

Abstract
Mixed corticomedullary adrenal tumours (MCMT) are rare. We describe the second reported case of a male patient presenting with hypertension and Cushing syndrome with MCMT. A man aged 48 years presented with hypertension and signs of Cushing syndrome. 24-hour urine cortisol was elevated, with detectable adrenocorticotropic hormone (ACTH). A high-dose dexamethasone suppression test indicated an adrenal or ectopic Cushing syndrome. Plasma metanephrines were normal. A 3 cm left adrenal mass was identified without potential ectopic sources of ACTH on imaging. After induction of anaesthesia for laparoscopic adrenalectomy, the patient developed resistant hypertension with stress-dose hydrocortisone administration. Surgery was cancelled and repeat testing revealed elevated plasma metanephrines. α-Blockade was administered for a presumed coexisting pheochromocytoma, and the patient underwent adrenalectomy. Pathology revealed an MCMT. This case highlights the importance of a thorough biochemical evaluation in patients with adrenal masses to rule out multiple hormone producing tumours.
Background
The adrenal gland is composed of an adrenal cortex and medulla, which arise from distinct embryological origins. The cortex is responsible for producing aldosterone, cortisol and androgens, whereas the medulla produces catecholamines. The clinical presentations of adrenal tumours vary accordingly to the hormones secreted. The rarely encountered mixed corticomedullary tumour (MCMT) is distinguished as a single tumour mass of the adrenal gland composed of intermixed cortex and medullary cells.1
The first reported case of a mixed adrenal corticomedullary tumour was published in 1969 by Mathison and Waterhouse.2 There have been 17 reported cases. Nearly, all patients are women presenting with signs of Cushing syndrome and/or hypertension, with benign MCMTs. To the best of our knowledge, we report the second case of a male patient diagnosed with Cushing syndrome and resistant hypertension, found to have an MCMT. This case highlights the importance of a thorough biochemical evaluation of an adrenal mass, as a missed diagnosis of pheochromocytoma can be catastrophic.
Case presentation
A man aged 48 years presented with 3 months of fatigue and hypertension. He also reported easy bruising and thigh weakness. He had a history of hyperlipidaemia, recent weight gain and a vertebral (T12) compression fracture. His mother and sister had a history of non-functioning benign adrenal adenomas. His medications included ramipril, carvedilol, spironolactone, potassium chloride and aspirin. On physical examination, he had classic Cushingoid features of facial plethora, moon facies, central obesity, dorsal cervical fat pad, abdominal striae and lower extremity oedema (figure 1). He was hypertensive with blood pressure (BP) of 180/120 mm Hg. Laboratory testing was performed to confirm suspicion of Cushing syndrome.
Figure 1.
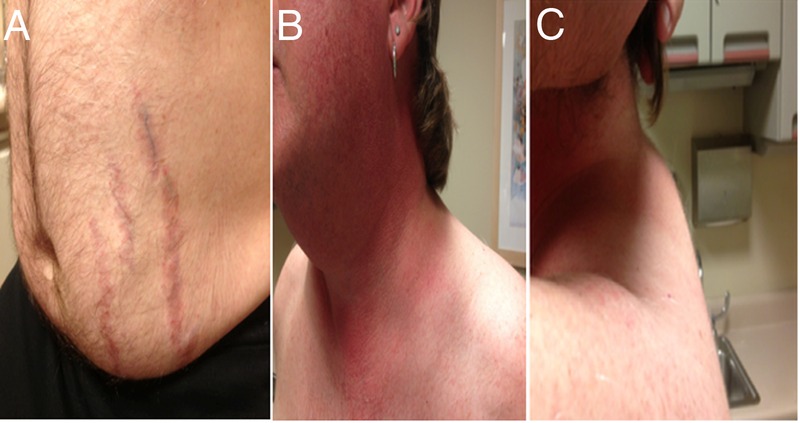
Physical examination: (A) purple abdominal striae, (B) facial plethora and (C) dorsal cervical fat pad.
