

Abstract
We report a case of acute oesophageal necrosis (AEN) and non-occlusive mesenteric ischaemia in an otherwise healthy 30-year-old man with cocaine and alcohol abuse. Although cocaine might be expected more frequently to cause oesophageal necrosis through sympathomimetic vasoconstriction, this is only the second known case report of AEN in a patient with cocaine abuse. His symptoms at presentation included epigastric abdominal pain, haematemesis and generalised weakness. He developed moderate neutropenia and severe lactic acidosis. Treatment consisted of intravenous proton-pump inhibitors, granulocyte colony stimulating factor, broad-spectrum antibiotics and ultimately exploratory laparotomy after his condition worsened. He died within 24 hours of presentation from a combination of systemic inflammatory response syndrome, acute respiratory distress syndrome and disseminated intravascular coagulation. AEN was discovered postmortem. We conclude that AEN should be suspected in any patient with haematemesis and substance abuse, and discovery of AEN should prompt a thorough evaluation for potentially lethal comorbid conditions.
Background
Acute oesophageal necrosis (AEN) or ‘black oesophagus’ is a relatively rare disease process with high associated mortality (up to 38%).1 The aetiology has been attributed to a combination of oesophageal ischaemia, reflux of gastric contents and impairment of mucosal defences usually due to poor nutritional status.2 Ischaemia typically affects the distal oesophagus and terminates abruptly at the gastro-oesophageal junction.3 Patients typically present with symptoms of upper gastrointestinal bleeding including haematemesis, coffee ground emesis or melena.1 AEN is typically associated with risk factors of diabetes, infection, medication, vascular disease, hypotension, malnutrition and advanced malignancies,4 and typically occurs in two general patient populations: elderly men and a mixed gender younger group with alcohol abuse.3
Cocaine abuse can lead to a variety of medical problems, principally through sympathomimetic and thrombogenic effects. Neurological sequelae include acute agitation, seizures and haemorrhagic, and ischaemic strokes. Cardiovascular complications include myocardial ischaemia and infarction as well as aortic dissection. Pulmonary toxicity primarily occurs with inhaled cocaine and includes bronchoconstriction and pneumothorax. Known gastrointestinal manifestations include mesenteric ischaemia, typically of the small bowel, but ischaemic colitis may also result.5 To the best of our knowledge, this is only the second case that reports AEN in a patient with cocaine use.
Case presentation
A 30-year-old-man presented to the emergency room (ER) with progressive epigastric pain of one-day duration, haematemesis and weakness. He had been taking calcium carbonate for reflux-related symptoms several times per day for the previous 2–3 months. His home medications included dextroamphetamine/amphetamine, bupropion and tadalafil. He consumed 6–10 beers per day but denied use of illicit substances. On his way home from work on the day of his presentation, he developed epigastric pain, nausea and haematemesis. He collapsed at home and 911 was called. He was transported to the ER where his blood pressure and heart rate were 105/73 mm Hg and 114 bpm, respectively.
Investigations
Notable laboratory tests performed in the ER were as follows: Cr 1.3 (0.7–1.2 mg/dL), CO2 21 (22–29 mmol/L), aspartate aminotransferase 373 (<41 U/L), alanine transaminase 136 (<42 U/L), amylase 41 (0–100 U/L), lipase 52 (13–60 U/L), anion gap 18 (7.0–16.0), white cell count 0.7 (4.0–9.8 K/mm3) and absolute neutrophil count 648 (>1500cells/microL). Abdominal X-ray showed normal bowel gas pattern without evidence of masses or free air. CT scan of the abdomen and pelvis with intravenous contrast showed gastric and small bowel thickening with a small amount of ascites (figure 1). Suspicion was raised for gastritis with an incomplete distal small bowel obstruction.
Figure 1.
