

Abstract
Dysregulation of the normal gene expression program is the cause of a broad range of diseases, including cancer. Detecting the specific perturbed regulators that have an effect on the generation and the development of the disease is crucial for understanding the disease mechanism and for taking decisions on efficient preventive and curative therapies. Moreover, detecting such perturbations at the patient level is even more important from the perspective of personalized medicine. We applied the Transcription Factor Target Enrichment Analysis, a method that detects the activity of transcription factors based on the quantification of the collective transcriptional activation of their targets, to a large collection of 5607 cancer samples covering eleven cancer types. We produced for the first time a comprehensive catalogue of altered transcription factor activities in cancer, a considerable number of them significantly associated to patient’s survival. Moreover, we described several interesting TFs whose activity do not change substantially in the cancer with respect to the normal tissue but ultimately play an important role in patient prognostic determination, which suggest they might be promising therapeutic targets. An additional advantage of this method is that it allows obtaining personalized TF activity estimations for individual patients.
Transcription factors (TFs) play a crucial role in the dynamic regulation of the gene expression program1. The knowledge cumulated in the last years on diverse cellular gene expression programs has drastically increased our understanding of the effects of dysregulation of gene expression in disease. In fact, a broad range of diseases and syndromes, including cancer2, are caused by mutations that affect TFs either directly or indirectly, by affecting cofactors, regulatory sequences, chromatin regulators, and noncoding RNAs that interact with these regions3. Specifically, dysregulations or changes in the activation status of distinct TFs are known to be linked to a number of cancers4,5,6. Actually, many oncogenes and tumour suppressor genes, including the well-known P53 gene7, are in fact8 TFs. Moreover, many cancer treatments are essentially transcriptional interventions9. Thus, hormonal therapies in breast and prostate cancers to block tumour progression are classical examples. More sophisticated interventions are the inhibition of global epigenomic regulators like BRD410. Consequently, understanding the determinants of the transcriptional changes leading to disease states in patients is a prerequisite to restore the normal functions of a cell or a tissue.
Alterations in the transcriptional regulatory network due to perturbed TF activity cause the dysregulation of gene expression observed during cancer progression. Different reverse engineering methods11,12,13,14,15,16,17 have been proposed to infer the specific TF activity that accounts for the observed differential expression across conditions. Reverse engineering methods use the transcription level of a TF to estimate its activity by calculating different types of correlation to its corresponding target genes. However, using TF expression levels as proxies of their activities can be misleading by several reasons. Firstly, the mRNA expression levels of many TFs are often relatively low compared to other genes, which increase the uncertainty of the corresponding measurements. Secondly, the regulation of TFs at the protein level has shown to be more relevant than changes at the mRNA level, as demonstrated for example in hypoxia-inducible factors18 and p5319. Moreover, the binding of a TF to the corresponding TFBS does not necessarily imply a transcriptional activity because post-transcriptional modifications and some extra co-factors may be required to promote gene expression20,21.
As a consequence of this, TF expression levels cannot be considered good descriptors of their activity. Contrarily, the expression levels of the TF’s targets, in which all the above mentioned effects are integrated, seem to be a more reasonable readout of TF activity. Despite the simplicity of this idea and its enormous potential, only a few algorithmic proposals have been made that exploit TF’s target expression levels to infer the corresponding TF activities, such as BASE22, RENATO23, REACTIN24, RABIT25 or others26. These methods have been applied to the study of survival in breast cancer27 or to obtain signatures of tumour stage in kidney renal clear cell carcinoma28.
Here we use a simple but efficient method to systematically detect TFs with altered activity by studying the activity of their corresponding target genes across a total of 5607 samples covering eleven cancer types. This study allowed us to produce the first comprehensive catalogue of TFs with activity altered across a broad spectrum of cancer types. Since the method used can also return personalized values of TF activity for each patient, we could also identify a number of TFs whose altered activity was significantly associated to patient’s survival, demonstrating their relevance in cancer progression and their potential as therapeutic targets.
Results and Discussion
Changes in TF activity across the different cancers
Raw RNA-seq counts for all the eleven cancers studied (Table 1) were normalized as described in Methods and tumour samples were compared to their normal tissue counterparts to obtain lists of genes differentially expressed. TF Target Enrichment Analysis (TFTEA) was applied to these lists ranked by value of the statistic. Figure 1 show changes in the activity of the different TFs when cancers are compared to their corresponding normal tissues. The predominant observed behaviour is the increase in TF activity. Actually, a set of TFs (E2F6, E2F4, MYC, MYC:MAX and NRF1) are always significantly more active in cancers than in normal tissues, and others (EGR1, ELF1, SP1, YY1, USF1, SP2, ZBTB33, MAX, CTCFL and NR2C2) are significantly active in almost all the cancers with a few exceptions, which suggest for them an important role in cancer development and progression. Actually, all of them appear in the COSMIC database29 and some of them are well-known oncogenes such as MYC30,31,32, MAX and MYC:MAX33, or proteins of the E2F family34, whose over-expression induces uncontrolled cell proliferation because they are TFs located upstream in pathways that control cell cycle35, being also considered prognostic factors36. The YY1 TF is a multifunctional protein that regulates various processes of development and differentiation and have a clear involvement in tumorigenesis, having been proposed as potential prognostic marker of diverse cancers37. SP1 and SP2 regulate many of the genes involved in the Warburg effect38, a well-known cancer hallmark39. Actually, high levels of SP1 protein are considered a negative prognostic factor for several cancers40,41.
