

Abstract
The effect of Helicobacter pylori infection on human and murine primary gastric cells was determined. CagA was phosphorylated following adherence of H. pylori to primary human gastric cells. However, it did not adhere to human primary duodenal cells or murine gastric cells, and CagA could not be detected in cell lysates. Identification of an easily available animal model of infection in which the organism adheres to gastric mucosal cells would enhance studies of the virulence of H. pylori.
Helicobacter pylori causes gastritis and peptic ulcer disease (8, 14, 20), and infection has been associated with an increased risk of developing gastric cancer (15, 24). Only humans and nonhuman primates are naturally infected by H. pylori. One of the most striking characteristics of H. pylori is that it adheres only to gastric mucus-secreting cells in vivo (32). It is not known why H. pylori exhibits such a strict species and tissue tropism. H. pylori can infect mice under experimental conditions, but it does not infect them naturally (16, 18, 19). Upon adherence of the bacteria to cultured cells of various gastric and nongastric origins, the CagA protein of the bacteria is translocated into the host cell and subsequently tyrosine phosphorylated (1, 3, 21, 29). This process has also been shown to occur in vivo (35). It is dependent on genes present in the cag pathogenicity island (PAI), which code for a type IV secretion system. Although CagA is considered an important virulence factor for H. pylori, the relevance of CagA translocation to the pathogenesis of the organism is not yet clear. Strains with cagE deleted have been associated with diminished gastric inflammation in Mongolian gerbils (22), but deletion of the entire cag PAI region had no effect on gastric inflammation in mice or piglets (9). Due to the lack of a suitable animal model, there is a heavy reliance on in vitro models to assess the relevance of adherence to gastric cells in mediating disease.
Using primary cells isolated from different sites along the gastrointestinal tract, we have previously shown that H. pylori is capable of exhibiting a tropism for gastric cells in vitro similar to that seen in vivo (5). This suggests that primary gastric cells are ideal for assessing factors that may play a role in H. pylori pathogenesis. The aim of this study was to determine the effect of CagA-positive and CagA-negative H. pylori infection of human primary gastric cells. Because of the species specificity of H. pylori, we also aimed to determine if CagA translocation and tyrosine phosphorylation occurred when H. pylori was incubated with primary murine gastric cells, since the mouse model of infection is used extensively to assess potential virulence factors of the organism and candidate vaccines.
H. pylori strain NCTC 11637, a CagA-positive strain with a functional cag PAI (1), was obtained from the National Collection of Type Cultures (Public Health Laboratory Service, London, England). Strain SS1 was obtained from Adrian Lee, University of New South Wales, Sydney, Australia. Strain SS1 has been adapted to colonize mice (18) and is CagA positive. It has a complete cag PAI, but it has been shown to not be fully functional (7). Strain PU44 was isolated from the gastric biopsy material of a child. This strain is CagA negative and has only a partial cag PAI. It was found to be negative for cagE, cagM, and cagT and positive for cagG (this study, see below). Therefore, this strain did not have a functional cag PAI. Bacteria were harvested from Columbia blood agar (Gibco, Paisley, United Kingdom) plates containing 7% (vol/vol) defibrinated horse blood. Quantitation of bacteria in suspension was done by optical density measurement at 600 nm and by viable counts. Harvested bacterial pellets were resuspended in 567 μl of TE buffer (l0 mM Tris-HCl [pH 8.0] 1 mM EDTA), and genomic DNA was extracted by an adaptation of the cetyltrimethyl ammonium bromide-NaCl method of Ausubel et al. (2). We looked for the cagA gene and cag PAI genes in H. pylori strain PU44 by using PCR. The sequences of the cagA oligonucleotides amplified a region spanning nucleotides 2549 to 3967 (12) and yielded a product of approximately 1.4 kb with H. pylori strain NCTC 11637, which was used as a positive control for PCR experiments. The primers used for amplification of cagE, cagG, cagM, and cagT were as previously described (33). One hundred-nanogram aliquots of genomic DNA from H. pylori strains were amplified in 100-μl reaction mixtures. PCR products were resolved in 1% (wt/vol) agarose gels and stained with ethidium bromide. Strain NCTC 11637 was found to be positive for all genes tested, whereas strain PU44 was negative for cagA, cagE, cagM, and cagT but positive for cagG.