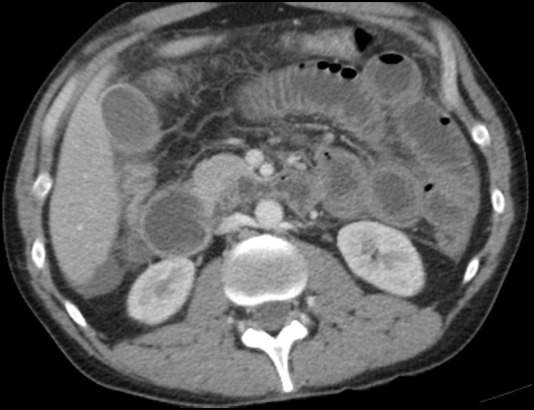
CT showing gastric and small bowel thickening.
Differential diagnosis
Acute pancreatitis
Acute hepatitis
Acute gastritis or gastroenteritis
Mallory-Weiss tear
Acute cholecystitis
Acute appendicitis
Gastric or duodenal ulcer with or without perforation
Sepsis from an intra-abdominal source
Acute mesenteric ischaemia
Bleeding oesophageal varices
Acute oesophageal necrosis
Small bowel obstruction
Gastrointestinal beriberi due to thiamine deficiency
Treatment
He was treated with intravenous proton-pump inhibitor, intravenous opioids and nasogastric tube insertion for presumptive small bowel obstruction. Esophagogastroduodenoscopy (EGD) was not considered emergently. Approximately 6 hours after triage intake, he became tachypnaeic and hypoxemic to 57% on room air, then developed carpopedal spasm and respiratory arrest. He briefly required cardiopulmonary resuscitation for pulselessness. He was intubated and transferred to the intensive care unit (ICU). Post arrest laboratory tests included Cr 2.6 (0.7–1.2 mg/dL), total bilirubin 2.1 (<1.3 mg/dL), anion gap 17 (7.0–16.0), lactic acid 11.2 (0.5–2.2 mmol/L) and arterial blood gas showed pH 6.96 (7.35–7.45), partial pressure of carbon dioxide 56 (35–45 mm Hg), partial pressure of oxygen in blood 146 (80–100 mm Hg) and bicarbonate 13 (24–28 meq/L). At this time he showed evidence of multiorgan failure as well as lactic acidosis, so sepsis and intestinal ischaemia were considered. A urine drug screen was submitted to rule out substance abuse, and was positive for cocaine, amphetamines and opiates. Subsequent treatment included granulocyte colony stimulating factor, broad-spectrum antibiotics, vasopressors and intravenous hydrocortisone.
His working diagnosis at this time was sepsis due to a gastrointestinal source. When his condition did not improve after another 4 hours, he was taken to the operating room for exploratory laparotomy to rule out intestinal perforation or intra-abdominal infection. Laparotomy revealed a dusky-coloured stomach and duodenum with a small amount of murky, purulent fluid around the first part of the duodenum and spleen. The spleen, liver, small and large intestines appeared healthy. The small bowel was run from the ligament of Treitz to the terminal ileum, and no obstruction or perforation was found. By this time, the colour of the stomach and duodenum improved, becoming pink and less dusky. The lesser sac was opened and no perforation was found in the posterior aspect of the stomach. The first part of the duodenum was inflamed with omental adhesions, so a microperforation was suspected in this area. A blue dye was placed down the nasogastric tube and no perforation was found. EGD was considered at this time, but a decision was made not to pursue this because of concern that the EGD might cause the opening of a sealed perforation. The patient's condition temporarily stabilised during surgery and vasopressors were reduced. The abdomen was left open with drains in place to prevent abdominal compartment syndrome.
Outcome and follow-up
He returned to the ICU where he later developed systemic inflammatory response syndrome (SIRS), disseminated intravascular coagulation, acute respiratory distress syndrome (ARDS) and worsening multiple organ failure. He died ∼6 hours after surgery.
Blood cultures were negative at 48 hours. After autopsy, the cause of death was determined to be oesophageal necrosis with enteric ischaemia due to acute intoxication by the combined effects of cocaine, bupropion and amphetamine. Autopsy also revealed evidence of upper gastrointestinal haemorrhage and fatty liver. Heart blood analysis revealed benzoylecgonine concentration (a cocaine metabolite) of 30±4 ng/mL. Postmortem review of the CT scan also revealed oesophageal thickening, in addition to the aforementioned findings (figure 2).
Figure 2.