Table 1. Cancer samples available for any cancer type selected.
| Cancer type | Tumour | Normal | Stage I | Stage II | Stage III | Stage IV | Alive | Deceased |
|---|---|---|---|---|---|---|---|---|
| Bladder Urothelial Cancer [BLCA] | 294 | 17 | 1 | 95 | 99 | 98 | 221 | 73 |
| Breast Cancer [BRCA] | 1039 | 113 | 177 | 591 | 237 | 17 | 937 | 98 |
| Colon Adenocarcinoma [COAD] | 428 | 41 | 73 | 168 | 120 | 58 | 374 | 53 |
| Head and Neck Squamous Cell Carcinoma [HNSC] | 480 | 42 | 26 | 74 | 72 | 245 | 320 | 158 |
| Kidney Renal Clear Cell Carcinoma [KIRC] | 517 | 72 | 256 | 56 | 125 | 81 | 358 | 159 |
| Kidney Renal Papillary Cell Carcinoma [KIRP] | 222 | 32 | 138 | 16 | 43 | 13 | 199 | 23 |
| Liver Hepatocellular carcinoma [LIHC] | 294 | 48 | 132 | 66 | 71 | 5 | 222 | 72 |
| Lung Adenocarcinoma [LUAD] | 473 | 55 | 255 | 116 | 81 | 24 | 355 | 118 |
| Lung Squamous Cell Carcinoma [LUSC] | 426 | 45 | 217 | 128 | 75 | 6 | 290 | 136 |
| Head and Neck Thyroid Carcinoma [THCA] | 500 | 58 | 282 | 54 | 110 | 52 | 481 | 14 |
| Uterine Corpus Endometrial Carcinoma [UCEC] | 508 | 23 | 318 | 49 | 114 | 27 | 464 | 43 |
Figure 1. Change of TF activity in the different cancers studied.

Cells in red indicate a significant increased activity of the TF in the cancer with respect to the corresponding normal tissue, according to the TFTEA, cells in blue indicate a significant decreased activity and cells in grey indicate that no significant change in activity was detected. Columns correspond to cancers and rows to TFs.
There are also a few TFs that show simultaneously significant, though opposite, behaviours across the studied cancers. This is the case, for example, of JUN:FOS, which induces anchorage-independent growth42 and SPI1, a known oncogene that increases the speed of replication43, which are deactivated in colon (COAD), uterine (UCEC), bladder (BLCA), lung (LUAD and LUSC) and prostate adenocarcinoma (PRAD) cancers, while are activated in the rest of cancers, suggesting the existence of different growing strategies in these two groups of cancers.
On the other hand, a few TFs systematically display a significant decrease in their activities. For example, two TFs with a largely unexplored role in human tumorigenesis, MEF2A and MEF2C, significantly reduce their activity in uterine (UCED), bladder (BLCA) and lung (LUSC) cancers. Supporting this observation, a significant down-regulation of MEF2A and MEF2C TFs was recently described in glioblastoma multiforme44. Actually, studies suggested that MEF2C is as target of miR-22345, an miRNA known to promote the invasion of breast cancer cells46.
Finally, other TFs display activations or deactivations shared by a few cancers and some of them present cancer-specific activities (See Fig. 1). Thus, FOS is activated in LIHC and THCA, or FOSL1 and FOSL2 are activated in KIRP, KIRC and THCA. Genes of the FOS family have been implicated as regulators of cell proliferation, differentiation, and transformation and are involved in many tumorigenic processes. Also REST gene, a transcriptional repressor that represses neuronal genes in non-neuronal tissues, is significantly activated in LIHC but significantly deactivated in COAD, maybe due to its dual role as a tumour suppressor and oncogene47.
Regarding TFs specific of cancers, JUNB, with a known role in liver regeneration48, but previously associated to different lymphomas such as Hodgkin49 or cutaneous T-cell50, seems to be also relevant in LIHC tumorigenesis. Thyroid carcinoma (THCA) presents a quite atypical pattern of TF activation. While it lacks some ubiquitous TFs, such as YY1, SP2, ZBTB33 or NR2C2, it presents significant activations in HNF4A, RXRA and RXR::RAR_DR5. Although HFN4A has traditionally been linked to diabetes, it has recently been suggested that this TF could be the link between ulcerative colitis and colorectal cancer51 and it has even be proposed as a biomarker of this cancer52 (colorectal cancer is not among the cancers included in this study). RXR and RAR are retinoid receptors that regulate cell growth and survival53, which have been proposed as cancer therapeutic targets54.
Cancers can be grouped in three main clusters according to their TF activity patterns. One of them is composed of uterine (UCEC), bladder (BLCA), lung (LUAD and LUSC) and prostate adenocarcinoma (PRAD) cancers. Another, more dispersed cluster is composed of breast (BRCA), kidney papillary cell (KIRP) head and neck squamous cell (HNSC) and liver (LIHC) cancers. Although showing a regulatory behaviour quite different among them, kidney clear cell (KIRC) and head and neck thyroid (THCA) carcinomas cluster together. Colon adenocarcinoma (COAD) maps closer to the first cluster but seems to be an outlier in terms of TF activity pattern.
Some cancers, however, display atypical activity patterns of activity for several TFs. For example, COAD shows a specific significant activity decrease of REST, CTCF (a known chromatin insulator protein that may play a central role in mediating long-range chromatin interactions, whose deregulation has an increasingly important role in the epigenetic imbalance in cancer55), EBF1 (identified as a tumour suppressor56) and TCF12. These regulatory differences might account, at least partially, for the different clinical behaviours of the distinct cancers analysed.
With respect to tissue of origin, both lung cancers, LUAD and LUSC, present quite similar TF activity profiles. Contrarily, kidney cancers KIRC and KIRP display remarkably different TF activity profiles. Interestingly, FOSL1 and FOSL2 TFs are specifically active almost uniquely in both cancers, while FOXA1 is significantly inactive. In particular, FOXA1, a TF involved in the differentiation of the pancreas and liver, is known to be expressed in breast cancer57 and others. Its remarkable down-activation in the two cancers originated in kidney could be part of the tumorigenesis in this organ.
