on each side of an operator-bound GalR dimer (Fig. 3). In principle, either of the two OE-bound GalR dimer interfaces can interact with any one of the OI-bound dimers. Thus, each pair of the four possible interfaces results in a different DNA trajectory in the DNA loop (Fig. 3). Our strategy to study the feasibility of different trajectories was to allow formation of only one of the four by orienting a heterodimer with one wild-type interface and the other mutant, in a specific way on each operator. Studying transcription of the P2 promoter by DNA looping in the four set-ups would determine the DNA trajectories that are feasible. The constructions are described below. The design of the heterodimers and the orientation on operators followed a modification of the principle reported by Zhou et al. (1993, 1994).
Isolation of GalR mutants with altered operator specificity
Orientation of GalR on the operator DNA requires the construction of hybrid operators and of GalR dimers from two subunits having different recognition specificities for the half-operators. To create GalR with altered DNA-binding specificity, we changed the critical first and/or second residues of the recognition helix (residues 15 and 16, respectively) of GalR to the corresponding residues of the homologous LacI. Operator sequences (GTGNNNNCGNNNNCAC), randomized at positions N, bound to the wild-type or mutant GalR proteins were enriched by using the SELEX method (Tuerk and Gold 1990). After four cycles the bound fraction was cloned into pSA850 plasmid, and several clones were sequenced (Table 1). Despite only a very limited difference between GalR and LacI in the recognition helix (V15A16TVSRVIN and Y17Q18TVSRVVN, respectively), none of the GalRV15Y,A16Q, GalRA16Q, and GalRV15Y hybrid homodimers were successful in DNA-binding experiments. However, the LacI protein, LacIY17V,Q18A, which carried the GalR recognition sequences, was shown to have DNA-binding specificity of GalR in in vitro DNA-binding assays as well as in in vivo transcription repression (Lehming et al. 1990; Barker et al. 1998). We found that another mutant, GalRA16T, successfully bound a fraction of the randomized operator sequences, examples of which are shown in Table 1, but not to the wild-type operator OE. To isolate an operator variant that GalRA16T does not recognize, we performed the same SELEX method of using the random pool of the operator sequences but with wild-type GalR protein. The bound fraction was amplified and then tested for binding of GalRA16T by electrophoretic mobility shift assay (EMSA). This time the nonbound fraction was further amplified and cloned into pSA850 plasmid. Sequences of randomly picked clones of the unbound fraction are shown in Table 1. Comparison of sequences from the GalRA16T-bound and -nonbound operator clones showed that GalRA16T has a higher affinity to an operator with the GTGGTANC half site than one with the GTGTAANC half site. This result was further confirmed by direct binding assays between GalRA16Tprotein and the two operators by EMSA (Fig. 4). Several other GalR mutants were also tested on these two DNA-binding sites to find one that has higher affinity to the operator with the GTGTAANC half site than to the one with GTGGTANC half site, exactly the opposite of the preference of GalRA16T. One such mutant was GalRV15T (Fig. 4). Based on these results we proposed that the heterodimer of GalRV15T and GalRA16T would bind to the hybrid GTGTAANCGNTACCAC operator site.
Table 1.
Operator preference of wild-type GalR and GalRA16T
| Bound by wild-type GalR | Bound by GalRA16T | Bound by wild-type GalR Not bound by GalRA16T |
|---|---|---|
| GTGTAATCGTTTTCAC | GTGGTACCGATACCAC | GTGTAAACGCTTTCAC |
| GTGTAAACGTTACCAC | GTGGTACCGTTCCCAC | GTGTAATCGATTACAC |
| GTGTAAACGATAACAC | GTGGTAGCGGTACCAT | GTGTAACCGTTACCAC |
| GTGTAAGCGGTACCAC | GTGGTACCGCTGCCAC | GTGTAAACGTTTTCAC |
| GTGTAATCGCTTCCAC | GTGGTAACGTTAACAC | GTGTAATCGATTACAC |
| GTGTAATCGATTACAC | GTGGTAGCGTTAACAC | GTGTAAACGTTCTCAC |
| GTGTAAACGCTCTCAC | GTGGTAACGTTTGCAC | GTGTAACCGTTTACAC |
| GTGTAACCGTTGTCAC | GTGGTAGCGTTACCAC | |
| GTGTAATCGTTAACAC | GTGGAACCGTTGCCAC | |
| GTGTAAACGCTTGCAC | GTGGGAGCGGTTTCAC | |
| GTGAAAGCGTTTCCAC | ||
| GTGAAAGCGTTACCAC | ||
| GTGAAAGCGCTAACAC | ||
| GTGGTACCGGTTCCAC | ||
| GTGGAAACGTTCTCAC | ||
| GTGGAAACGGTACCAC |
Figure 4.