Whole-cell lysates of H. pylori cells were run on sodium dodecyl sulfate (SDS)-polyacrylamide gels (10% acrylamide). Proteins were transferred to nitrocellulose and membranes were probed with a whole-cell rabbit-raised CagA antiserum (1/5,000 dilution; generously provided by A. Covacci). Antigen-antibody complexes were detected with goat anti-rabbit immunoglobulin G (IgG) (Sigma) conjugated to peroxidase. Blots were developed with an enhanced chemiluminesence (ECL) system according to the manufacturer's instructions (Amersham Corp). Strain PU44 was negative for CagA. Strain NCTC 11637 was used as a positive control.
Human primary gastric and duodenal epithelial cells were isolated from biopsy specimens obtained from children undergoing endoscopy at Our Lady's Hospital for Sick Children, Dublin, Ireland. Ethical approval for this study was obtained from the Ethics Committee in the hospital. Biopsy tissue was checked for infection with H. pylori by Campylobacter-like organism testing, culture, and histology. Only tissue that was negative by all three criteria was used in this study. Cells were isolated from the biopsy tissue as previously described (5). Once isolated, cells were resuspended in growth medium comprising 50:50 Dulbecco's modified Eagle's medium-Ham's F-12 containing insulin (8 μg/ml), hydrocortisone (50 μg/ml), 10% (vol/vol) fetal calf serum, and 10-μg/ml each penicillin, streptomycin, and gentamicin. A total of 5 ×105 cells were plated onto sterile glass coverslips, which were placed in six-well plates, and grown for 2 days in a 5% CO2 incubator at 37°C prior to addition of the bacteria. Mice of the CD1 strain were fasted overnight and sacrificed by cervical dislocation, and the stomachs were removed. Whole mouse stomachs were chopped into small pieces with sterile scalpels, and cells were isolated and cultured as described above. Primary cell cultures stained positive with antipancytokeratin antibody (Sigma) revealed by fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibody confirming the epithelial origin of the cells.
Before inoculation, the plates were washed in prewarmed phosphate-buffered saline (PBS) (37°C) to remove any nonadherent cells. Antibiotic-free growth medium (2.5 ml) was then added to the cells. A total of 107 H. pylori bacteria were added to different wells, and culture medium was added to control cells. From this point onward, cells were maintained under microaerophilic conditions generated by use of Campygen Paks (Oxoid, Basingstoke, United Kingdom) at 37°C to provide suitable growth conditions for H. pylori. After 2 h, the coverslips were washed in PBS to remove nonadherent bacteria. Cells were stained in order to localize bacteria and tyrosine-phosphorylated proteins. After fixation in 2% (vol/vol) formaldehyde, cells were washed with PBS, blocked with 10% normal goat serum diluted in PBS containing 0.02% (wt/vol) saponin (Sigma), and then stained with a monoclonal antiphosphotyrosine antibody (PY99; Santa Cruz), and an anti-mouse Ig conjugated to Texas Red (Molecular Probes). Cells were then stained with a rabbit whole-cell anti-H. pylori antibody (raised in house) (5) and a secondary anti-rabbit FITC-conjugated antibody (Sigma). All antibodies were used at 1/100 dilutions, and the diluent was 10% (vol/vol) goat serum in PBS containing 0.02% (wt/vol) saponin. Slides were examined with a confocal laser-scanning microscope (Bio-Rad MRC 1024). Cells were counted, and the percentage of cells with adherent bacteria was calculated. The effect of genistein, a phosphotyrosine kinase inhibitor, on H. pylori adherence to primary gastric cells was examined by supplementing the cell culture medium with 100 μM genistein for 1 h before and during infection.