Investigations
Laboratory studies revealed hypercortisolaemia with an AM cortisol of 31.2 µg/dL (nl<22 µg/dL) and a non-suppressed adrenocorticotropic hormone (ACTH) of 18 pg/mL. Twenty-four-hour urine-free cortisol was elevated to 787 μg/dL (nl<60 μg/dL); as was midnight salivary cortisol to 1.71 μg/dL (nl<0.09 μg/dL). An 8 mg dexamethasone suppression test was performed, revealing an elevated AM cortisol of 31 μg/dL, and non-suppressed ACTH of 24 pmol/L. Calcitonin and CEA were within normal limits. Corticotropin-releasing hormone (CRH) stimulation test failed to stimulate cortisol and ACTH. Plasma metanephrines, renin and aldosterone were within normal limits. Cortisol levels were measured with a competitive antibody immunoassay; ACTH via a non-competitive antibody immunoassay.
Abdominal CT revealed a 3.2 cm left adrenal nodule with washout consistent with a lipid poor adenoma (figure 2). MRI of the abdomen and pelvis revealed a 3 cm left adrenal nodule with indeterminate imaging characteristics, consistent with either a lipid poor adenoma or an adrenal tumour. There was no atrophy or hyperplasia of the contralateral adrenal gland. A brain MRI and CT of the chest, abdomen and pelvis revealed no other potential ectopic sources of ACTH production.
Figure 2.
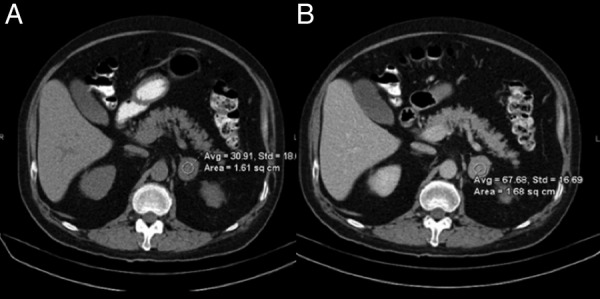
Abdominal CT with and without contrast. The left adrenal mass measures 3.2 cm, with (A) precontrast density 31 HU, (B) and postcontrast density 68 HU, with 15 min washout of 67%, compatible with a lipid-poor adenoma. No contralateral adrenal gland hyperplasia.
Differential diagnosis
Adrenal Cushing syndrome was suspected, given the adrenal mass on imaging a non-suppressed ACTH, and an absence of bilateral adrenal hyperplasia. The non-suppressed ACTH value indicated an adrenal or ectopic source of ACTH, as dexamethasone would be expected to suppress ACTH more than 50% if arising from a pituitary source. As calcitonin and CEA were within normal limits, medullary thyroid cancer as a source of ACTH was unlikely. CRH stimulation test was consistent with a non-pituitary Cushing syndrome.
Treatment
We proceeded with a left laparoscopic adrenalectomy. After induction of anaesthesia, the patient received a stress dose of hydrocortisone succinate 100 mg intravenous, and the patient developed hypertensive crisis to a BP of 220/120 mm Hg. The surgery was aborted and the patient was stabilised. Repeat plasma and 24-hour urine metanephrines were performed, and found to be elevated: 24-hour urine metanephrines: 405 μg/24 hours (nl<261μg/24 hours), and repeat plasma metanephrines 146 pg/mL (nl<57 pg/mL). MIBG scintigraphy was not performed as we did not feel it would alter patient management. The patient was treated with α-blockade with phenoxybenzamine, as well as nifedipine and liberal PO fluid intake. The BP was titrated to orthostatic hypotension for 1 week in preparation for surgery. We then performed an uneventful left laparoscopic adrenalectomy. The patient tolerated intraoperative stress-dose steroids and did not experience severe BP fluctuations during surgery, and overall, his perioperative course was uneventful. Frozen section of the specimen revealed an MCMT.
The gross specimen revealed a well circumscribed, soft brown encapsulated adrenal mass, measuring 3.9 cm (figure 3). Histologically, the mass comprised an intimate admixture of adrenal cortical cells and pheochromocytes/adrenal medullary cells (figure 4A). There was no capsular or lymphovascular invasion, or necrosis. There were 0 mitoses/50 HPF. The cortical component stained strongly for inhibin-A and melan-A (figure 4B, C) on immunohistochemistry. The medullary component stained positive for chromogranin-A and synaptophysin, and sustentacular cells were S100 positive (figure 4D). An ACTH immunohistochemical stain was negative. Ki-67 labelling index was low (<1%).
Figure 3.