EMSA of mutant operators in the presence of GalRV15T (top) and GalRA16T (bottom). Complexes were formed with 0.1 nM DNA and 0.1, 0.5, 1, and 10 nM GalR protein.
Subunit exchange of GalR dimers in solution
StrepII-tagged GalRA16T and HIS6-tagged GalRV15T proteins were purified as described in Materials and Methods. GalRV15T also had the T322R mutation, which prevented the tetramerization of the protein (Geanacopoulos and Adhya 2002). The purified proteins were mixed in different ratios and incubated overnight at 4°C. The difference in length of the HIS6 (longer) and StrepII (shorter) tags enabled us to distinguish the nature of the dimeric composition of the proteins simply by EMSA, when bound to DNA. The results of EMSA of binding of the hybrid operator sites GTGTAAACGATACCAC (“OE”) and GTGTAAACGATACCAT (“OI”) to different combinations of GalR mutants are shown in Figure 5. It was clear that whereas GalRA16T homodimers did not show significant binding, the GalRV15T variant bound efficiently to both hybrid operator sites. More importantly, at a 5:1 ratio of GalRA16T/GalRV15T, both the hybrid operators were occupied predominantly by the GalR heterodimers. At this ratio the estimated composition of the mixture is ∼69% GalRA16T homodimer, ∼30% GalRA16T/GalRV15T heterodimer, and ∼1% GalRV15T homodimer.
Figure 5.

Binding of GalR+, GalRA16T, and GalRV15T,T322R to hybrid operator sequences. Free (F) and GalR-bound (B) operators were separated by EMSA as described in Materials and Methods. Mobility of the StrepII-tagged GalR (designated as GalRS) complex was different from the mobility of the HIS6-tagged complex (designated as GalRH). In lanes 4 and 5, a ratio of the GalR-bound operators migrates at an intermediate position, showing that most of the bound operators are complexed with a heterodimer protein.
Transcription repression in different loop conformations
Operator sequences in plasmid pSA850 were substituted by the corresponding hybrid operator sequence to orient the interacting surfaces of GalR heterodimers in a given way (Fig. 3). Mutations in the operators did not have any effect on DNA looping-mediated transcription repression. In vitro transcription on the resulting plasmid DNAs in the presence of wild-type GalR protein showed that GalR homodimers could form repressosome and repress the gal promoters in the presence of HU (Fig. 3, lanes 3,6,9,12). In a similar experiment, GalRA16T/GalRV15T, T322R heterodimers repressed P2 promoter effectively only when the orientation of the functional tetramerization surfaces would allow the AL1 loop formation (Fig. 3, cf. lanes 2 and 5,8,11). The lack of repression in the case of PL1 and PL2 loops is entirely consistent with the DNA elastic energy calculations (Geanacopoulos et al. 2001), demonstrating that the energies required for the DNA deformations in these loops are rather large and would exceed the energy yield of the GalR tetramerization. To further investigate the reason for failure of repression in the AL2 loop, we constructed stereochemical models of the transient GalR–DNA loops formed in the absence of HU.
Modeling the AL1 and AL2 loops
DNA elastic energy calculations were performed in both the AL1 and AL2 configurations (Table 2). The AL1 loop has slightly lower deformation energy than that of the AL2 loop, with the difference not exceeding 2 kcal/mole. Most of the deformation energy (75%) is associated with the DNA bending, whereas the energy cost of the twist angle deviations from their equilibrium values is moderate in both loop configurations. The optimized loops are presented in Figure 6. Although of overall similar configurations, the loops have certain differences.
Table 2.
Energy of DNA bending and twisting for the antiparallel loops
| Etotal | Etwist | Ebend | |
|---|---|---|---|
| Linear DNA + GalR | |||
| AL1 loop | 26.0 | 6.4 | 21.2 |
| AL2 loop | 28.0 | 7.6 | 22.2 |
| Linear DNA + GalR + HU bound to hbs (+6.5) | |||
| AL1 loop | 9.4 | 8.7 | 5.8 |
| AL2 loop | 25.5 | 14.9 | 13.6 |
Values for the loops formed by transient interaction of operator-bound GalR dimers were calculated as described in Materials and Methods. Energy values (kcal/mole) obtained in the presence of HU, bound at hbs (+6.5), are as described in Geanacopoulos et al. (2001).