For immunoprecipitation studies, cells were grown in six-well dishes to 50% confluence. They were washed with antibiotic-free medium and infected with H. pylori for 2 h under microaerophilic conditions. The cells were washed with ice-cold PBS containing 2 mM Na3VO4 to remove nonadherent bacteria and lysed in 0.5 ml of ice-cold lysis buffer (10 mM Tris HCl [pH 7.4], 150 mM NaCl, 1% (vol/vol) Triton X-100, 1 mM EDTA, 1 mM EGTA, complete mini EDTA-free protease inhibitor cocktail tablet [Boehringer Mannheim], 2 mM Na3VO4, 0.5% (vol/vol) NP-40). The lysate was precleared by the addition of normal mouse Igs (Santa Cruz) together with 20 μl of protein A/G agarose (Santa Cruz) and incubated with end-over-end rotation at 4°C for 30 min. The agarose beads were pelleted by centrifugation, and the supernatant was immunoprecipitated with antiphosphotyrosine PY99 (Santa Cruz) for 1 h at 4°C, after which protein A/G agarose was added overnight at 4°C. The precipitates were washed four times with lysis buffer and then boiled in 40 μl of electrophoresis sample buffer (62.5 mM Tris HCl [pH 6.8], 2% (wt/vol) SDS, 10% (vol/vol) glycerol, 5% (vol/vol) mercaptoethanol, 0.0003% bromophenol blue) for 5 min. Samples were stored at −20°C. Twenty-microliter volumes of whole-cell lysates or immunoprecipitates containing 2.5 μg of protein were separated by SDS-polyacrylamide gel electrophoresis (PAGE) on gels containing 8% acrylamide, unless otherwise stated, and blotted onto Immobilon P membranes (Millipore Corp.) The membranes were blocked with 2% (wt/vol) bovine serum albumin (BSA) (Sigma) in T-PBS (PBS containing 0.05% [vol/vol] Tween 20) and incubated with a primary antibody in T-PBS containing 1% (wt/vol) BSA overnight at 4°C, and then membranes were washed in T-PBS and incubated in horseradish peroxidase-conjugated secondary antibody diluted in T-PBS containing 1% (wt/vol) BSA for 1.5 h at room temperature. Antigen-antibody complexes were detected with an ECL detection system as directed by the manufacturer (Amersham Corp.). The primary antibodies used were (i) mouse monoclonal antiphosphotyrosine antibody PY99 (Santa Cruz) (1/1,000 dilution); (ii) CagA antiserum, as described above (however, for these blots it was used at a 1/2,000 dilution instead of 1/5,000); and (iii) a rabbit-raised whole-cell H. pylori antibody. The secondary antibodies used were peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Sigma), both used at a 1/1,000 dilution.
The amount of protein in samples for SDS-PAGE was measured with a Micro bicinchoninic acid protein assay reagent kit (Pierce Reagents), a detergent-compatible bicinchoninic acid formulation for the colorimetric detection and quantitation of total protein, according to the manufacturer's instructions.
In order to assess the role of adherence of H. pylori to epithelial cells in inducing tyrosine phosphorylation, we used cells that H. pylori adheres to and cells it doesn't adhere to. In this study, we assessed the adherence of H. pylori to cultured primary epithelial cells isolated from (i) human gastric tissue (tissue H. pylori colonizes naturally in vivo and causes inflammation), (ii) human duodenal tissue (tissue H. pylori does not colonize in vivo), and (iii) murine gastric tissue (tissue H. pylori colonizes only under experimental conditions in vivo and in which it only causes a low degree of inflammation). The CagA-positive strain NCTC 11637 adhered to primary human gastric cells but not to human duodenal cells or murine gastric cells (Fig. 1 and 2A, D, and E). H. pylori strain SS1, which has been specifically adapted to colonize mice, also adhered minimally to primary murine gastric tissue (Fig. 2F). Confocal microscopy demonstrated that some H. pylori organisms adhering to primary gastric cells colocalized with areas of tyrosine phosphorylation. This occurred with both CagA-positive and CagA-negative isolates; however, the amount of CagA-positive bacteria colocalizing with tyrosine phosphorylation was greater than the amount of CagA-negative bacteria that colocalized with tyrosine phosphorylation (Fig. 2A and C). Organisms were also detected adhering to gastric cells in areas without tyrosine phosphorylation (Fig. 2A and C). Pretreatment of gastric cells with genistein, an inhibitor of tyrosine phosphorylation, abolished tyrosine phosphorylation but had no effect on adherence of H. pylori NCTC 11637 to gastric cells (Fig. 2B).
FIG. 1.

Adherence of H. pylori strain NCTC 11637 to cultured primary cells isolated from human gastric biopsy specimens, human duodenal biopsy specimens, and mouse stomach. Primary cells were incubated with H. pylori for 2 h. The cells were fixed and stained for bacteria by using an anti-H. pylori antibody and a secondary antibody conjugated to FITC. Cells with and without adherent bacteria were counted. The results are expressed as the mean results of three experiments ± the standard deviation.