It is worth noticing that, as previously mentioned, the expression level of TFs in the tissues according to The Protein Human Atlas database58 is uncorrelated with the corresponding activity detected from the expression of the corresponding targets (Supplementary Figure 1). This reinforces the usefulness of this approach, given that the direct observation of TF expression would have not rendered detectable changes in their behaviours.
Supplementary Table 1 contains the complete list of p-values obtained for all the TFs in all the cancers studied.
Changes in TF activity across cancer stages
The availability of clinical information, such as cancer stage allowed the stratification of cancer samples into their different stages. In any cancer type, the samples in any stage were compared to the corresponding normal samples. Figure 2 summarizes the changes in the activity status of TFs with respect to the normal situation in all the stages of the cancers analysed. Although the profiles of TF activity observed in this analysis are overall similar to the results produced by the comparison of cancer versus normal gene expressions, this analysis renders a more detailed picture of the changes in TF activity across stages in the different cancers. In fact, all the TFs present some activity change in some stages, even if this activity was not detected in the general cancer-control comparison. Thus, for example, RXRA, that was significantly active only in THCA in the cancer – control comparison, here presents a complex activation pattern across stages in BLCA as well. Other TFs, for which no significant change in the activity was previously found comparing cancer versus normal tissues, present however significant stage-specific activations, such as PPARG::RXRA activated in THCA, TAL::GATA1, down-activated in LUSC, ECR::USP, down-activated in several stages of LIHC and COAD.
Figure 2. Change of activity in all TF included in this study across cancer stages in the different cancers studied.

Each panel corresponds to a single TF, with stages in rows and cancers in columns. The colour scale in the figure ranges from red, indicating a significant increased activity of the TF in the stage of the cancer with respect to the corresponding normal tissue, according to the TFTEA, to blue, indicating a significantly decreased activity. The colour scale represents −log10 (adjusted p-value). Cells in grey indicate that no significant change in activity was detected. Cells in white correspond to stages in cancers with very few individuals (see Table 1) in which the analysis could not be carried out.
Supplementary Table 1 contains the complete list of p-values obtained for all the comparisons of TF activities across stages in all the cancers studied.
TF activity and survival
The availability of survival data for the cancers analysed (Table 1) allows testing hypotheses on the contribution of distinct TF activities in the cancers to the disease outcome by validating their association with patient’s survival. Since TFTEA can be applied in a personalized way to individual samples (see Methods) it is possible to know what TFs are active in any particular sample. Therefore, it is straightforward to test the relationship between TF activity and patient’s survival using Kaplan-Meier (K-M) curves59. Figure 3 summarises the K-M plots representing TF activities significantly associated to patient survival (See detailed plots in Supplementary Figure 2 and Supplementary Table 2). As expected, more significant results were found in the cancers with more detailed data on survival, which are KIRC, BRCA and HNSC (See Table 1). A total of 19 TFs presented a strong significant (adjusted p-values < 0.05) association between its activity and patient survival in BRCA, HNSC and KIRC. The number of TFs in the figure increases to 92 if we consider significant nominal p-values, and cover all the cancers (Fig. 3, Table 2 and Supplementary Table 3). Some of the TFs highly associated to survival have been detected in the study of TF activity across cancers. Examples in KIRC are: JUN:FOS, known to be correlated to KIRK survival28 and probably related to metastatic proliferation42, SPI1, whose activation has been linked to survival in gastric cancer41 in agreement with our observation (Fig. 4A), JUND, whose upregulation is significantly related to bad prognostic (Fig. 4B) and it has been described that can collaborate with NF-κB to increase antiapoptotic gene expression60 and also NRF1, EGR1, ETS1, ZEB1, MAX and FOSL1. Previous studies of TF activity in KIRK reveal a number of them significantly correlated to survival28. Among the TFs that overlap with this study, FOS, JUN::FOS, REST and TCF12 are found to be significantly related to survival, while GATA1 did not reached the significance threshold. In HNSC, JUND and ELF1 are differentially activated between the cancer and the normal tissue and also significantly associates to survival. In agreement with our results (Fig. 3, Table 2 and Supplementary Table 3), it has already been described that TAL1 was significantly correlated with breast cancer survival27.
Figure 3. K-M plots representing TF activities significantly associated to patient survival in all the cancers analysed.

TFs in bold present a significant association with adjusted p-values < 0.05 and TFs in italics have nominal p-values < 0.05.
Table 2. TFs significantly associated to survival.