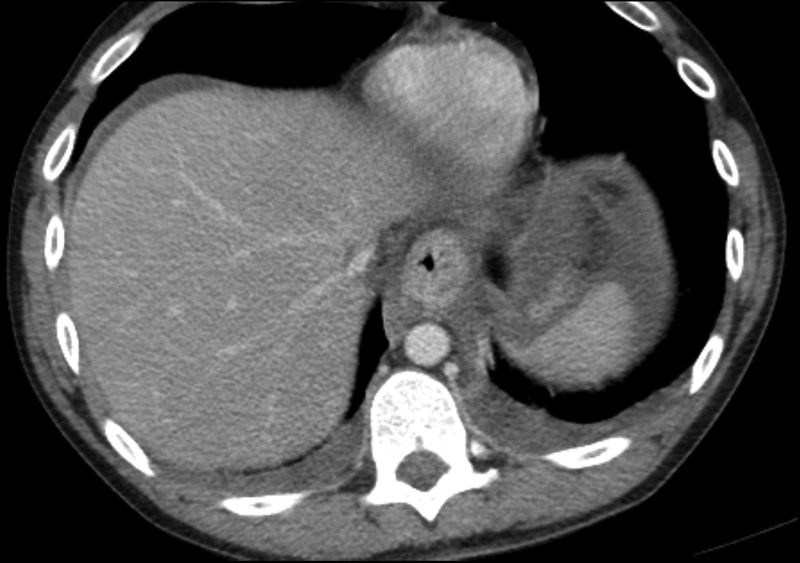
Postmortem review of CT also showing esophageal thickening.
Discussion
AEN is a disease process that is only recently becoming better understood. It was first described while performing a postmortem by Brennan in 19676 while the first endoscopic case report was published in 1990 by Goldenberg et al.7 The incidence of this condition is difficult to determine, but it ranges between 0.01% and 0.0125% of Caucasians undergoing EGD4 and up to 0.2% in autopsy reports and clinical trials.8 The mortality rate associated with AEN in case series is ∼32–38%,1 9 most of which has been attributed to serious comorbid conditions. Consequently, the disease specific mortality has been estimated as low as 6%.2 Treatment is usually conservative, involving the administration of high-dose proton-pump inhibitors3 and serial EGDs.9 Immediate surgical intervention for AEN is usually only required when necrosis has led to oesophageal perforation, which occurs in ∼6% of cases and usually requires partial or complete esophagectomy.2 When conservative treatment is effective, oesophageal stricture or stenosis may result in ∼10% of cases,2 which may require subsequent endoscopic intervention.
While it may be straightforward to infer that cocaine ingestion could cause the necessary oesophageal ischaemia to induce AEN, the literature is relatively silent on this possible association. A PubMed literature review ((‘acute oesophageal necrosis’ or ‘black oesophagus’) and cocaine) on 28 June 2016 revealed only one case report by Altenburger et al10 involving a patient with a history of alcohol and cocaine abuse in whom AEN was found postmortem. In contrast, many cases of AEN have been reported in patients with active alcohol abuse, and this risk factor was present in our case as well as in Altenburger's case. Interestingly, alcohol can react with cocaine to form cocaethylene, which has a longer half-life and greater toxicity.5 Perhaps the combination of alcohol and cocaine may further increase the risk of AEN, but further research is required to investigate this hypothesis.
As previously mentioned, much of the mortality associated with AEN has been attributed to comorbid conditions. In this case, death was likely a direct result of severe lactic acidosis that led to hypotension, multiple organ failure and fatal cardiac arrhythmia. The lactic acidosis may have also caused the AEN.11 Lactic acidosis, in turn, might have been caused by non-occlusive mesenteric ischaemia (NOMI) secondary to cocaine use12 and the use of prescribed stimulants. In contrast to AEN, NOMI carries a very high mortality rate of 70–100% because many cases lead to ARDS and SIRS.13 This patient developed similar complications, and it is likely that NOMI, as a comorbid condition to the AEN, was a significant contributor to his death. If NOMI is suspected and confirmed early, it can sometimes be treated effectively with intravenous vasodilators (eg, papaverine or prostaglandin E1).14 15
This patient's lactic acidosis also could have been caused by severe thiamine deficiency,16 although thiamine was not checked in this patient. Thiamine deficiency, often caused by alcohol abuse (but not always),17 can cause a syndrome known as gastrointestinal beriberi. Symptoms include abdominal pain, vomiting, leg oedema, weakness and paresthesias. Gastrointestinal beriberi may also lead to a severe lactic acidosis (pH has been reported as low as 6.82)18 which can be fatal.16 This can be corrected easily with thiamine administration, which is quite safe and can lead to rapid improvement of symptoms.18 Thiamine deficiency can present in a variety of ways, and should therefore be considered in most patients with active alcohol abuse regardless of their presenting symptoms. Thiamine supplementation should be strongly considered early in these patients, and the supplementation order can be added to emergency room protocols to improve consistency of use.