FIG. 2.

Immunofluorescent microscopy of primary cells stained for adherent bacteria and tyrosine phosphorylation. Cells and bacteria were incubated for 2 h under microaerophilic conditions. They were fixed, permeabilized, and stained for bacteria with an anti-H. pylori antibody and a secondary antibody conjugated to FITC. They were stained for phosphotyrosine with a monoclonal antiphosphotyrosine antibody and an anti-mouse antibody conjugated to Texas Red. Bacteria appear green, tyrosine-phosphorylated proteins appear red, and colocalization of the two stains appears yellow. (A) Human primary gastric cells incubated with H. pylori strain NCTC 11637. (B) Human primary gastric cells treated with genistein prior to incubation with H. pylori strain NCTC 11637. (C) Human primary gastric cells incubated with H. pylori strain PU44. (D) Human primary duodenal cells incubated with H. pylori strain NCTC 11637. (E) Murine primary gastric cells incubated with H. pylori strain NCTC 11637. (F) Murine primary gastric cells incubated with H. pylori strain SS1. Panels A, B, E, and F were viewed with a 63× oil immersion lens. Panels C and D were viewed with a 40× water immersion lens.
SDS-PAGE and Western immunoblotting demonstrated that adherence of H. pylori strain NCTC 11637 to primary gastric cells resulted in the tyrosine phosphorylation of a 145-kDa protein. This was identified as the CagA protein of the bacteria by probing the Western immunoblot with a specific CagA antibody (Fig. 3). No other tyrosine-phosphorylated proteins were detected in infected gastric cells that were absent in uninfected cells or vice versa. Western immunoblotting did not detect either the CagA protein or a 145-kDa tyrosine-phosphorylated protein in lysates of primary human duodenal cells or primary murine gastric cells that had been incubated with H. pylori strain NCTC 11637, but no adherence of the bacteria to the cells occurred (Fig. 3). Therefore, even though murine gastric and human duodenal cells were exposed to H. pylori organisms, translocation and subsequent tyrosine phosphorylation of the bacterial CagA protein occurred only with human gastric cells, cells that the organism actually adhered to.
FIG. 3.

Immunoblot analysis of primary human gastric cells, primary human duodenal cells, and primary murine gastric cells infected with H. pylori strain NCTC 11637. Primary cells were infected for 2 h with H. pylori strain NCTC 11637. Cell lysates were subjected to immunoblot analysis with anti-CagA serum and antiphosphotyrosine antibodies. Blots A, C, and E were probed with an anti-CagA (α CagA) serum, and blots B, D, and F were probed with antiphosphotyrosine antibodies (α PY). Human gastric cells were used in blots A and B. Lanes 1, cell lysate of primary gastric cells infected with H. pylori; lanes 2, cell lysate of uninfected primary gastric cells. The CagA protein is clearly present in H. pylori-infected cells (blot A, lane 1), and a protein with identical molecular mass reacts with antiphosphotyrosine antibody (blot B, lane 1). The CagA protein was not detected by the anti-CagA serum (blot A, lane 2) or the antiphosphotyrosine antibody in H. pylori-uninfected cells (blot B, lane 2). Human duodenal cells were used in blots C and D. Lane 1 in blot C contained whole-cell lysate of H. pylori strain NCTC 11637. Lane 2 in blot C and lane 1 in blot D contained cell lysates from infected cells, and lane 3 in blot C and lane 2 in blot D contained lysates from uninfected cells. Murine gastric cells were used in blots E and F. Lane 1 in blot E contained whole-cell lysate of H. pylori strain NCTC 11637. Lane 2 in blot E and lane 1 in blot F contained cell lysates from infected cells, and lane 3 in blot E and lane 2 in blot F contained lysates from uninfected cells. The amount of protein loaded in each lane was 2.5 μg.
Adherence of strain PU44 to primary gastric cells induced tyrosine phosphorylation of two proteins with molecular masses of 73 and 65 kDa. Expression of the 65-kDa protein appears to be upregulated in infected cells, and the 73-kDa protein was not detected in uninfected cells. In addition, upregulation of the two proteins was not detected in the cell lysate of cells that were infected with the CagA-positive isolate NCTC 11637 (Fig. 4). H. pylori proteins could be detected by Western immunoblotting in the cell lysates of infected cells, but none of the immunoprecipitated proteins could be detected with the anti-H. pylori serum (Fig. 5). These experiments were performed twice with the same results each time.