| Cancer type | Variables (TFs and PURITY) selected by the Cox model | Total | TFs with individual effect on survival (K-M) | Total |
|---|---|---|---|---|
| BLCA | SRF, YY1, CTCFL, POU2F2, ZNF263, USF1, EBF1, THAP1, MYC, MYC::MAX, NR1H3::RXRA, JUN, REST, JUN::FOS, GATA1, PPARG::RXRA, E2F6, ZEB1, BHLHE40, FOS, RXR::RAR_DR5, ECR::USP | 22 | EBF1, MEF2C, PAX5, SRF | 4 |
| BRCA | RXRA, E2F4, USF1, MYC::MAX, MAX, ZBTB33, FOXA1, PPARG::RXRA, RXR::RAR_DR5, TAL1::GATA1, IRF4, SPI1, YY1, JUND, SP2, ZEB1, GATA1, PURITY | 18 | EBF1, FOSL1, GATA2, TAL1::GATA1, ZBTB33 | 5 |
| COAD | PAX5, CTCFL, E2F6, EGR1, THAP1, MYC, HNF4A, NRF1, JUN, JUN::FOS, ZEB1, ZBTB33, FOXA1, TCF12, HNF4G, PBX3, SPI1, JUND, CTCF, E2F4, PURITY | 21 | MYC, E2F6, EBF1, FOXA1, FOXA2, HNF4G, MAX | 7 |
| HNSC | SPI1, ELF1, EGR1, EBF1, RXRA::VDR, BHLHE40, RXR::RAR_DR5, JUN::FOS, GATA2 | 9 | ELF1, JUND, FOS, JUN, MYC, CTCF, CTCFL, ETS1, FOSL1, FOSL2, JUN::FOS, NRF1, RXR::RAR_DR5, SP1, SP2, NR2C2, YY1 | 17 |
| KIRC | RXRA, E2F6, MEF2A, ELF1, POU2F2, USF1, MYC, MYC::MAX, GATA1, MEF2C, HNF4G, RXR::RAR_DR5, FOXA2, CTCFL, FOXA1, TCF12, PURITY | 17 | EGR1, JUN::FOS, JUN, MEF2A, NRF1, SPI1, SRF, USF1, YY1, ZEB1, FOS, MYC, CTCF, CTCFL, E2F4, E2F6, ELF1, ETS1, FOSL1, FOXA2, HNF4G, IRF4, JUNB, JUND, MAX, MYC::MAX, REST, PAX5, PBX3, RXR::RAR_DR5, RXRA::VDR, SP1, SP2, TCF12, NR2C2, ZBTB33, ZNF263 | 38 |
| KIRP | — | 0 | RXRA | 1 |
| LIHC | YY1, JUND, ELF1, USF1, THAP1, HNF4A, MAX, NRF1, ZBTB33, JUNB, MEF2C, HNF4G, TAL1::GATA1, RXRA, SP2, ETS1, NR1H3::RXRA, REST, ECR::USP, IRF4 | 20 | EGR1 | 1 |
| LUAD | PURITY, ELF1, SP2, MAX, REST, FOSL2, GATA1, NR2C2, JUND, E2F6, BHLHE40, JUNB | 12 | JUND, MEF2C, TAL1::GATA1, TCF12 | 4 |
| LUSC | PURITY, E2F4, MEF2A, EGR1, SP1, POU2F2, ETS1, NR1H3::RXRA, NRF1, RXRA::VDR, JUN, JUN::FOS, FOSL1, NR2C2, ELF1, ZEB1, BHLHE40, HNF4G, PBX3 | 19 | FOS, ELF1, NRF1, REST | 4 |
| THCA | — | 0 | EBF1, ECR::USP, HNF4A, PAX5, RXRA, RXRA::VDR | 6 |
| UCEC | PURITY, SRF, E2F6, ELF1, EGR1, SP1, USF1, SP2, MYC::MAX, HNF4A, FOSL2, ZEB1, MEF2C, HNF4G, TAL1::GATA1, ECR::USP, FOXA2, CTCF, E2F4, MAX, JUN::FOS, FOSL1 | 22 | E2F4, E2F6, ELF1, GATA2, HNF4G, PAX5 | 6 |
The first column denoted the cancer type analysed. The second column contains the variables included in the Cox multiple regression model, which can be TFs and tumour purity. The third column contains the total number of TFs included in the Cox model. The fourth column shows TFs that show a significant association to survival by themselves. The fifth column contains the number of TFs significant in the K-M analysis. TFs in bold are significant with an adjusted p-value < 0.05. TFs in grey and in italic are significant with a nominal p-value < 0.05.
Figure 4. K-M plots representing TF activities significantly associated to patient survival.

Survival curves are represented as solid lines and their corresponding confidence intervals as dotted lines. (A) High activity (green curve) of SPI1 in KIRC is significantly associated to patient survival (FDR-adjusted p-value = 2.09 × 10−6); (B) High activity of JUND in HNSC is significantly associated to bad prognostic (FDR-adjusted p-value = 3.42 × 10−5); (C) Low activity of CTCF in KIRC is significantly associated to bad prognostic (FDR-adjusted p-value = 2.81 × 10−4); (D) Low activity of MEF2C in KIRC is significantly associated to bad prognostic (FDR-adjusted p-value = 1.84 × 10−5).
Interestingly, there are TFs whose activity does not change significantly between the cancer and the normal tissue (see Fig. 1), but play, however, an unquestionable role in survival. These are the cases of EBF1 in BRCA, which is a tumour suppressor56 and its lower activity is associated to higher mortality or CTCF in KIRC61 (see Fig. 4C). The case of MEF2A and MEF2C is similar: lower activity is significantly associated to worst prognostic (Fig. 4D), which is supported by the fact that its inhibition by miR-223 promotes the invasion of breast cancer cells45,46. These observations suggest that TFs whose activity is not especially relevant in the cancer tissue are however important in the determination of the prognostic of the patients and might be interesting therapeutic targets.
A complete list of p-values obtained for all the relationships of TF activities with survival in all the cancers studied can be found in Supplementary Table 3.
Combined contribution of TF activity to survival and the impact of tumour purity
Despite the obvious impact of individual TF activities in patient survival, it is clear that such a complex phenotype cannot be the effect of unique TF activities but rather will require of the interplay of several TFs. In order to capture at least part of the complexity of this interplay of TF activities that will ultimately affect patient survival we used a multivariate procedure. Conceptually, increasing levels of TF activity, as reported by TFTEA, accounts for higher expressions of increasingly larger number of targets of the TF. This continuous variable is modelled for multiple TFs with respect to the event of death in the patients by applying Cox multiple regression models and using a stepwise algorithm (see details in Methods).
