

Abstract
Myoclonus-dystonia syndrome (MDS) is a rare disease manifesting myoclonus as the only neurological symptom which may be accompanied by dystonia. It usually starts in the first or second decade of life. It has a benign course with spontaneous remissions but can cause functional disability in some patients. In this paper, we report a patient diagnosed as probable MDS on the basis of clinical and electrophysiological features who showed marked improvement under levetiracetam treatment.
Keywords: Myoclonus, dystonia, treatment
Myoclonus-dystonia syndrome (MDS) is a rare, sometimes sporadically encountered autosomal dominant hereditary disease occasionally manifesting symptoms of isolated myoclonus potentially accompanied with dystonia, and frequently seen in the first two decades of life [1]. Myoclonic contractions mostly, and predominantly involve upper extremities, and especially their proximal parts. Alcohol intake typically suppresses myoclonic jerks [1, 2]. Dystonic episodes (storms) of mild-moderate severity can accompany myoclonic jerks [1].
For ıts symptomatic treatment, benzodiazepines, anticholinergics, levodopa, dopamine agonists, amantadine, serotonergic agents, beta blockers, neuroleptics, and antiepileptic agents have been tried. In some patients, with myoclonic seizures favourable responses to levetiracetam, piracetam, and zonisamide have been obtained [3, 4]. In refractory cases, deep brain stimulation constitutes an alternative treatment option [5].
In our article, we have presented a case with a probable diagnosis of MDS based on clinical, and electrophysiologic examinations who also responded to levetiracetam therapy.
CASE REPORT
A-14-year-old female patient with a right hand dominancy consulted to us with complaints of involuntary jerks of her left arm especially when she were moving her arm or writing something. Her complaints started when she was 7 years of age, and her complaints continued without demonstrating any progression. Her complaints worsened especially when she tried to write with her left hand, and consequently she consulted to a neurologist in her home city who referred the patient to us without initiating any therapy. Her medical history was unremarkable. Her family history revealed only consanguineous marriage with a third-degree relative. Neurological examination detected presence of only involuntary movements without any other pathologic finding. Involuntary movements were characterized by brief, focal dysrhythmic jerks worsened by certain postures, and movements involving all along the affected left arm, but being more predominant at the distal part of this extremity. Biochemical analysis couldn’t detect any significant characteristic. Her electroencephalographic (EEG) examination did not demonstrate cortical discharges associated with myoclonic jerks. On her electromyograms (EMGs) myoclonic activities correlated with clinically observed involuntary movements with varying monophasic or polyphasic amplitudes recurring at irregular intervals were noted (Figure 1). During examination of her somatosensory evoked potentials (SEPs) tibial SEP was evaluated as within normal limits. On examination of median SEP, SEPs with relatively longer latency, and higher amplitude were detected (Figure 2). C reflex was absent. With these findings, we thought that our patient were experiencing myoclonic jerks of subcortical origin. Then she was evaluated based on the criteria suggested by Grunewald et al., and diagnosis of probable MDS was made. The patient was started on escalating levetiracetam therapy (1000 mg/day). At the control visit in our outpatient clinic, a marked decrease in her myoclonic episodes were observed.
FIGURE 1.
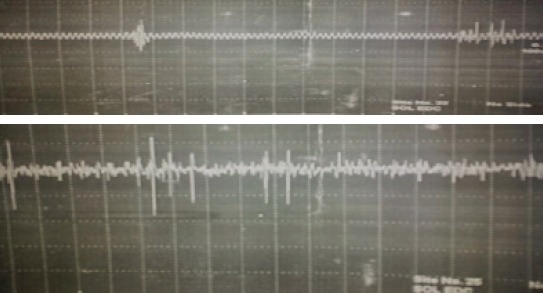
EMG during involuntary movement.
FIGURE 2.