FIG. 4.
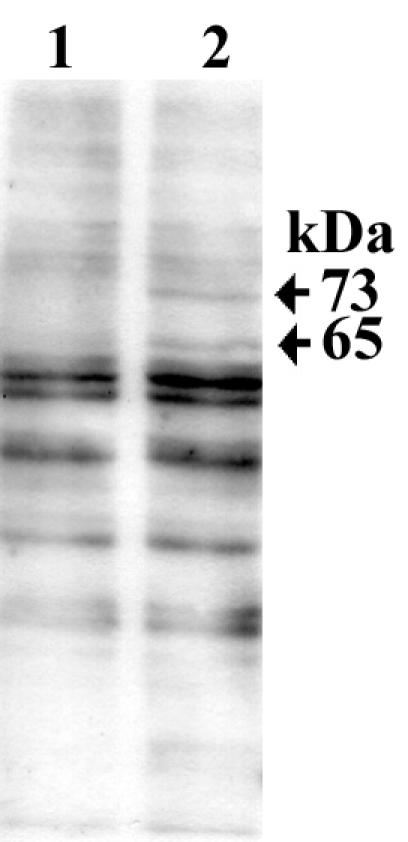
Immunoblot analysis of tyrosine-phosphorylated proteins in primary human gastric cells infected with H. pylori strain PU44. The blot was probed with antiphosphotyrosine antibody: lane 1, cell lysate of primary human gastric cells; lane 2, cell lysate of H. pylori-infected primary human gastric cells. Arrows indicate the positions of two proteins with molecular masses of 73 and 65 kDa present in H. pylori-infected cells. The 73-kDa protein is absent in uninfected cells, and the expression of the 65-kDa protein appears to be upregulated in infected cells. The amount of protein loaded in each lane was 2.5 μg.
FIG. 5.
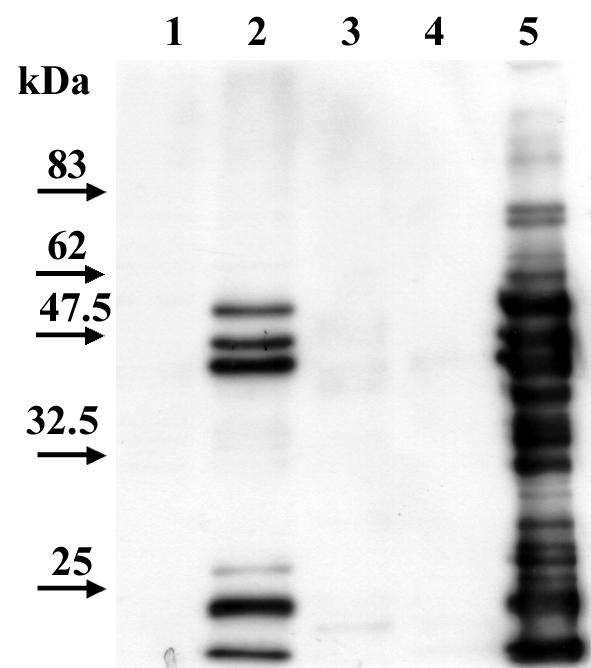
Immunoblot analysis of primary human gastric cells infected with H. pylori strain PU44. The blot was probed with a rabbit-raised whole-cell anti-H. pylori antibody. Lanes: 1, cell lysate of primary human gastric cells; 2, cell lysate of H. pylori-infected primary human gastric cells; 3, proteins from primary human gastric cells that were immunoprecipitated with antiphosphotyrosine antibody; 4, proteins from H. pylori-infected primary human gastric cells immunoprecipitated with antiphosphotyrosine antibody; 5, whole-cell lysate of H. pylori strain PU44. The amount of protein loaded in each lane was 2.5 μg.