Figure 6.

Stereoviews of the minimal energy configurations of the loop modes AL1 (A, back view) and AL2 (B, front view). Two GalR dimers (purple and teal blue) are shown as ribbons. The OE operator is highlighted with red color, and the OI is highlighted with yellow. The -10 element of the P2 promoter is colored blue and orange. The AL1 and AL2 loops have similar overall structure. However, the direction of local DNA bending is different in two modes; the DNA surface that is inside at the AL1 loop apex is turned halfway outside in the AL2 loop (cf. orientation of the DNA grooves in the colored DNA fragments). The experimentally observed HU-binding site (hbs, position +6.5, 6 bp are colored magenta and orange) and putative binding site (“hbs”, position -14.5) occupy structurally equivalent segments of the AL1 and AL2 loops relative to the corresponding apex (indicated with dashed white arrows).
It follows from the simulations that only the AL1 loop has DNA configuration favorable for HU binding at the position +6.5, corresponding to the experimentally observed hbs locus (Aki and Adhya 1997). In contrast, hbs is located too far from the loop apex to act as a site of the sharp DNA bending in the AL2 loop. Accordingly, the results of energy calculations indicate that the AL2 loop is not stabilized by HU binding at the position +6.5 (Table 2; Geanacopoulos et al. 2001).
Rather, a DNA fragment with its center located around position -14 in the AL2 loop is a structural equivalent of hbs in the AL1 loop (Fig. 6). First, it is the most strongly bent DNA fragment in the AL2 loop. Second, the DNA minor groove in the center of this fragment is oriented inward toward the loop, which is similar to the minor groove orientation at hbs in the AL1 loop. Such an orientation would provide the proper phasing of this putative HU-binding site relative to the loop apex and operators. At the same time, structural details of the two loop apexes differ somewhat. The position -13.5 in the AL2 loop is analogous to the position to the position +6.5 (hbs) in the loop AL1 in terms of the distances to the operators (measured in base pairs). However, the total twist between the center of the closest operator, OI, and position +6.5 in the AL1 loop is about 30° less than the corresponding twist between the closest operator, OE, and position -13.5 in the AL2 loop (Table 3). Instead, the position -14.5 in the AL2 loop is analogous to the position +6.5 in the AL1 loop in terms of phasing between HU-binding site and operators as follows from the calculations (Table 3).
Table 3.
Distances and twist angles between operators and putative HU binding sites in the antiparallel loops formed in the absence of HU
| AL1 loop
|
AL2 loop
|
||||
|---|---|---|---|---|---|
| OI to +6.5 bp/deg | OE to +6.5 bp/deg | OE to -13.5 bp/deg | OI to -13.5 bp/deg | OE to -14.5 bp/deg | OI to -14.5 bp/deg |
| 47 bp/108.7° | 66 bp/-46.8° | 47 bp/138.5° | 66 bp/-84.2° | 46 bp/101.5° | 67 bp/-47.2° |
The experimentally observed position of HU binding, +6.5, is favorable only in the AL1 loop, as shown by the energy calculations (Geanacopoulos et al. 2001), and it provides implausible HU binding site in the loop AL2 (Fig. 6). Accordingly, the distance and twist angles have not been calculated between this position and the operators in the AL2 loop. Rather, the position -13.5 in the AL2 loop is an equivalent of the position +6.5 in the AL1 loop in terms of the distance from the operators, and the position -14.5 in terms of phasing relative to the operators (see text for further discussion).
All twist angles were brought to the range -180° to 180° by subtracting the integer number of complete turns, n × 360, where n is the nearest integer to the (Total twist)/360°.
Discussion
In transcription repression in prokaryotes, sequence-specific regulatory proteins prevent RNA polymerase binding by steric occlusion or modify RNA polymerase action by contacting one of its subunits (Choy et al. 1995a; Schlax et al. 1995; Monsalve et al. 1997; Yamamoto and Ishihama 2003). It is commonly believed that, unlike histones in eukaryotes, nucleoid associated histone-like proteins do not have a general inhibitory role in gene repression in prokaryotes. The Gal repressosome is a complex prokaryotic nucleoprotein structure that likely combines transcription repression and chromosome compaction (Virnik et al. 2003). Understanding the assembly and defining the structure may provide valuable information on the interaction of specific and nonspecific DNA-binding proteins and the effect of their concerted action on the structural organization of the bacterial nucleoid. Although the kinetic and thermodynamic parameters of the GalR repressosome have been determined (Lia et al. 2003), the mechanism of its assembly is largely unknown. In one model, the strong binding of GalR or GalR–HU complex to the operator sequences precedes HU binding to DNA. The interaction between GalR and HU brings HU to the vicinity of the gal regulatory region and facilitates its DNA binding (Kar and Adhya 2001).