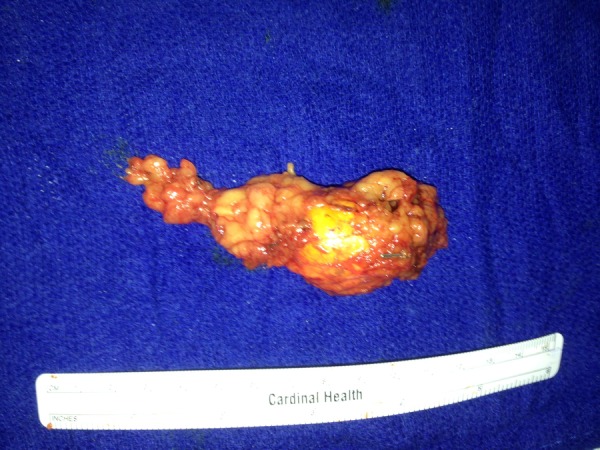
Gross adrenal specimen. (Mixed corticomedullary tumour visible at the right end of the specimen.)
Figure 4.
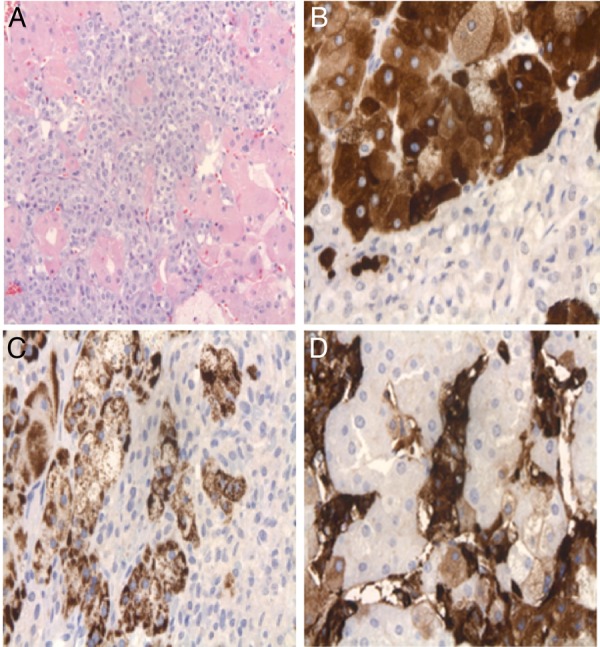
Histology of mixed corticomedullary tumour. (A) Pheochromocytes (centre, basophilic) mixed with adrenal cortical cells (with eosinophilic cytoplasm). (B) Adrenal cortical cells stain (+) for inhibin-A (upper left); pheochromocytes staining (−) (lower right). (C) Adrenal cortical component expressing melan-A. (D) Pheochromocytes stain (+) for chromogranin A; adrenal cortical cells stain (−).
Outcome and follow-up
Postoperatively, the patient was administered a steroid taper with hydrocortisone. One week later, he remained normotensive on nifedipine and enalapril. Plasma metanephrines normalized and were <25 pg/mL (nl<57) and plasma normetanephrines were 126 pg/mL (nl<148). Random cortisol also normalised to 12.0 μg/dL (nl<22) with an undetectable ACTH of <5 pg/mL (nl <50) on the steroid taper. ACTH was now undetectable after adrenalectomy on the replacement steroid taper, indicating that the MCMT was likely the source of the elevated ACTH levels. Over the following year, he was tapered off nearly all BP medications. At 1 year, laboratory results confirmed biochemical cure with normal cortisol and ACTH levels: AM cortisol of 5.8 μg/dL (nl 5–25 μg/dL), and ACTH of 21 pg/mL (nl<69 pg/mL). The patient's BP was normal at 126/84 mm Hg, controlled with a single agent of ramipril daily. On physical examination, he had complete resolution of stigmata of Cushing syndrome. He has no evidence of disease at 34 months.
Discussion
MCMT of the adrenal gland is a rare entity, with only 17 reported cases in the literature (table 1). We report the second case of a male patient presenting with Cushing syndrome and hypertension, who was discovered to have an MCMT.
Table 1.