Studies demonstrating translocation and tyrosine phosphorylation of CagA have all been done with various gastric and nongastric cell lines, such as AGS, Kato III, MKN45, HEp2, HEL, and CHO (1, 3, 21, 29). Human primary gastric cells have been shown to be ideal for assessing in vitro the adherence properties of H. pylori (5, 13, 27). This study shows that tyrosine phosphorylation of the CagA protein of H. pylori occurs with human primary gastric cells but not with human primary duodenal cells or murine primary gastric cells. Adherence appears to be essential in translocation of the CagA protein into gastric cells and subsequent tyrosine phosphorylation, since mere exposure of either human duodenal or murine gastric cells to the bacteria did not result in translocation of the CagA protein. However, tyrosine phosphorylation is not an event that is necessary for adherence to occur, since inhibition of tyrosine phosphorylation with genistein did not inhibit adherence of the organism. This is in agreement with other work in which genistein has been shown to inhibit invasion of AGS cells by H. pylori but not to have an effect on adherence of extracellular bacteria (17, 30). Segal et al. (28) presented evidence that in addition to tyrosine phosphorylation of CagA a number of other proteins in AGS cells were tyrosine phosphorylated when the cells were infected with CagA-positive bacteria. We could not identify any other tyrosine-phosphorylated proteins besides CagA present in primary gastric cells infected with H. pylori strain NCTC 11637 that were absent in uninfected cells.
Studies of the mouse model of H. pylori have included a wide range of murine strains, including BALB/c, C57BL/6, FVB/N, HSD/ICR, and CD1 (4, 6, 10, 11, 19, 34). H. pylori strain SS1 has been adapted to colonize mice and is both cagA and vacA positive (18). In this study, CagA-positive H. pylori cells (including strain SS1) did not adhere to cultured primary gastric cells isolated from CD1 mice, and even strain NCTC 11637, which has an active cag PAI, did not induce tyrosine phosphorylation in these cells. The cag PAI of SS1 has been shown to be functionally impaired and it does not induce interleukin-8 (IL-8) expression in epithelial cells (7, 9). There appears to be an adaptation of H. pylori isolates for growth in the mouse that attenuates the ability of the organism to induce inflammation (25). The identification of an easily available animal model of H. pylori infection that mimics human disease would greatly enhance studies on the virulence of H. pylori. Animal models in which the organism adheres to the gastric mucosa are essential if we are to determine the relevance of adherence and subsequent translocation and tyrosine phosphorylation of CagA in inducing gastric disease. In vitro studies of gerbil primary gastric cells should be undertaken in order to determine if H. pylori infection of the gerbil is a more representative model of H. pylori infection in humans (6, 22, 23, 31).
While there has been intense interest in the consequences of CagA translocation into eukaryotic cells and tyrosine phosphorylation, the effect of adherence of CagA-negative isolates of H. pylori to gastric cells has been largely ignored. In this study, the pattern of tyrosine phosphorylation induced was different, depending on the presence or absence of CagA. When the CagA-positive strain was used, tyrosine-phosphorylated host cell proteins could not be detected. We noted two tyrosine-phosphorylated proteins induced by infection of human primary gastric cells with a CagA-negative isolate of H. pylori. This strain also lacked cagE (a gene involved in IL-8 secretion and similar to the virB4 gene of Agrobacterium tumefaciens), cagM (a gene involved in IL-8 secretion and similar to the gene coding for hook-associated protein type 3 of Vibrio parahaemolyticus) and cagT (similar to the IPAC surface antigen of Shigella flexneri). The tyrosine-phosphorylated proteins induced are possibly of host cell origin rather than bacterial origin, since (i) they were not recognized by a whole-cell anti-H. pylori serum and (ii) one of them could be detected, but at much lower intensity, in uninfected cells. Puls et al. (26) speculated that a possible consequence of CagA tyrosine phosphorylation could be that CagA competes with host cell proteins as a substrate for tyrosine phosphorylation, which might explain the lack of phosphorylation of host cell proteins. However, they concluded that this is unlikely to happen, since even with very weak tyrosine phosphorylation of CagA phosphorylation of host cell proteins was not noted. In contrast, our findings would indicate that perhaps CagA phosphorylation does inhibit phosphorylation of other host cell proteins. The identification of two distinct signaling mechanisms induced by adherence of CagA-positive and CagA-negative isolates of H. pylori to gastric cells could help to elucidate the differences in the spectrum of diseases that have been shown in some studies to be induced by these two types of strains.
Acknowledgments
This work was supported by a grant from The Children's Medical and Research Foundation, Crumlin, Dublin, Ireland.