We demonstrated the ability of GalR to repress the P2 promoter by specific interaction of repressor dimers in the absence of HU (Fig. 2). These results suggest that HU is not essential for loop closure. We assume that under normal conditions the DNA loop is formed by transient interaction of operator-bound GalR dimers, and HU plays an architectural role by stabilizing the repressosome structure. Increasing the loop size (Virnik et al. 2003) or strengthening the tetramerization interface of GalR dimers (Geanacopoulos and Adhya 2002; Semsey et al. 2002) enhanced nonassisted loop formation, resulting in reduced dependency on the architectural contribution of HU. In this model, the transient GalR–DNA loop structure is recognized by HU specifically. HU binding was not observed by LacI binding to gal DNA carrying two lac instead of gal operator sequences (Aki and Adhya 1997). LacI forms tetramers by flexible C-terminal heptad repeats. The latter loop did not show a kinked DNA by electron microscopy (Mandal et al. 1990). Similarly, tetramerization of hybrid GalR via the LacI flexible C-terminal heptad repeats results in poor repression compared to dimeric GalR in vivo (Barker et al. 1998). Hence, the rigid stacking interaction of wild-type GalR dimers may play a key role in creating a high-affinity kinked HU-binding site in the DNA loop.
Based on DNA wrapping in LacI–DNA interactions, the parallel mode was suggested as a general model of DNA loop formation (Tsodikov et al. 1999). However, in AFM studies of the Gal repressosome, the parallel geometry of DNA loop was not observed (Virnik et al. 2003). Evaluation of the four different models of repressosome structures, containing a V-shaped GalR tetramer and HU bound at position +6.5, also indicated that parallel looping is energetically more expensive (Geanacopoulos et al. 2001). Comparison of the DNA bending energy in the antiparallel geometries suggested that the transient AL1 or AL2 GalR–DNA loops can form with similar chances in the absence of HU (Table 2). However, efficient looping repression was observed experimentally only in the AL1 conformation, consistent with the predictions from energy evaluation of HU-assisted DNA loops (Geanacopoulos et al. 2001). Two models could explain the failure to observe repression in the AL2 loop scenario: (1) Loop closing by GalR in the AL2 configuration does not lead to the formation of a high-affinity HU-binding site, thus preventing a stable repressosome formation. Although structures of the putative HU-binding sites are similar in AL1 and AL2 loops, the sequences at these positions are different (Fig. 6). Although the sequence dependence of HU binding has never been reported, such a possibility cannot be ruled out completely. (2) HU recognizes the transient AL2 GalR–DNA loop and a stable complex is formed but the latter is not able to repress the transcription. Similar energy values were obtained for the AL2 loop in the absence or presence of HU binding to hbs, suggesting that HU binding at position +6.5 does not significantly contribute to the stability of the AL2 loop (Table 2; Geanacopoulos et al. 2001). The structural analog of hbs in the potential AL2 loop is centered at position -13.5/-14.5. In this structure RNA polymerase may open up the closed complex, facilitating transcription (Choy et al. 1995b) or oust HU from the -13.5/-14.5 site, which overlaps the -10 element (Fig. 6.), thus destabilizing the loop and reactivating the promoter. We are currently testing why the set-up with the AL2 mode of looping failed to repress transcription.
In summary, we propose that direct interaction of operator-bound GalR dimers can close the gal DNA loop on its own and create a high-affinity HU-binding site. Occupation of this site by HU results in repressosome formation. The assembled repressosome contains a loop with an undertwisted antiparallel DNA trajectory.
Materials and methods
DNA manipulation methods
Plasmid manipulations followed protocols described in Sambrook and Russel (2001). Transformations were performed with XL-1 Blue competent cells (Stratagene). Restriction endonucleases and DNA oligonucleotide primers were purchased from Invitrogen, PCR (GeneAmp XL) and sequencing (ABI Prism) kits from Applied Biosystems, and DNA purification kits from QIAGEN. DNA sequencing reactions were analyzed in a PerkinElmer/Applied Biosystems (model 373A) automated sequencer.