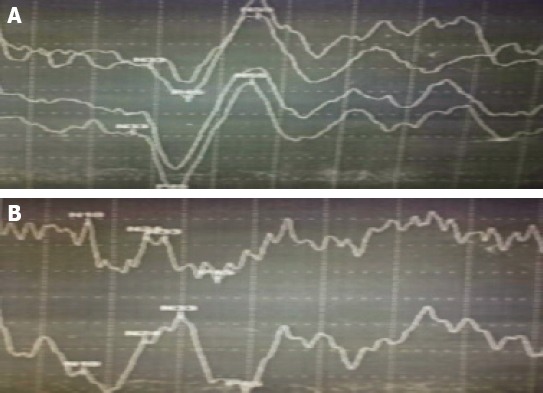
(A) Right median SEP response (B) Left median SEP response.
DISCUSSION
Myoclonus can be physiologic, central or peripheral or it can emerge during the course of many different disease states. Clinical findings, imaging modalities, and electrophysiologic examinations aid in the determination of etiology of myoclonus.
MDS is a rare, sometimes sporadically encountered, generally autosomal dominant hereditary disease manifesting symptoms of isolated myoclonus potentially accompanied with only dystonia, and frequently seen in the first two decades of life which also dramatically alleviates with alcohol intake [1, 2].
In the year 2008 yılında Grunewald et al. classified MDS as definite MDS (early-onset myoclonus, and dystonia episodes or isolated myoclonus occurring predominantly in the upper extremity with a positive family history for myoclonus and/or dystonia), probable MDS (early-onset myoclonus, and dystonia or isolated myoclonus occurring predominantly in the upper extremity), possible MDS (cervical dystonia or isolated jolting movements involving various parts of the body or dystonia and/or myoclonus affecting lower half of the body or alcohol unresponsive–MDS). MDS manifesting before 26 years of age is considered as early-onset MDS [6]. In 2009 Kinugava et al. proposed five diagnostic criteria for definite MDS based on available clinical data, and comprehensive literature review:
1) Age of onset ≤20 years; 2) Myoclonus with or without dystonia, 3) Positive family history, 4) Absence of any other neurological comorbidity 5) Normal cerebral magnetic resonance imaging finding [1]. According to Kinugava classification our patient did not meet definite MDS criteria due to lack of any positive family history, and she was included in probable MDS category based on Grunewald classification.
In some cases diagnosed as autosomal dominant hereditary MDS, mutations in the epsilon-sarcoglycan gene (SGCE) was identified at locus 7q21 on the long arm of the 7. chromosome. Grunewald et al. identified SCGE mutations in patients with diagnosis of definite (6/10; 60%), and probable (3/15; 2%) MDS [6]. Carrecchio et al. (2013) also detected. SCGE mutations in patients diagnosed as definite (7/23; 30.4%), and probable MDS (8/52; 15.4%). In the study performed by Carrecchio et al. more than half of the patients with mutations had not positive family history for MDS. Lack of positive family history was correlated with reduced penetrance of SCGE gene, and existence of de novo mutations [7]. Similarly, Ritz et al. couldn’t demonstrate presence of mutations in half of their patients diagnosed as definite MDS [8]. Because of our restricted facilities, we couldn’t perform genetic tests on our patient whose family history was unremarkable for MDS Studies performed hitherto have demonstrated that criteria defined for MDS are not 100% sensitive, and specific for SCGE mutations. On the other hand, in some patients with SCGE mutation atypical phenotype (adult-onset, lower extremity involvement, predominant dystonia) has been observed [9].
In MDS, myoclonic contractions predominantly affect upper extremities. More rarely, lower extremity, face, and vocal chord can be involved. DMS tends to affect proximal parts of the extremities, however, as is seen in our patient, cases of myoclonuc jerks predominantly affecting distal parts of the extremities have been also reported [1, 2]. Isolated cases of myoclonus can be seen or mild or moderate degrees of dystonia can accompany myoclonic episodes. Dystonia is more frequently seen as writer’s cramp, and cervical dystonia [10]. In our patient, myoclonus manifested itself as irregular, brief, focal jerks which increase in frequency with change of posture, and movement, and observed all along the left arm, but being more predominant at the distal part of the extremity. Repetitive neurological examinations performed on our hospitalized patient during her hospital stay, and follow-up period did not reveal any evidence of dystonia.