Editor: J. T. Barbieri
REFERENCES
- 1.Asahi, M., T. Azuma, S. Ito, Y. Ito, H. Suto, Y. Nagai, M. Tsubokawa, Y. Tohyama, S. Maeda, M. Omata, T. Suzuki, and C. Sasakawa. 2000. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 191:593-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1987. Current protocols in molecular biology. John Wiley and Sons, Chichester, United Kingdom.
- 3.Backert, S., E. Ziska, V. Brinkmann, U. Zimny-Arndt, A. Fauconnier, P. R. Jungblut, M. Naumann, and T. F. Meyer. 2000. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2:155-164. [DOI] [PubMed] [Google Scholar]
- 4.Bercik, P., R. De Giorgio, P. Blennerhassett, E. F. Verdu, G. Barbara, and S. M. Collins. 2002. Immune-mediated neural dysfunction in a murine model of chronic Helicobacter pylori infection. Gastroenterology 123:1205-1215. [DOI] [PubMed] [Google Scholar]
- 5.Clyne, M., and B. Drumm. 1993. Adherence of Helicobacter pylori to primary human gastrointestinal cells. Infect. Immun. 61:4051-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Court, M., P. A. Robinson, M. F. Dixon, and J. E. Crabtree. 2002. Gastric Helicobacter species infection in murine and gerbil models: comparative analysis of effects of H. pylori and H. felis on gastric epithelial cell proliferation. J. Infect. Dis. 186:1348-1352. [DOI] [PubMed] [Google Scholar]
- 7.Crabtree, J. E., R. L. Ferrero, and J. G. Kusters. 2002. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter 7:139-141. [DOI] [PubMed] [Google Scholar]
- 8.Drumm, B., P. Sherman, E. Cutz, and M. Karmali. 1987. Association of Campylobacter pylori on the gastric mucosa with antral gastritis in children. N. Engl. J. Med. 316:1557-1561. [DOI] [PubMed] [Google Scholar]
- 9.Eaton, K. A., D. Kersulyte, M. Mefford, S. J. Danon, S. Krakowka, and D. E. Berg. 2001. Role of Helicobacter pylori cag region genes in colonization and gastritis in two animal models. Infect. Immun. 69:2902-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falk, P. G., L. Bry, J. Holgersson, and J. I. Gordon. 1995. Expression of a human alpha-1,3/4-fucosyltransferase in the pit cell lineage of FVB/N mouse stomach results in production of Leb-containing glycoconjugates: a potential transgenic mouse model for studying Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 92:1515-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foynes, S., N. Dorrell, S. J. Ward, R. A. Stabler, A. A. McColm, A. N. Rycroft, and B. W. Wren. 2000. Helicobacter pylori possesses two CheY response regulators and a histidine kinase sensor, CheA, which are essential for chemotaxis and colonization of the gastric mucosa. Infect. Immun. 68:2016-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Go, M. F., and D. Y. Graham. 1996. Presence of the cagA gene in the majority of Helicobacter pylori strains is independent of whether the individual has duodenal ulcer or asymptomatic gastritis. Helicobacter 1:107-111. [DOI] [PubMed] [Google Scholar]
- 13.Heczko, U., V. C. Smith, R. Mark Meloche, A. M. Buchan, and B. B. Finlay. 2000. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes Infect. 2:1669-1676. [DOI] [PubMed] [Google Scholar]
- 14.Hentschel, E., G. Brandstatter, B. Dragosics, A. M. Hirschl, H. Nemec, K. Schutze, M. Taufer, and H. Wurzer. 1993. Effect of ranitidine and amoxicillin plus metronidazole on the eradication of Helicobacter pylori and the recurrence of duodenal ulcer. N. Engl. J. Med. 328:308-312. [DOI] [PubMed] [Google Scholar]
- 15.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. 1994. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 61:1-241. [PMC free article] [PubMed] [Google Scholar]
- 16.Karita, M., Q. Li, D. Cantero, and K. Okita. 1994. Establishment of a small animal model for human Helicobacter pylori infection using germ-free mouse. Am. J. Gastroenterol. 89:208-213. [PubMed] [Google Scholar]