Electrophoretic mobility shift assay (EMSA)
32P-labeled DNA fragments and GalR were mixed in the same buffer used for in vitro transcription assays (see below) together with 10% glycerol and 2.5 ng/μL sonicated salmon sperm DNA. After incubation for 10 min at room temperature, reaction mixtures were loaded on a 4% polyacrylamide gel and electrophoresed with 1.5 mA/cm of gel in 1× TBE buffer at room temperature.
Synthesis of partially random gal operator sequences
gal operators with partially random DNA sequences used in the first round of selection were synthesized in vitro from two partially complementary oligonucleotides (5′-TCTACGGGGTCT GACGCTCAGTGTGNNNNCGNNNNCACTGACAATTAA TCATCGGCATAG-3′ and 5′-ATGCCGATATACTATGCCG ATGATTAATTGTCA-3′). Oligonucleotides (3 nM) were mixed in 50 μL 1× B buffer (Promega) and annealed by heating to 95°C and slow cooling to 30°C. After addition of 1 mM dNTPs and 5 μCi [α-32P]-dATP, the 3′ ends were extended with Klenow DNA polymerase (Invitrogen) at 37°C for 1 h. The resulting fragments were extracted with phenol, then with chloroform, and finally precipitated with ethanol.
In vitro selection of operator sequences
GalR-bound and nonbound 32P-labeled random operator sequences were separated by EMSA as described above. After electrophoresis was completed, GalR-bound DNA were extracted from gel with a QIAquick Gel Extraction Kit (QIAGEN). The isolated operator pool was amplified in a PCR reaction using oligonucleotides 5′-ATGCCGATATACTATGCCGATGATTA ATTGTCA-3′ and 5′-CCTTTGATCTTTTCTACGGGGTCTG ACGCTCAGT-3′ as primers.
In vitro transcription
Transcription reactions were performed as described (Geanacopoulos et al. 1999). The reaction mixture (50 μL) contained 20 mM Tris acetate at pH 7.8, 10 mM magnesium acetate, 200 mM potassium glutamate, 2 nM DNA template, and 20 nM RNA polymerase. After incubation of the reactions at 37°C for 5 min, transcription was started by the addition of 1.0 mM ATP, 0.1 mM GTP, 0.1 mM CTP, 0.01 mM UTP, and 5 μCi of [α-32P]UTP (3000 Ci/mmole). Reactions were terminated after 10 min by addition of an equal volume of transcription loading buffer (0.025% bromophenol blue, 0.025% xylene cyanol, 0.01 M EDTA, and 90% deionized formamide). After heating at 90°C for 3 min, the samples were loaded onto 8% polyacrylamideurea DNA sequencing gels. DNA bands were quantified using the ImageQuant™ PhosphorImager (Molecular Dynamics). PhosphorImager readings were analyzed using the GraphPad Prism 2.0 program (GraphPad Software).
Assay of β-glucuronidase activity
Cells were grown overnight in LB medium and diluted 50-fold for further growth in M63 supplemented with 0.4% (w/v) D-fructose, 0.1% (w/v) casamino acids, and 0.004% (w/v) vitamin B1. At various times, aliquots of cells were removed, pelleted, and resuspended in M63 medium containing 100 μg/mL chloramphenicol and stored on ice (Wilson et al. 1992). The optical density of cells was measured at 600 nm. The activity of β-glucuronidase from the galP2∼gusA fusion was determined by the SOFTMAX microplate spectrophotometer system. Aliquots of 50 μL of permeabilization buffer (100 mM Tris at pH 7.8, 32 mM sodium phosphate, 8 mM dithiothreitol, 8 mM CDTA, and 4% Triton X-100) containing 200 μg/mL polymyxin B (Schupp et al. 1995) were placed in the wells of a microtiter plate, followed by the addition of 100 μL of cells. Cells were allowed to permeabilize at room temperature for 15 min before 50-μL aliquots of GUS assay buffer (0.5 mM dithiothreitol, 1 mM EDTA, 50 mM sodium phosphate at pH 7.0) containing 1.25 mM α-p-nitrophenyl β-D-glucuronide (PNPG) were added (Wilson et al. 1992). The rate of β-glucuronide hydrolysis was determined at 405 nm at 37°C.