Recently, the importance that the non-cancerous components of the tumour (that include immune cells, fibroblasts, endothelial cells and normal epithelial cells) may have in cancer biology has been described62. Actually, it has been shown in some circumstances, the presence of these cells may alter the results of genomic analyses, including survival62. In order to check potential alterations in the TF activities inferred from the datasets studied here, we have compared the mean tumour purities with the outcome of the application of the method to see if there was any relationship between the mean purity of the cancer and the potential sensibility of the method in detecting TF activations (measured as the number of significant TF activity changes detected). Supplementary Figure 3 clearly shows that there is no observable trend between both variables, which strongly suggests that the application of the method to the analysed datasets was not significantly affected by the mean cancer purity. However, the fact that TF activity estimations are not affected by tumour purity does not discard a possible confounding effect of the non-cancerous component of the cancer in this measurement. To study this potential confounding effect, the value of tumour purity was introduced in the Cox model as another variable.
The results obtained, listed in Table 2 and summarized in Fig. 5, clearly demonstrate a significant connection between multiple TF activity and patient survival for all the cancers analysed. The influence of TF activity in bad prognostic of the tumour seems to be a complex process in which different TF act cooperatively to activate (or deactivate) a large number of cell programmes that initiate and/or progress distinct cancer hallmarks39 in the tumour cells. The results depict a relevant contribution of tumour purity to patient survival in three out of the eleven cancers analysed (both lung cancers LUAD and LUSC, and the endometrial carcinoma, UCEC). Although non-significant, tumour purity is still selected by the Cox model in another three cancers (BRCA, COAD and KIRK), where probably plays a more marginal role.
Figure 5. Combinations of TFs significantly associated to patient survival in the different cancers when a Cox model is applied.
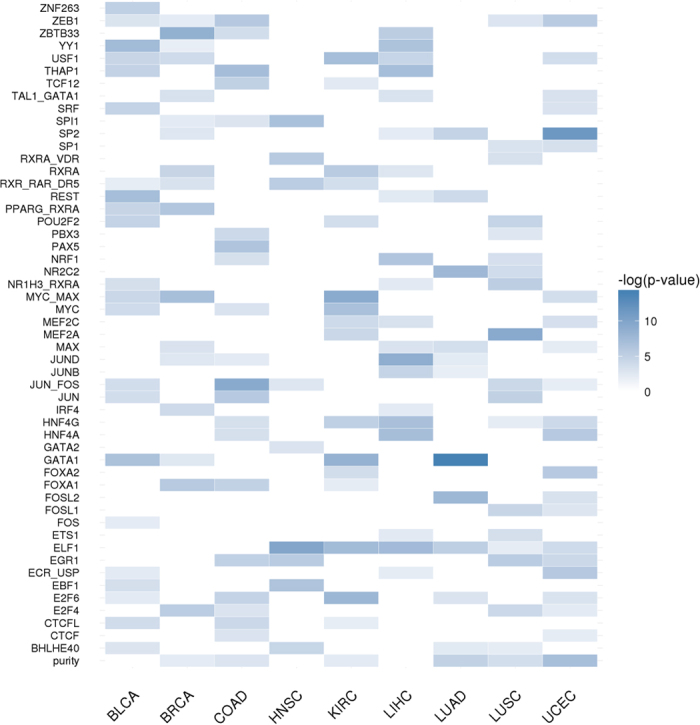
Cancers are represented in columns and TFs in rows. For each cancer, several TFs and sometimes tumour purity were included in a cox model. The colour intensity is related to the significance of this association (p-value).
Potential limitations of the method
It must be taken into account that the information on TFBSs might contain a non-negligible number of false positives along with the true TFBSs in the real TF targets. This reduces the power of detection of the method given that, if a TF is activating their real targets, and a number of genes with random activity are considered to be part of the gene set of the TF, the complete gene set will show an activity lower than the actual activity. In addition, only a relatively low number of TFs are well characterized in terms of target genes they activate.
Another potential problem that can reduce the sensitivity of the analysis is the fact that many TFs need of a combination of factors to properly carry out transcription.
Finally, only KIRC (and to a lesser extent HNSC) had enough data on deceased patients to carry out robust survival analysis. Supplementary Figure 4 clearly depicts this trend. The survival is best detected in KIRC and HNSC because a higher number of deceased patients is present in the dataset analysed (which also explains the high values of these cancers, unrelated with cancer purity, observed in Supplementary Figure 3).
In spite of these problems that reduce the potential of discovery of the proposed methodology in the current datasets, we have discovered a reasonable amount of significant associations of TF activity with cancer progression and with survival. Despite limited, the results obtained, which can be considered the “tip of the iceberg”, and are quite encouraging.
Conclusions
The availability of survival and other relevant clinical data makes the analysis of pan-cancer Big Data repositories (ICGC and others) especially compelling, given that new unexpected associations of genomic data to relevant clinical outcomes can be found. Despite the relevance of regulation in cancer this seems to be the first pan-cancer analysis carried out to date. We have applied the TFTEA, a simple but robust methodology, to detect significant changes in the TF activity status when two groups of individuals are compared. In addition, the methodology also provides TF activity values per individual. This interesting property allows detecting TF-mediated deregulations specific for individuals, thus opening the door to possible personalized therapeutic interventions.
Regardless of the expectable reduction in the detection power that the current definitions of TF target gene sets could produce in methods that rely on this knowledge, the TFTEA still discovered a considerable number of significant associations between TF activity and the acquisition of cancer, the progression of cancer across stages or the survival of patients. Actually, many of the altered activities in TFs found were described in the literature either directly as causal alterations or, at least, linked to cancer, providing an extra support to the validity of the proposed methodology. Moreover, statistical modelling allowed detecting an important role of tumour purity in survival. This suggests that, in some cases, the TF activity related to survival detected by the test could be due in part to other non-cancerous components of the tumour (probably immune cells, but also fibroblasts, endothelial cells and normal epithelial cells).
Actually, the findings of this work constitute most probably an underestimation of the total number of TFs linked to bad prognostic, due to the lack of enough survival data among the samples that precluded obtaining significant results for more TFs. This suggests that more detailed results would be obtained by the application of TFTEA to patient cohorts with richer clinical annotations.