This patient also had a moderate neutropenia, which is typically caused by infection, medication, or a haematological condition. While his signs and symptoms appeared consistent with sepsis, his blood cultures remained negative, making bloodstream infection less likely as a cause for his decompensation. Alternatively, his neutropenia may have been caused by levamisole, which is found as an adulterant in up to 69% of cocaine confiscated in the USA.19 Levamisole can cause neutropenia, thrombocytopenia, fever and vasculitis.19 Whether this may have contributed to his mortality remains unclear.
Overall, the pathophysiology of this patient's disease process is still uncertain. He presented with symptoms of severe epigastric pain and haematemesis, which could be directly attributable to oesophageal necrosis and sloughing of the oesophageal epithelium. On the other hand, his epigastric pain at presentation also may have been an indicator of the relatively recent onset of mesenteric ischaemia. This theory is supported by the fact that he had a mildly low carbon dioxide at presentation with an elevated anion gap of 18 (although lactate was not checked until his resuscitation 6 hours later). His heavy alcohol use predisposed him to AEN by increasing reflux of gastric contents and decreasing his mucosal defences through poor nutritional status. Also, heavy alcohol use put him at risk for thiamine deficiency, which could have contributed to the lactic acidosis. Cocaine use was his major risk factor for NOMI through the process of splanchnic vasoconstriction, and we hypothesise that cocaine, in a similar way, increased his risk of AEN by causing or contributing to oesophageal ischaemia.
Acute oesophageal necrosis is a rare diagnosis that carries with it significant morbidity and mortality, most often due to comorbid conditions. It should be considered in patients who present with haematemesis—especially in older men with comorbid conditions and in younger patients with alcohol abuse. Diagnosis by early EGD is essential, although early EGD may not have changed the outcome in this rapidly progressing and fatal case. After reviewing this case and that of Altenburger et al, we propose a hypothesis that cocaine abuse may be an independent risk factor for AEN. This hypothesis could be evaluated using a case–control study comparing cases of AEN to controls and analysing a potential association with prior exposure to cocaine. While management of AEN is generally conservative using proton-pump inhibitors and avoiding oral intake, comorbid conditions (in this case, NOMI) may require aggressive treatment to prevent morbidity and mortality. In contrast, aggressive surgical management of the AEN itself is usually not warranted (in the absence of oesophageal perforation) and would have been unlikely to change the outcome in this case. The presence of AEN should prompt an early and thorough aetiological evaluation—including evaluation for substance abuse—to identify and treat these potentially lethal comorbid conditions.
Patient's perspective.
As the patient's surviving spouse, I appreciate the opportunity to offer my perspective on my husband's medical condition and sudden death.
Acute onset and rapid decline
First, from my perspective his condition was very acute and his death absolutely surprising to both the family and the medical team. He was a seemingly healthy 30-year-old in good physical condition and with no significant medical history.
The day prior to being admitted to the hospital, he reported of generally feeling unwell, and specifically of suffering from the ‘feeling of infection,’ noting inflammation of minor cuts, hangnails and clogged pores.
The day he was admitted, he awoke feeling unwell but still went to work. By early afternoon, he left work to rest at his parent's home, reporting of a debilitating headache as well as diarrhoea and vomiting. By early evening he drove himself home and collapsed on entering the door, reporting severe stomach pain and blood in his vomit. I called 911, and he was taken by ambulance to the hospital, being admitted around 19:30.