Protein purification
Hexahistidine-tagged GalR was purified as described (Semsey et al. 2002). Briefly, harvested cells were resuspended in 1/40 volume of Lysis I buffer (50 mM sodium phosphate at pH 8.0, 0.5 mg/mL lysozyme) and stored on ice for 30 min. An equal volume of Lysis II buffer (50 mM sodium phosphate at pH 8.0, 2 M NaCl, 8 mM imidazole, 20% glycerol, 0.01% Triton X-100) was added and incubated for 30 min on ice. The cell debris was removed by centrifugation at 10,000g for 1 h. Addition of 3% Ni-NTA slurry (QIAGEN) to the solution was followed by 1-h incubation at 4°C. A Poly-Prep Chromatography Column (Bio-Rad) was used to collect the protein bound to Ni-NTA agarose from the mixture. Twenty column-volumes of washing buffer (50 mM sodium phosphate at pH 8.0, 600 mM NaCl, 60 mM imidazole, and 10% glycerol) was allowed to flow through the column. GalR was eluted by four column-volumes of elution buffer (50 mM sodium phosphate at pH 8.0, 600 mM NaCl, 10% glycerol) containing 250 mM imidazole and fraction containing GalR were stored at -80°C in 100-μL aliquots. StrepII-tagged protein was purified similarly. Cell lysate was incubated with Strep-Tactin Sepharose (IBA) for 1 h and loaded on a Poly-Prep Chromatography Column. Twenty column-volumes of washing buffer (50 mM sodium phosphate at pH 8.0, 600 mM NaCl, and 10% glycerol) was allowed to flow through the column. GalR was eluted by five column-volumes of elution buffer (50 mM sodium phosphate at pH 8.0, 600 mM NaCl, 10% glycerol, and 2.5 mM desthiobiotin).
HU protein was purified according to the method described by Aki et al. (1996).
DNA elastic energy calculations
The energy of the DNA elastic deformations was calculated similar to the procedure described by Geanacopoulos et al. (2001). Namely, the total energy of the loop was modeled as a sum of the energies of the deformations in the DNA dimeric steps not occupied by GalR or HU:
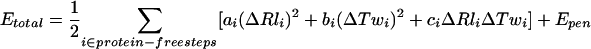 |
1 |
Here, i varies from 1 to 112 in the case of HU free GalR–DNA loop, and from 1 to 73 in the case of complete GalR–DNA–HU nucleoprotein assemblage; ΔRli and ΔTwi are the deviations of the Roll and Twist angles at the i-th dimeric step from the equilibrium values obtained from the analysis of the measurements of the DNA twisting in solution (Kabsch et al. 1982) and analysis of the DNA deformability in the complexes with proteins (Olson et al. 1998); ai, bi, and ci are the elastic constants impeding bending and twisting deformations in the step (Olson et al. 1998). In the analysis, the equilibrium values for the Twist and Roll angles and the values of the elastic constants are sequence-dependent. The bend-twist cross-term accounts for the correlation between the Twist and Roll angles (Olson et al. 1998). The penalty term, Epen, secures the fast closure of the loop in the course of simulation. The standard energy minimization procedure was applied to optimize the DNA loop configuration under the structural constrains imposed by the proteins. It included several consecutive runs of 10,000 linear searches for each loop configuration. In addition to the total energy, the bending and twisting energies (the first and second terms in Eq. 1) were documented as well.
The models for GalR bound to DNA were taken from the crystal structures of PurR–DNA complex (Schumacher et al. 1994), and the model for HU bound to DNA was taken from the complex based on the affinity cleavage data (Lavoie et al. 1996).
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1209404.