Reported cases of mixed corticomedullary tumours
| Reference | Age/gender | Clinical presentation | Hormones secreted | Tumour size (cm) |
|---|---|---|---|---|
| Mathison and Waterhouse,2 1969 | 39 F | Cushing syndrome, intraoperative HTN | Cortisol, ACTH*, catecholamines | 4 |
| Akai et al,3 1993 | 61 F | HTN, hyperglycaemia | Cortisol, ACTH*, catecholamines | 3.5 |
| Ohta et al,4 1995 | 32 M | Cushing syndrome, intraoperative HTN | Cortisol, ACTH* | 4.5 |
| Michal and Havlicek5 1996 | 56 F | Cushing syndrome, HTN | Cortisol, aldosterone, catecholamines | 8 |
| 32 F | Cushing syndrome, HTN | Cortisol, aldosterone, catecholamines | 9 | |
| Wieneke et al,1 2001 | 34 F | HTN, hair loss, amenorrhoea | Cortisol | 4.5 |
| 52F | Flank pain | None | 2.5 | |
| Chu et al,6 2003 | 55 F | Mild Cushing syndrome | Cortisol, ACTH* | 2.5 |
| Ma et al,7 2007 | 41 F | Cushing syndrome, intraoperative HTN | Cortisol, ACTH* | 4 |
| Lee et al,8 2008 | 25 F | Cushing syndrome, gestational diabetes, HTN | Cortisol, catecholamines | 3.2 |
| Kimura et al,9 2009 | 54 F | HTN, diabetes | Cortisol, catecholamines | 4.9 |
| Alexandraki et al,10 2009 | 66 F | Subclinical Cushing syndrome | Cortisol, ACTH* | 4.2 |
| Singh et al,11 2010 | 48 F | Abdominal distention, pedal oedema | Catecholamines | 8 |
| Lau et al,12 2011 | 64 F | HTN | Catecholamines | 3.6 |
| Donatini et al,13 2013 | 53 M | Right flank pain | Catecholamines | 5.5 |
| Turk et al,14 2012† | 78 F | Hypertensive urgency | Catecholamines | 10 |
| Michalopoulos et al,15 2013† | 63 M | Right upper quadrant palpable mass | Cortisol, ACTH* | 8 |
| Total number of cases: 17 | Median age 53.5 years (range 25–78) Females 82% (14/17) Males 18% (3/17) |
Cushing syndrome 47% (8/17) Intraop HTN 18% (3/17) HTN 47% (8/17) |
Cortisol 71% (12/17) Aldosterone 12% (2/17) Catecholamines 59% (10/17) ACTH* 88% (7/8) |
Median size 4.5 cm (range: 2.5–10) |
*ACTH was detectable (non-suppressed) in the setting of hypercortisolaemia.
†Mixed corticomedullary carcinoma (MCMC).
HTN, hypertension; Intraop, intraoperative.
MCMT was first reported in 1969 by Mathison and Waterhouse.2 The exact aetiology of this hybrid tumour remains unclear, as the adrenal cortex and medulla arise from distinct embryological origins. The medulla arises from migrating ectodermal neural crest cells, while the cortex arises from the dorsal mesoderm. Within this composite tumour, the two tissue types are intimately intermixed in a similar distribution. Wieneke et al proposed that ACTH-induced cortical hyperplasia may result from ACTH production from the pheochromocytoma itself, or from catecholamine-induced secretion from the pituitary. They further hypothesise that the neoplastic cortical and medullary components collide or combine together to form an MCMT. Lee et al speculated that neoplastic changes of adrenal cortical cells result in hypersecretion of cortisol, causing hyperplasia of surrounding pheochromocytes, thereby producing an MCMT.
Immunohistochemical studies indicating cortical cells include inhibin-A and melan-A, while medullary cells are characterised by positive staining for synaptophysin, S-100, and chromogranin A, all of which were positive in our case.3
In the 17 reported cases, the median age for patients was 54 years, and the median tumour size was 4.5 cm (table 1). The majority of patients were female (82%), and almost half (47%) of these patients presented with Cushing syndrome and/or hypertension. Similar to our case, three patients (18%) experienced hypertensive crises during the perioperative period due to missed diagnosis of a multihormone-secreting tumour.2 4 7 The majority (71%) of patients had elevated cortisol, 12% had elevated aldosterone and 59% had elevated catecholamines. Of the 12 previous cases with elevated cortisol, 8 report ACTH levels and the vast majority (7/8, 88%) had unsuppressed ACTH levels, despite a hypercortisolaemic state.2–4 6–8 10 15
Most patients (82%) underwent an open adrenalectomy and 12% underwent a laparoscopic adrenalectomy. One patient underwent bilateral laparoscopic adrenalectomy for bilateral lesions.8 Not surprisingly, in all cases, the diagnosis of MCMT in all patients was incidental, after adrenalectomy. The vast majority of tumours (88%) were benign, and 12% were malignant.