Records of myoclonic episodes, and neurophysiologic examinations including EEG, and SEP aid in the diagnosis of MDS. On EMG, myoclonic discharges each lasting for 25-250 ms are recorded during resting, and tonic contractions.. Myoclonic contractions have a dysrhythmic pattern with a frequency not exceeding 10 Hz [1, 2]. On EEG examination of cortical myoclonus, frequently negative waves are observed before onset of myoclonic jerks. In subcortical myoclonus, any EEG activity is not seen. During recording of SEPs, giant somatosensory evoked potentials are not accompanied by C-reflex. On our patient’s EMG, we observed occasional mono-, or polyphasic myoclonic activities of different amplitudes recurring at irregular intervals which also correlated with clinically observed involuntary movements. During EEG examination cortical discharges associated with myoclonic activities were not observed. On SEP examination, giant somatosensory evoked potentials were not observed and also C-reflex was not recognized. In the light of all these data myoclonic contractions of our patient were interpreted as nonepileptic subcortical myoclonus. Within this context, when patient’s age at the onset of the disease, normal cranial magnetic resonance imaging (MRI) findings, and persistence of isolated myoclonus as the only neurological pathology affecting solely her upper extremity during 7 years of the disease process were taken into consideration, juvenile Huntington disease (Westphal variant), early-onset Parkinson’s disease, Wilson’s Disease, and pantothenate -kinsase associated neurodegeneration which are contemplated in the differential diagnosis of the disease were ruled out.
Pathophysiology of the disease is not completely understood. However in some studies, primary dysfunction of basal ganglions have been implicated in its pathophysiology. Nonobservance of C-reflex, premyoclonic critical discharges, and giant somatosensory evoked potentials on EEG, indicates subcortical origin of myoclonic jerks [3, 11]. Dramatic response obtained with deep brain stimulation (DBS) applied on globus pallidus internus (GPI) in patients with MDS suggests involvement of GPI among subcortical structures [5].
Nowadays, a definitive treatment for MDS does not exist. Wide spectrum of symptomatic pharmacological treatment modalities have been tried including benzodiazepines, anticholinergics, L-dopa, dopamine agonists, amantadine, neuroleptics, serotonergic agents, and beta-blockers [3, 4]. Apart from oral medications, botulinum toxin is being used for the treatment of dystonic posture [12, 13]. In refractory cases leading to serious disabilities, DBS which targets GPI is a treatment alternative [5]. Effectiveness of antiepileptic drugs as levetiracetam, piracetam, and zonisamide in symptomatic treatment of MDS has been demonstrated [1, 2, 14]. In our case levetiracetam at daily oral doses of 1000 mg provided marked symptomatic relief.
MDS is a rarely seen disease with a potential of causing serious disabilities. Antiepileptic levetiracetam can achieve symptomatic improvement, and raise quality of life of some patients. In the 21th century where genetic evaluation methods have been revolutionized, we foresee gene therapy as an effective treatment alternative for MDS.