References
- Aki T. and Adhya, S. 1997. Repressor induced site-specific binding of HU for transcriptional regulation. EMBO J. 12: 3666-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aki T., Choy, H.E., and Adhya, S. 1996. Histone-like protein HU as a specific transcriptional regulator: Cofactor role in repression of gal transcription by Gal repressor. Genes Cells 1: 179-188. [DOI] [PubMed] [Google Scholar]
- Antson A.A., Burns, J.E., Moroz, O.V., Scott, D.J., Sanders, C.M., Bronstein, I.B., Dodson, G.G., Wilson, K.S., and Maitland, N.J. 2000. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 430: 805-809 [DOI] [PubMed] [Google Scholar]
- Barker A., Fickert, R., Oehler, S., and Muller-Hill, B. 1998. Operator search by mutant Lac repressors. J. Mol. Biol. 278: 549-558. [DOI] [PubMed] [Google Scholar]
- Choy H.E. and Adhya, S. 1992. Control of gal transcription through DNA looping; inhibition of the initial transcribing complex. Proc. Natl. Acad. Sci. 89: 11264-11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy H.E., Park, S.W., Aki, T., Parrack, P., Fujita, N., Ishihama, A., and Adhya, S. 1995a. Repression and activation of transcription by Gal and Lac repressors: Involvement of α subunit of RNA polymerase. EMBO J. 14: 4523-4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy H.E., Park, S.W., Parrack, P., and Adhya, S. 1995b. Transcription regulation by inflexibility of promoter DNA in a looped complex. Proc. Natl. Acad. Sci. 92: 7327-7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer T., Fang, J., Ortega, M., Yang, Q., Maes, L., Duffy, C., Berton, N, Sippy, J., Overduin, M., Feiss, M., et al. 2002. Insights into specific DNA recognition during the assembly of a viral genome packaging machine. Mol. Cell 9: 981-991. [DOI] [PubMed] [Google Scholar]
- Dodd I.B., Perkins, A.J., Tsemitsidis, D., and Egan, J.B. 2001. Octamerization of lambda CI repressor is needed for effective repression of P(RM) and efficient switching from lysogeny. Genes & Dev. 15: 3013-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols H. 1990. Nucleoprotein structures initiating DNA replication transcription, and site-specific recombination. J. Biol. Chem. 265: 14697-14700. [PubMed] [Google Scholar]
- Friedman A.M., Fischmann, T.O., and Steitz, T.A. 1995. Crystal structure of Lac repressor core tetramer and its implications for DNA looping. Science 268: 1721-1727. [DOI] [PubMed] [Google Scholar]
- Geanacopoulos M. and Adhya, S. 2002. Genetic analysis of GalR tetramerization in DNA looping during repressosome assembly. J. Biol. Chem. 277: 33148-33152. [DOI] [PubMed] [Google Scholar]
- Geanacopoulos M., Vasmatzis, G., Lewis, D.E.A., Roy, S., Lee, B.K., and Adhya, S. 1999. GalR mutants defective in repressosome formation. Genes & Dev. 13: 1251-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geanacopoulos M., Vasmatzis, F., Zhurkin, V.B., and Adhya, S. 2001. Gal repressosome contains an antiparallel DNA loop. Nat. Struct. Biol. 8: 432-436. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Sander, C., and Trifonov, E.N. 1982. The ten helical twist angles of B-DNA. Nucleic Acids Res. 10: 1097-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S. and Adhya, S. 2001. Recruitment of HU by piggyback: A special role of GalR in repressosome assembly. Genes & Dev. 15: 2273-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie B.D., Shaw, G.S., Millner, A., and Chaconas, G. 1996. Anatomy of a flexer–DNA complex inside a higher-order transposition intermediate. Cell 85: 761-771. [DOI] [PubMed] [Google Scholar]
- Lehming N., Sartorius, J., Kisters-Woike, B., von Wilcken-Bergmann, B., and Muller-Hill, B. 1990. Mutant lac repressors with new specificities hint at rules for protein–DNA recognition. EMBO J. 9: 615-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D.E.A. and Adhya, S. 2002. In vitro repression of the gal promoters by GalR and HU depends on the proper helical phasing of the two operators. J. Biol. Chem. 277: 2498-2504. [DOI] [PubMed] [Google Scholar]
- Lewis D.E.A., Geanacopoulos, M., and Adhya, S. 1999. Roles of HU and DNA supercoiling in transcription repression: Specialized nucleoprotein repression complex at gal promoters in Escherichia coli. Mol. Microbiol. 31: 451-462. [DOI] [PubMed] [Google Scholar]
- Lia G., Bensimon, D., Croquette, V., Allemand, J.F., Dunlap, D., Lewis, D.E., Adhya, S., and Finzi, L. 2003. Supercoiling and denaturation in Gal repressor/heat unstable nucleoid protein (HU)-mediated DNA looping. Proc. Natl. Acad. Sci. 100: 11373-11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal N., Su, W., Haber, R., Adhya, S., and Echols, H. 1990. DNA looping in cellular repression of transcription of the galactose operon. Genes & Dev. 4: 410-418. [DOI] [PubMed] [Google Scholar]
- Monsalve M., Calles, B., Mencia, M., Salas, M., and Rojo, F. 1997. Transcription activation or repression by phage psi 29 protein p4 depends on the strength of the RNA polymerase–promoter interactions. Mol. Cell. 1: 99-107. [DOI] [PubMed] [Google Scholar]
- Muller J., Barker, A., Oehler, S., and Muller-Hill, B. 1998. Dimeric lac repressors exhibit phase-dependent co-operativity. J. Mol. Biol. 284: 851-857. [DOI] [PubMed] [Google Scholar]
- Nash H.A. 1996. The E. coli HU and IHF proteins; accessory factors for complex protein–DNA assemblies. In Regulation of gene expression in E. coli (ed. E.C.C. Lin and A.S. Lynch), pp. 149-179. R.G. Landes Company, Georgetown, TX.