In our patient, the MCMT produced cortisol, catecholamines and likely ACTH. Levels of ACTH were undetectable postoperatively when the patient was placed on a steroid taper. Cortisol and catecholamines also normalised postoperatively. Similar to our patient, in seven of eight published cases, patients presented with a detectable serum ACTH, despite a hypercortisolaemic state from an adrenal source, in which ACTH is usually suppressed.2–4 6–7 10 15 The detectable ACTH level is not grossly elevated, as is usually seen from a pituitary or ectopic source.16 Pheochromocytomas are a known possible ectopic source of ACTH. In patients with detectable ACTH from pure pheochromocytoma, the mean ACTH is 344 ng/L, higher than levels reported in patients with MCMT.17 The pheochromocytes are likely the source of ACTH in patients with MCMT and a detectable ACTH; however, this cannot be definitively concluded in our case, as the pheochromocytes did not stain positive for ACTH. Since the levels of ACTH are not as elevated in MCMT patients compared with those with pure pheochromocytomas, it is possible that the ACTH stain may not cross threshold for detection by standard microscopy. Of the seven of eight reported cases in the literature with a detectable serum ACTH, three reported testing for ACTH staining with immunohistochemistry.3–4 6 Of those three cases, only one stained positive for ACTH.3 In cases of negative IHC for ACTH, further testing with mRNA of POMC and in situ hybridisation technique could be considered. Staining for DHEA-ST in the adjacent normal adrenal tissue can also help to discern ACTH-dependent from ACTH-independent Cushing syndrome.
It should also be noted that in patients with an adrenal mass, pheochromocytoma should be ruled out prior to performance of a DST, as glucocorticoids can induce the production of catecholamines via induction of biosynthetic enzymes, resulting in hypertensive crisis.
All of the described MCMTs in the literature have been unilateral, and most are benign (88%, 15/17 cases). Two cases of malignant MCMT, or mixed corticomedullary carcinoma (MCMC), have recently been reported.14–15 When evaluating these mixed tumours histologically, features concerning for malignancy include tumour necrosis, atypical mitotic activity, nuclear pleomorphism, lymphovascular invasion and a high Ki-67 proliferative index.14 Both patients with MCMC reported in the literature were treated with adjuvant etoposide and platinum-based chemotherapy. Both developed distant hepatic metastases at 4 and 12 months, respectively.14–15 One patient died 18 months after surgery and follow-up information was not reported for the other patient.
MCMT remains a rare entity with unclear pathogenesis. In our patient, an MCMT was responsible for production of cortisol, catecholamines and likely ACTH. He has no evidence of recurrence after laparoscopic adrenalectomy at 34 months. In the majority of reported cases of MCMT, patients present with hypertension and classic signs of Cushing syndrome. MCMTs are more common in women and are likely to be benign. Of the two reported malignant cases, the prognosis was dismal. Although this is a rare entity, this case highlights the importance of a thorough biochemical evaluation in patients presenting with an adrenal mass, to rule out multiple hormone production, as a missed diagnosis of pheochromocytoma can be catastrophic.
Learning points.
Mixed corticomedullary tumours are rare with protean clinical presentations.
Hormonal activity of these lesions is variable. Cortisol, catecholamines and adrenocorticotropic hormone are the most frequently produced. A thorough biochemical evaluation is necessary prior to any intervention in all patients with adrenal masses.
Adrenalectomy for benign mixed corticomedullary tumour (MCMT) is effective with resolution of presenting symptoms and signs. Two reported cases of malignant MCMTs had a dismal prognosis, despite adjuvant treatment.