Methods
Cancer samples used
Eleven cancer types amounting to 5607 samples (Table 1) were selected on the basis of the simultaneous availability of paired samples (transcriptome analysis from both tumour sample and adjacent healthy tissue) and clinical data (tumour stage and survival). Raw read count data files were downloaded from the ICGC data portal63 and clinical data were downloaded from the TCGA data portal64 using sample IDs to cross-reference patient’s data.
Gene expression data processing
The trimmed mean of M-values normalization (TMM)65 was the method of choice and was applied using the edgeR package66, using the default parameters. The differential expression analysis between cases and controls was carried out using the limma package67,68. Firstly, the voom function69 is applied to weight and transform TMM normalized values to make them suitable for lineal model analysis. Then, the lmFit function is used to adjust a lineal model and an empirical Bayes method is used to estimate differential expression values.
The Human Protein Atlas58,70 was used as a reference for the gene expression levels of TFs in normal tissues.
Transcription factors used in the study
We have used a total of 52 TF available in ENSEMBL (GRCh38.p3), which are: RXRA, SRF, SPI1, YY1, PAX5, JUND, CTCF, CTCFL, E2F4, E2F6, MEF2A, ELF1, EGR1, SP1, POU2F2, ZNF263, USF1, SP2, ETS1, EBF1, THAP1, MYC, MYC::MAX, HNF4A, NR1H3::RXRA, MAX, NRF1, RXRA::VDR, JUN, REST, FOSL2, JUN::FOS, ZEB1, ZBTB33, GATA2, GATA1, BHLHE40, FOXA1, JUNB, FOS, FOSL1, NR2C2, TCF12, MEF2C, HNF4G, PPARG::RXRA, RXR::RAR_DR5, TAL1::GATA1, PBX3, ECR::USP, IRF4 and FOXA2.
Any of these TFs activates a set of genes. Here we consider that a gene can potentially be activated by a TF if it includes possible binding sites for it, located between 5000 bp upstream from the most external transcription origin and the first exon. TFBSs have been mapped by Ensembl71,72 along the genome. Briefly, for any TF which has both a ChIP-seq data and a JASPAR73 publicly available position weight matrix (PWM), Ensembl annotates the position of putative TFBSs within the ChIP-seq peaks (details can be found in specific Ensembl web pages74). This information is accessible in a more efficient way in different publicly available resources, such as CellBase75, whose web services76 were used here. Supplementary Table 4 shows the list of target genes for each TF.
Estimation of significant transcription factor activities in a cancer datasets
Since direct inference of TF activity from its own gene expression level is problematic, in this work we indirectly infer its activity from the collective activity of their gene targets. The method used here is an analysis of Gene Set Enrichment (GSE) that we call TF Target Enrichment Analysis (TFTEA). In this approach, each TF has an associated gene sets composed by the all their target genes (those containing a TFBS for the TF located between 5000 bp upstream from the transcription origin and the first exon of the gene).
Like other GSE methods, the TFTEA algorithm detects asymmetrical distributions of targets of TFs in the top (or the bottom) of a list of ranked genes. When two conditions are compared and the genes are ranked by differential expression (or fold change or any other related parameter), the detection of a significant accumulation of targets of a given TF in the upper (or lower) part of the ranked list indicates that such TF has significantly increased (or decreased) its activity in one of the conditions with respect to the other one. Here, differential expression in calculated by means of a limma test67, and the results of the statistic are used to define the ranked list of genes. A logistic regression is the most efficient methodology used for the detection of gene sets with a significant systematic over- or under-expressed77,78. Specifically, the association of a gene set composed by the targets of a specific TF to high or low values of the ranked list of genes is tested by means of the value of the slope of the logistic regression. The null hypothesis, slope = 0, is tested against the alternative slope ≠ 0 based on the maximum likelihood parameter estimates and the Wald test. For testing slope = 0, the Wald statistic can be shown to follow a chi-square distribution with one degree of freedom and the p-value is calculated assuming this null distribution77,78.
Since many TFs were tested with the logistic regression across the eleven cancers, multiple testing effects need to be corrected- Here we have used the popular FDR method79 for this purpose. Figure 6 schematizes the application of the method.
Figure 6. Schema of the TFTEA method to obtain TFs differentially activated between two conditions compared.

The method uses gene expression values and compares two conditions (A and B) by means of any test to obtain a rank of differentially expressed genes (Rank DE) based on the statistic. Then, for each TF, a logistic regression78 is applied to discover associations of the TF targets to high or low values of the rank (lower panel). Thus, targets of TF1 show a clear association to high values of the statistic, meaning that have significantly higher expression in condition (A) than in condition ( ), which demonstrated the differential activity of TF1. TF2 is the opposite case, in which the TF is significantly less active in (B) than in (A). TF3 have their targets active or inactive in both conditions, meaning that these activities are not a collective property and consequently are not due to TF3, but maybe to other regulators.
Estimation of personalized transcription factor activities per individual
We also used TFTEA to relate individual survival events to TF activity. Since this method requires of a ranked list of genes, each normalized patient sample needs to be compared to a reference. This reference value was obtained as the average normalized expression value across all the normal samples (see Fig. 7). For each gene (g) of any cancer sample (c), its expression value (vcg) is compared with the corresponding average expression value for this gene in all the healthy samples (mg) and the resulting value is divided by the standard deviation of the gene expression value in the healthy samples. This comparison provides for each cancer sample a value per gene that can be interpreted as a fold change (Fcg) with respect to the average healthy expression:
Figure 7. Schema of the TFTEA method to obtain personalized values of survival.