He spent several hours in the emergency room with severe stomach pain that could not be controlled with medication, so intense that he could barely talk. By 1:30, he was incorrectly diagnosed with a bowel obstruction; as they inserted a nasogastric tube he lost consciousness and required resuscitation.
Shortly thereafter, he tested positive for cocaine and the medical team began to suspect gastric ischaemia, which exploratory surgery confirmed around 8:00. The results of the surgery were inconclusive but hopeful; the surgeon reported a necrotic stomach that started to pink up during the surgery. Further surgery was planned once my husband's condition stabilised.
However, after the surgery my husband declined rapidly, developing both acute respiratory distress syndrome and disseminated intravascular coagulation. He was pronounced dead by 16:00, <24 hours from the time he called me with a headache from his parents' house.
Gastrointestinal history
At least twice in the 10 years before his death, my husband reported stomach pain that we believed to be an ulcer, though it was never officially diagnosed. He was a heavy drinker and also frequently took ibuprofen, which in addition to stress were what we believed to be the contributing factors for an ulcer. In the 3 months prior to his death, the office notes from his general practitioner indicate a diagnosis of gastro-oesophageal reflux disease, with ranitidine prescribed to treat. He frequently used over-the-counter antiacid tablets as well as the home remedy apple cider vinegar to treat what he described as ‘acid stomach’. He also reported being lactose intolerant.
Prescribed medications
The medical examiner attributed his cause of death to cocaine as well as bupropion and amphetamine, the latter two being prescribed medications for anxiety and attention deficit hyperactivity disorder. I believe he began taking these about a year before his death. He switched from bupropion to lexapro several months before his death, and then back to bupropion 1 week prior to his death. He did not disclose his alcohol use to the prescribing physician.
Alcohol and drug use
In terms of alcohol use, I would estimate my husband averaged 3–4 drinks per day (primarily beer) for at least 10 years prior to his death. By a few weeks before his death he had increased this amount to 6–10 drinks per day, with the increase primarily occurring over the 6 months prior.
Evidence from close friends and secret text/email correspondence suggests he was not a long-term drug user; prior to his death I had no knowledge of any drug use. In the 2 weeks before his death, my best guess is that he tried heroin once and cocaine once, the latter most likely used the day before he was admitted to the hospital. He started using an e-cigarette occasionally about 3 months before he died. He also used tadalafil without medical supervision, as well as caffeine, both of which were present in his system at the time of death (in addition to the bupropion, amphetamine and cocaine).
Conclusion
My husband never regained consciousness postresuscitation. He never knew he had tested positive for cocaine, and never had the opportunity to answer questions about the extent of his drug use or provide details about his medical history. The reasons he developed such a rare medical condition and then experienced multiple fatal complications are just among the many secrets he has taken to the grave.
Learning points.
Acute oesophageal necrosis is a rare condition that should be suspected in patients with haematemesis and alcohol abuse.
Identification of acute oesophageal necrosis should prompt a search for potentially lethal comorbid conditions.
A thorough substance abuse history is imperative for the young patient with acute abdominal pain, including a low threshold for urine toxicology.
Intestinal ischaemia should be considered early in the patient with acute abdominal pain, especially when imaging shows bowel wall thickening without a transition point.
Thiamine supplementation should be considered for any patient presenting to the emergency room with a history of alcohol abuse.
Footnotes
Contributors: CEP is the primary author and guarantor of this case report. He wrote the abstract, introduction and discussion. TZP contributed the narrative of the case report, which was then edited by CEP.