Footnotes
Contributors: JLM, TML, and NG reviewed the literature, analysed and interpreted the data and drafted revisions of the manuscript. SG analysed the slides of the tissue samples and contributed histological commentary. JLM was responsible for the conception of the case report, supervised data analysis and interpretation, and revised it critically. All authors approve of the final version of the submitted manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wieneke JA, Thompson LDR, Heffess CS. Corticomedullary mixed tumor of the adrenal gland. Ann Diagn Pathol 2001;5:304–8. 10.1053/adpa.2001.28297 [DOI] [PubMed] [Google Scholar]
- 2.Mathison DA, Waterhouse CA. Cushing's syndrome with hypertensive crisis and mixed adrenal cortical adenoma-pheochromocytoma (Corticomedullary adenoma). Am J Med 1969;47:635–41. 10.1016/0002-9343(69)90193-4 [DOI] [PubMed] [Google Scholar]
- 3.Akai H, Sanoyama K, Namai K et al. . [A case of adrenal mixed tumor of pheochromocytoma and adrenocortical adenoma presenting diabetes mellitus and hypertension]. Nihon Naibunpi Gakkai Zasshi 1993;69:659–69. [DOI] [PubMed] [Google Scholar]
- 4.Ohta T, Motoyama T, Imai T et al. . Cortico-medullary mixed tumor (pheochromocytoma and cortical adenoma) of the adrenal gland. J Urol Pathol 1995;3:157–64. [Google Scholar]
- 5.Michal M, Havlicek F. Corticomedullary tumors of the adrenal glands. Report of two cases. Association of corticomedullary tumor with spindle cell sarcoma. Pathol Res Pract 1996;192:1082–9. 10.1016/S0344-0338(96)80023-9 [DOI] [PubMed] [Google Scholar]
- 6.Chu AY, LiVolsi VA, Fraker DL et al. . Corticomedullary mixed tumor of the adrenal gland with concurrent adrenal myelolipoma. Arch Pathol Lab Med 2003;127:e329–32. [DOI] [PubMed] [Google Scholar]
- 7.Ma W-Y, Yang A-H, Chang Y-H et al. . Coexistence of adrenal Cushing syndrome and pheochromocytoma in a ‘corticomedullary adenoma’: a case report and review of the literature. Endocrinologist 2007;17:341 10.1097/TEN.0b013e3181596219 [DOI] [Google Scholar]
- 8.Lee P, Bradbury RA, Sy J et al. . Phaeochromocytoma and mixed corticomedullary tumour—a rare cause of Cushing's syndrome and labile hypertension in a primigravid woman postpartum. Clin Endocrinol (Oxf) 2008;68:492–4. 10.1111/j.1365-2265.2007.03038.x [DOI] [PubMed] [Google Scholar]
- 9.Kimura T, Usui T, Inamoto S et al. . Image in endocrinology. Pheochromocytoma with subclinical Cushing's syndrome caused by corticomedullary mixed tumor of the adrenal gland. J Clin Endocrinol Metab 2009;94:746–7. 10.1210/jc.2008-2013 [DOI] [PubMed] [Google Scholar]
- 10.Alexandraki KI, Michail OP, Nonni A et al. . Corticomedullary mixed adrenal tumor: case report and literature review. Endocr J 2009;56:817–24. 10.1507/endocrj.K09E-010 [DOI] [PubMed] [Google Scholar]
- 11.Singh M, Mandal S, Kakkar AK et al. . Mixed corticomedullary tumour with myelolipoma: a rare coexistence. Pathology 2010;42:589–91. 10.3109/00313025.2010.508741 [DOI] [PubMed] [Google Scholar]
- 12.Lau SK, Chu PG, Weiss LM. Mixed cortical adenoma and composite pheochromocytoma-ganglioneuroma: an unusual corticomedullary tumor of the adrenal gland. Ann Diagn Pathol 2011;15:185–9. 10.1016/j.anndiagpath.2010.02.005 [DOI] [PubMed] [Google Scholar]
- 13.Donatini G, Slycke SV, Aubert S et al. . Corticomedullary mixed tumor of the adrenal gland—a clinical and pathological chameleon: case report and review of literature. Updates Surg 2013;65:161–4. 10.1007/s13304-011-0132-1 [DOI] [PubMed] [Google Scholar]
- 14.Turk AT, Asad H, Trapasso J et al. . Mixed corticomedullary carcinoma of the adrenal gland: a case report. Endocr Pract 2012;18:e37–42. 10.4158/EP11222.CR [DOI] [PubMed] [Google Scholar]
- 15.Michalopoulos N, Pazaitou-Panayiotou K, Boudina M et al. . Mixed corticomedullary adrenal carcinoma. Surg Today 2013;43:1232–9. 10.1007/s00595-012-0458-4 [DOI] [PubMed] [Google Scholar]
- 16.Isidori AM, Kaltsas GA, Grossman AB. Ectopic ACTH syndrome. Front Horm Res 2006;35:143–56. 10.1159/000094323 [DOI] [PubMed] [Google Scholar]
- 17.Ballav C, Naziat A, Mihai R et al. . Mini-review: pheochromocytomas causing the ectopic ACTH syndrome. Endocrine 2012;42:69–73. 10.1007/s12020-012-9646-7 [DOI] [PubMed] [Google Scholar]