The method uses gene expression values and compares two conditions (A and B). However, in this case all the samples in the (B) condition are used to produce an average expression value for any of the genes in this condition (m vector). Then, each sample in the (A) condition (vc) can be compared to the average expression in the (B) condition and a rank of fold change (Fcg) is generated for each sample. Then, this ranking is used in the same way that the rank of differential expression was used in Fig. 6 to find differentially activated TFs in samples from the condition (A) with respect to the average (B) condition. Is samples are paired the Fcg value can be derived from the direct comparison between them.
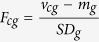 |
Fcg values can thus be used to rank genes in a unique individual according to its relative expression with respect to the average expression values of their counterparts in a normal tissue.
Once a list of genes ranked by decreasing Fcg values is obtained for each patient, the TFTEA can be applied in a personalized manner to detect those TFs significantly activated (or deactivated) in each particular individual.
If samples are paired, the Fcg rank can be generated by direct comparison or each pair.
Correlation between transcription factor activity and patient survival
Kaplan-Meier (K-M) curves59 are used to relate TF activity to survival in the different cancers. The value of the statistic of each TF in each individual was used as a proxy of its activity. Only FT-cancer pairs with 10 events (deaths) or more were taken into account and the multiple testing adjustments were made taking into account only the pairs analysed. Calculations were carried out using the function survdiff from the survival R package80.
Cox regression analysis81 is used to relate combined TF activity to survival in the different cancers. Since tumour purity has been involved in survival62, we used individual tumour purity values as an extra variable in the cox regression. Calculations were carried out using the function coxph from the survival R package80. A stepwise algorithm, implemented in the step function from the R package stats82,83, is used to add or remove TFs or the tumour purity value according to the significance of their contributions to explain survival in the multiple regression model. In this way a final list of variables (TFs and cancer purity) whose combination is significantly related to survival is obtained. The step package uses Akaike Information Criterion (AIC) to select the best model by iteratively adding and removing variables.
Tumour purity estimation
There are different approaches to estimate tumour purity values, such as ESTIMATE, based on gene expression profiles of known immune stromal genes84; ABSOLUTE, based on somatic copy-number data85; LUMP (leukocytes unmethylation for purity), based on averages of non-methylated immune-specific CpG sites62.
Consensus measurement of purity estimations (CPE) is the median purity level after normalizing levels from all methods to give them equal means and s.d.’s (75.3 ± 18.9%).
Here, the per individual purity values provided in Supplementary Data 1 in the Aran’s paper62 are used to study the contribution of tumour purity to survival in the Cox regression.
Code availability
The code is open and available at: https://github.com/babelomics/TFTEA.
Additional Information
How to cite this article: Falco, M. M. et al. The pan-cancer pathological regulatory landscape. Sci. Rep. 6, 39709; doi: 10.1038/srep39709 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work is supported by grants BIO2014-57291-R from the Spanish Ministry of Economy and Competitiveness (MINECO), Plataforma de Recursos Biomoleculares y Bioinformáticos PT13/0001/0007 from the ISCIII, both co-funded with European Regional Development Funds (ERDF), PROMETEOII/2014/025 from the Conselleria de Educacio of the Valencian Community, FP7-PEOPLE-2012-ITN MLPM2012 318861 and H2020-INFRADEV-1-2015-1 ELIXIR-EXCELERATE (ref. 676559) from the EU FP7; and Fundació la Marató TV3 [20133134].
Footnotes
Author Contributions M.M.F. has carried out the analysis; J.C.C. has helped with analysis and supervised the development of the work; M.B. developed the original algorithm; J.D. conceived the work and wrote the paper.
References
- Hobert O. Gene regulation by transcription factors and microRNAs. Science 319, 1785–1786, doi: 10.1126/science.1151651 (2008). [DOI] [PubMed] [Google Scholar]
- Furney S. J., Higgins D. G., Ouzounis C. A. & Lopez-Bigas N. Structural and functional properties of genes involved in human cancer. BMC Genomics 7, 3, doi: 10.1186/1471-2164-7-3 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. I. & Young R. A. Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancafort P. et al. Genetic reprogramming of tumor cells by zinc finger transcription factors. Proceedings of the National Academy of Sciences of the United States of America 102, 11716–11721 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura C. et al. Frequent downregulation of the runt domain transcription factors RUNX1, RUNX3 and their cofactor CBFB in gastric cancer. International journal of cancer 113, 221–228 (2005). [DOI] [PubMed] [Google Scholar]
- Gilliland D. G. The Diverse Role of the ETS Family of Transcription Factors in Cancer Commentary re: B. Davidson, Ets-1 Messenger RNA Expression Is a Novel Marker of Poor Survival in Ovarian Carcinoma. Clin. Cancer Res., 7: 551–557, 2001. Clinical Cancer Research 7, 451–453 (2001). [PubMed] [Google Scholar]
- Strano S. et al. Mutant p53: an oncogenic transcription factor. Oncogene 26, 2212–2219 (2007). [DOI] [PubMed] [Google Scholar]
- Introna M. & Golay J. How can oncogenic transcription factors cause cancer: a critical review of the myb story. Leukemia (08876924) 13 (1999). [DOI] [PubMed] [Google Scholar]
- Darnell J. E. Transcription factors as targets for cancer therapy. Nature Reviews Cancer 2, 740–749 (2002). [DOI] [PubMed] [Google Scholar]
- Dawson M. A. & Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012). [DOI] [PubMed] [Google Scholar]