Footnotes
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
REFERENCES
- 1.Kinugawa K, Vidailhet M, Clot F, Apartis E, Grabli D, Roze E. Myoclonus-dystonia: an update. Mov Disord. 2009;24:479–89. doi: 10.1002/mds.22425. [DOI] [PubMed] [Google Scholar]
- 2.Roze E, Apartis E, Clot F, Dorison N, Thobois S, Guyant-Marechal L, et al. Myoclonus-dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology. 2008;70:1010–6. doi: 10.1212/01.wnl.0000297516.98574.c0. [DOI] [PubMed] [Google Scholar]
- 3.Obeso JA, Rothwell JC, Lang AE, Marsden CD. Myoclonic dystonia. Neurology. 1983;33:825–30. doi: 10.1212/wnl.33.7.825. [DOI] [PubMed] [Google Scholar]
- 4.Quinn NP. Essential myoclonus and myoclonic dystonia. Mov Disord. 1996;11:119–24. doi: 10.1002/mds.870110202. [DOI] [PubMed] [Google Scholar]
- 5.Kuncel AM, Turner DA, Ozelius LJ, Greene PE, Grill WM, Stacy MA. Myoclonus and tremor response to thalamic deep brain stimulation parameters in a patient with inherited myoclonus-dystonia syndrome. Clin Neurol Neurosurg. 2009;111:303–6. doi: 10.1016/j.clineuro.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grünewald A, Djarmati A, Lohmann-Hedrich K, Farrell K, Zeller JA, Allert N, et al. Myoclonus-dystonia: significance of large SGCE deletions. Hum Mutat. 2008;29:331–2. doi: 10.1002/humu.9521. [DOI] [PubMed] [Google Scholar]
- 7.Carecchio M, Magliozzi M, Copetti M, Ferraris A, Bernardini L, Bonetti M, et al. Defining the epsilon-sarcoglycan (SGCE) gene phenotypic signature in myoclonus-dystonia: a reappraisal of genetic testing criteria. Mov Disord. 2013;28:787–94. doi: 10.1002/mds.25506. [DOI] [PubMed] [Google Scholar]
- 8.Ritz K, Gerrits MC, Foncke EM, van Ruissen F, van der Linden C, Vergouwen MD, et al. Myoclonus-dystonia: clinical and genetic evaluation of a large cohort. J Neurol Neurosurg Psychiatry. 2009;80:653–8. doi: 10.1136/jnnp.2008.162099. [DOI] [PubMed] [Google Scholar]
- 9.Hartmann CJ, Leube B, Wojtecki L, Betz B, Groiss SJ, Bauer P, et al. A novel mutation of the SGCE-gene in a German family with myoclonus-dystonia syndrome. J Neurol. 2011;258:1186–8. doi: 10.1007/s00415-011-5911-6. [DOI] [PubMed] [Google Scholar]
- 10.Koukouni V, Valente EM, Cordivari C, Bhatia KP, Quinn NP. Unusual familial presentation of epsilon-sarcoglycan gene mutation with falls and writer’s cramp. Mov Disord. 2008;23:1913–5. doi: 10.1002/mds.21935. [DOI] [PubMed] [Google Scholar]
- 11.Marelli C, Canafoglia L, Zibordi F, Ciano C, Visani E, Zorzi G, et al. A neurophysiological study of myoclonus in patients with DYT11 myoclonus-dystonia syndrome. Mov Disord. 2008;23:2041–8. doi: 10.1002/mds.22256. [DOI] [PubMed] [Google Scholar]
- 12.Costa J, Borges A, Espírito-Santo C, Ferreira J, Coelho M, Moore P, et al. Botulinum toxin type A versus botulinum toxin type B for cervical dystonia. Cochrane Database Syst Rev. 2005:1–CD004314. doi: 10.1002/14651858.CD004314.pub2. [DOI] [PubMed] [Google Scholar]
- 13.Costa J, Espírito-Santo C, Borges A, Ferreira JJ, Coelho M, Moore P, et al. Botulinum toxin type A therapy for cervical dystonia. Cochrane Database Syst Rev. 2005:1–CD003633. doi: 10.1002/14651858.CD003633.pub2. [DOI] [PubMed] [Google Scholar]
- 14.Matur Z, Bebek N, Gürses C, Baykan B, Gökyiğit A. Miyoklonus-distoni sendromu: Olgu sunumu Epilepsi. 2007;13:94–6. [Google Scholar]