Competing interests: A potential conflict of interest exists in that both authors of this report are brothers to the patient described in this case. While this relationship may bias us toward potentially overestimating the importance of this case to the medical community, we do not believe that this bias has negatively affected the quality of the report or our review of the relevant literature.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Day A, Sayegh M. Acute oesophageal necrosis: a case report and review of the literature. Int J Surg 2010;8:6–14. 10.1016/j.ijsu.2009.09.014 [DOI] [PubMed] [Google Scholar]
- 2.Gurvits GE, Shapsis A, Lau N et al. Acute esophageal necrosis: a rare syndrome. J Gastroenterol 2007;42:29–38. 10.1007/s00535-006-1974-z [DOI] [PubMed] [Google Scholar]
- 3.Gurvits GE, Cherian K, Shami MN et al. Black esophagus: new insights and multicenter international experience in 2014. Dig Dis Sci 2015;60:444–53. 10.1007/s10620-014-3382-1 [DOI] [PubMed] [Google Scholar]
- 4.Zacharia GS, Sandesh K, Ramachandran T. Acute esophageal necrosis: an uncommon cause of hematemesis. Oman Med J 2014;29:302–4. 10.5001/omj.2014.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmerman JL. Cocaine intoxication. Crit Care Clin 2012;28:517–26. 10.1016/j.ccc.2012.07.003 [DOI] [PubMed] [Google Scholar]
- 6.Brennan JL. Case of extensive necrosis of the oesophageal mucosa following hypothermia. J Clin Pathol 1967;20:581–4. 10.1136/jcp.20.4.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldenberg SP, Wain SL, Marignani P. Acute necrotizing esophagitis. Gastroenterology 1990;98:493–6. 10.1016/0016-5085(90)90844-Q [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin CW, Person TD, Denlinger CE. Management of acute esophageal necrosis syndrome. J Thorac Cardiovasc Surg 2011;141:e23–4. 10.1016/j.jtcvs.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 9.Worrell SG, Oh DS, Greene CL et al. Acute esophageal necrosis: a case series and long-term follow-up. Ann Thorac Surg 2014;98:341–2. 10.1016/j.athoracsur.2013.09.023 [DOI] [PubMed] [Google Scholar]
- 10.Altenburger DL, Wagner AS, Li S et al. A case of black esophagus with histopathologic description and characterization. Arch Pathol Laboratory Med 2011;135:797–8. 10.1043/2010-0128-C.1 [DOI] [PubMed] [Google Scholar]
- 11.Endo T, Sakamoto J, Sato K et al. Acute esophageal necrosis caused by alcohol abuse. World J Gastroenterol 2005;11:5568–70. 10.3748/wjg.v11.i35.5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sudhakar CB, Al-Hakeem M, MacArthur JD et al. Mesenteric ischemia secondary to cocaine abuse: case reports and literature review. Am J Gastroenterol 1997;92:1053–4. [PubMed] [Google Scholar]
- 13.Yasuhara H. Acute mesenteric ischemia: the challenge of gastroenterology. Surg Today 2005;35:185–95. 10.1007/s00595-004-2924-0 [DOI] [PubMed] [Google Scholar]
- 14.Herskowitz MM, Gillego V, Ward M et al. Cocaine-induced mesenteric ischemia: treatment with intra-arterial papaverine. Emerg Radiol 2002;9:172–4. 10.1007/s10140-002-0218-7 [DOI] [PubMed] [Google Scholar]
- 15.Mitsuyoshi A, Obama K, Shinkura N et al. Survival in nonocclusive mesenteric ischemia: early diagnosis by multidetector row computed tomography and early treatment with continuous intravenous high-dose prostaglandin E(1). Ann Surg 2007;246:229–35. 10.1097/01.sla.0000263157.59422.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein M, Weksler N, Gurman GM. Fatal metabolic acidosis caused by thiamine deficiency. J Emerg Med 2004;26:301–3. 10.1016/j.jemermed.2003.11.014 [DOI] [PubMed] [Google Scholar]
- 17.Duca J, Lum CJ, Lo AM. Elevated lactate secondary to gastrointestinal beriberi. J Gen Intern Med 2016;31:133–6. 10.1007/s11606-015-3326-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donnino M. Gastrointestinal beriberi: a previously unrecognized syndrome. Ann Intern Med 2004;141:898–9. 10.7326/0003-4819-141-11-200412070-00035 [DOI] [PubMed] [Google Scholar]
- 19.Martinez E, Alvi R, Venkatram S et al. Recurrent febrile neutropenia and thrombocytopenia in a chronic cocaine user: a case of levamisole induced complications. Case Rep Crit Care 2015;2015:303098 10.1155/2015/303098 [DOI] [PMC free article] [PubMed] [Google Scholar]