- 17.Kwok, T., S. Backert, H. Schwarz, J. Berger, and T. F. Meyer. 2002. Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect. Immun. 70:2108-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, A., J. O'Rourke, M. C. De Ungria, B. Robertson, G. Daskalopoulos, and M. F. Dixon. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112:1386-1397. [DOI] [PubMed] [Google Scholar]
- 19.Marchetti, M., B. Arico, D. Burroni, N. Figura, R. Rappuoli, and P. Ghiara. 1995. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science 267:1655-1658. [DOI] [PubMed] [Google Scholar]
- 20.Marshall, B. J., and J. R. Warren. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311-1315. [DOI] [PubMed]
- 21.Odenbreit, S., J. Puls, B. Sedlmaier, E. Gerland, W. Fischer, and R. Haas. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287:1497-1500. [DOI] [PubMed] [Google Scholar]
- 22.Ogura, K., S. Maeda, M. Nakao, T. Watanabe, M. Tada, T. Kyutoku, H. Yoshida, Y. Shiratori, and M. Omata. 2000. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J. Exp. Med. 192:1601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osawa, H., K. Sugano, M. Iwamori, M. Kawakami, M. Tada, and M. Nakao. 2001. Comparative analysis of colonization of Helicobacter pylori and glycolipids receptor density in Mongolian gerbils and mice. Dig. Dis. Sci. 46:69-74. [DOI] [PubMed] [Google Scholar]
- 24.Parsonnet, J., G. D. Friedman, D. P. Vandersteen, Y. Chang, J. H. Vogelman, N. Orentreich, and R. K. Sibley. 1991. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325:1127-1131. [DOI] [PubMed] [Google Scholar]
- 25.Philpott, D. J., D. Belaid, P. Troubadour, J. M. Thiberge, J. Tankovic, A. Labigne, and R. L. Ferrero. 2002. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylori isolates. Cell Microbiol. 4:285-296. [DOI] [PubMed] [Google Scholar]
- 26.Puls, J., W. Fischer, and R. Haas. 2002. Activation of Helicobacter pylori CagA by tyrosine phosphorylation is essential for dephosphorylation of host cell proteins in gastric epithelial cells. Mol. Microbiol. 43:961-969. [DOI] [PubMed] [Google Scholar]
- 27.Richter-Dahlfors, A., U. Heczko, R. M. Meloche, B. B. Finlay, and A. M. Buchan. 1998. Helicobacter pylori-infected human antral primary cell cultures: effect on gastrin cell function. Am. J. Physiol. 275:G393-G401. [DOI] [PubMed] [Google Scholar]
- 28.Segal, E. D., S. Falkow, and L. S. Tompkins. 1996. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc. Natl. Acad. Sci. USA 93:1259-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein, M., R. Rappuoli, and A. Covacci. 2000. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl. Acad. Sci. USA 97:1263-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su, B., S. Johansson, M. Fallman, M. Patarroyo, M. Granstrom, and S. Normark. 1999. Signal transduction-mediated adherence and entry of Helicobacter pylori into cultured cells. Gastroenterology 117:595-604. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe, T., M. Tada, H. Nagai, S. Sasaki, and M. Nakao. 1998. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology 115:642-648. [DOI] [PubMed] [Google Scholar]
- 32.Wyatt, J. I., B. J. Rathbone, M. F. Dixon, and R. V. Heatley. 1987. Campylobacter pyloridis and acid induced gastric metaplasia in the pathogenesis of duodenitis. J. Clin. Pathol. 40:841-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaoka, Y., S. Kikuchi, H. M. el-Zimaity, O. Gutierrez, M. S. Osato, and D. Y. Graham. 2002. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology 123:414-424. [DOI] [PubMed] [Google Scholar]
- 34.Yamaoka, Y., M. Kita, T. Kodama, S. Imamura, T. Ohno, N. Sawai, A. Ishimaru, J. Imanishi, and D. Y. Graham. 2002. Helicobacter pylori infection in mice: role of outer membrane proteins in colonization and inflammation. Gastroenterology 123:1992-2004. [DOI] [PubMed] [Google Scholar]
- 35.Yamazaki, S., A. Yamakawa, Y. Ito, M. Ohtani, H. Higashi, M. Hatakeyama, and T. Azuma. 2003. The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to SHP-2 in human gastric mucosa. J. Infect. Dis. 187:334-337. [DOI] [PubMed] [Google Scholar]