- Olson W.K., Gorin, A.A., Lu, X.J., Hock, L.M., and Zhurkin, V.B. 1998. DNA sequence-dependent deformability deduced from protein–DNA crystal complexes. Proc. Natl. Acad. Sci. 95: 11163-11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. and Russel, D.D. 2001. Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Santero E., Hoover, T.R., North, A.K., Berger, D.K., Porter, S.C., and Kustu, S. 1992. Role of integration host factor in stimulating transcription from the sigma 54-dependent nifH promoter. J. Mol. Biol. 227: 602-620. [DOI] [PubMed] [Google Scholar]
- Schlax P.J., Capp, M.W., and Record Jr., M.T. 1995. Inhibition of transcription initiation by lac repressor. J. Mol. Biol. 245: 331-350. [DOI] [PubMed] [Google Scholar]
- Schumacher M.A., Choi, K.Y., Zalkin, H., and Brennan, R.G. 1994. Crystal structure of LacI member, PurR, bound to DNA: Minor groove binding by α helices. Science 266: 763-770. [DOI] [PubMed] [Google Scholar]
- Schupp J.M., Travis, S.E., Price, L.B., Shand, R.F., and Keim, P. 1995. Rapid bacterial permeabilization reagent useful for enzyme assays. Biotechniques 19: 18-20. [PubMed] [Google Scholar]
- Semsey S., Geanacopoulos, M., Lewis, D.E.A., and Adhya, S. 2002. Operator-bound GalR dimers close DNA loops by direct interaction: Tetramerization and inducer binding. EMBO J. 21: 4349-4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J.O. and Travers, A.A. 2001. HMG1 and 2, and related `architectural' DNA-binding proteins. Trends Biochem. Sci. 26: 167-174. [DOI] [PubMed] [Google Scholar]
- Tsodikov O.V., Saecker, R.M., Melcher, S.E., Levandoski, M.M., Frank, D.E., Capp, M.W., and Record Jr., M.T. 1999. Wrapping of flanking nonoperator DNA in lac repressor–operator complexes: Implications for DNA looping. J. Mol. Biol. 294: 639-655. [DOI] [PubMed] [Google Scholar]
- Tuerk C. and Gold, L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505-510. [DOI] [PubMed] [Google Scholar]
- Virnik K., Lyubchenko, Y.L., Karymov, M.A., Dahlgren, P., Tolstorukov, M.Y., Semsey, S., Zhurkin, V.B., and Adhya, S. 2003. `Antiparallel' DNA loop in Gal repressosome visualized by atomic force microscopy. J. Mol. Biol. 334: 53-63. [DOI] [PubMed] [Google Scholar]
- Wassem R., deSose, E.M., Yates, M.G., Pedrosa, F.D., and Buck, M. 2000. Two roles for integration host factor at an enhancer-dependent nifA promoter. Mol. Microbiol. 35: 756-764. [DOI] [PubMed] [Google Scholar]
- Wilson K.J., Hughes, S.G., and Jefferson, R.A. 1992. The Escherichia coli gus operon: Induction and expression of the gus operon in Escherichia coli and the occurrence and use of the gus in other bacteria. In Gus protocols: Using the gus gene as a reporter of gene expression (ed. S.R. Gallagher), pp. 7-22. Academic Press, San Diego, CA.
- Yamamoto K. and Ishihama, A. 2003. Two different modes of transcription repression of the Escherichia coli acetate operon by IclR. Mol. Microbiol. 47: 183-184 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Busby, S., and Ebright, R.H. 1993. Identification of the functional subunit of a dimeric transcription activator protein by use of oriented heterodimers. Cell 73: 375-379. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Pendergrast, P.S., Bell, A., Williams, R., Busby, S., and Ebright, R.H. 1994. The functional subunit of a dimeric transcription activator protein depends on promoter architecture. EMBO J. 13: 4549-4557. [DOI] [PMC free article] [PubMed] [Google Scholar]