- Jang I. S., Margolin A. & Califano A. hARACNe: improving the accuracy of regulatory model reverse engineering via higher-order data processing inequality tests. Interface focus 3, 20130011, doi: 10.1098/rsfs.2013.0011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin A. A. et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7 Suppl 1, S7, doi: 10.1186/1471-2105-7-S1-S7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Foat B. C. & Bussemaker H. J. Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data. BMC Bioinformatics 5, 1 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith J. J. et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol 5, e8, doi: 10.1371/journal.pbio.0050008 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal M., Belcastro V., Ambesi-Impiombato A. & Di Bernardo D. How to infer gene networks from expression profiles. Molecular systems biology 3, 78 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roven C. & Bussemaker H. J. REDUCE: An online tool for inferring cis-regulatory elements and transcriptional module activities from microarray data. Nucleic acids research 31, 3487–3490 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pournara I. & Wernisch L. Factor analysis for gene regulatory networks and transcription factor activity profiles. BMC Bioinformatics 8, 61 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J. H. et al. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 296, 1886–1889, doi: 10.1126/science.1073440 (2002). [DOI] [PubMed] [Google Scholar]
- Harris M. L., Baxter L. L., Loftus S. K. & Pavan W. J. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res 23, 496–513, doi: 10.1111/j.1755-148X.2010.00711.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filtz T. M., Vogel W. K. & Leid M. Regulation of transcription factor activity by interconnected post-translational modifications. Trends in pharmacological sciences 35, 76–85, doi: 10.1016/j.tips.2013.11.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tootle T. L. & Rebay I. Post-translational modifications influence transcription factor activity: a view from the ETS superfamily. Bioessays 27, 285–298, doi: 10.1002/bies.20198 (2005). [DOI] [PubMed] [Google Scholar]
- Cheng C., Yan X., Sun F. & Li L. M. Inferring activity changes of transcription factors by binding association with sorted expression profiles. BMC Bioinformatics 8, 1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleda M. et al. Inferring the regulatory network behind a gene expression experiment. Nucleic Acids Res 40, W168–172, doi: 10.1093/nar/gks573 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., Liu C.-C. & Cheng C. REACTIN: regulatory activity inference of transcription factors underlying human diseases with application to breast cancer. BMC Genomics 14, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P., Freedman M. L., Liu J. S. & Liu X. S. Inference of transcriptional regulation in cancers. Proceedings of the National Academy of Sciences 112, 7731–7736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacht T., Oswald M., Eils R., Eichmuller S. B. & Konig R. Estimating the activity of transcription factors by the effect on their target genes. Bioinformatics 30, i401–407, doi: 10.1093/bioinformatics/btu446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Li L. M., Alves P. & Gerstein M. Systematic identification of transcription factors associated with patient survival in cancers. BMC Genomics 10, 1 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Su P.-F., Zhao S. & Shyr Y. Transcriptome-wide signatures of tumor stage in kidney renal clear cell carcinoma: connecting copy number variation, methylation and transcription factor activity. Genome medicine 6, 1–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- The COSMIC database, http://cancer.sanger.ac.uk/cosmic (2015).
- Gabay M., Li Y. & Felsher D. W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor perspectives in medicine 4, doi: 10.1101/cshperspect.a014241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E., Lin C. Y., Eilers M. & Levens D. L. Taming of the beast: shaping Myc-dependent amplification. Trends Cell Biol 25, 241–248, doi: 10.1016/j.tcb.2014.10.006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W., Halasz M., Granovskaya M. & Kholodenko B. N. The dynamic control of signal transduction networks in cancer cells. Nat Rev Cancer 15, 515–527, doi: 10.1038/nrc3983 (2015). [DOI] [PubMed] [Google Scholar]
- Amati B. & Land H. Myc—Max—Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Current opinion in genetics & development 4, 102–108 (1994). [DOI] [PubMed] [Google Scholar]
- Nevins J. R. The Rb/E2F pathway and cancer. Hum Mol Genet 10, 699–703 (2001). [DOI] [PubMed] [Google Scholar]
- Chen H.-Z., Tsai S.-Y. & Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nature Reviews Cancer 9, 785–797 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaleel S. S., Andrews E. H., Ung M., DiRenzo J. & Cheng C. E2F4 regulatory program predicts patient survival prognosis in breast cancer. Breast Cancer Res 16, 486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui G. The regulation of YY1 in tumorigenesis and its targeting potential in cancer therapy. Molecular and Cellular Pharmacology 1, 157–176 (2009). [Google Scholar]
- Archer M. C. Role of sp transcription factors in the regulation of cancer cell metabolism. Genes & cancer 2, 712–719 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- Vizcaíno C., Mansilla S. & Portugal J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacology & therapeutics 152, 111–124 (2015). [DOI] [PubMed] [Google Scholar]
- Wang L. et al. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clinical Cancer Research 9, 6371–6380 (2003). [PubMed] [Google Scholar]
- Van Dam H. & Castellazzi M. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene 20 (2001). [DOI] [PubMed] [Google Scholar]
- Rimmelé P. et al. Spi-1/PU. 1 oncogene accelerates DNA replication fork elongation and promotes genetic instability in the absence of DNA breakage. Cancer research 70, 6757–6766 (2010). [DOI] [PubMed] [Google Scholar]
- Laddha S. V. et al. Genome-wide analysis reveals downregulation of miR-379/miR-656 cluster in human cancers. Biology direct 8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnnidis J. B. et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451, 1125–1129 (2008). [DOI] [PubMed] [Google Scholar]
- Yang M. et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Molecular cancer 10, 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman A. M. How much REST is enough? Cancer Cell 13, 381–383 (2008). [DOI] [PubMed] [Google Scholar]
- Hsu J., Bravo R. & Taub R. Interactions among LRF-1, JunB, c-Jun, and c-Fos define a regulatory program in the G1 phase of liver regeneration. Molecular and cellular biology 12, 4654–4665 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathas S. et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-κB. The EMBO journal 21, 4104–4113 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X. et al. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 101, 1513–1519 (2003). [DOI] [PubMed] [Google Scholar]
- Barrett J. C. et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nature genetics 41, 1330–1334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappa K., Robertson G. R. & Sladek F. M. HNF4α: a new biomarker in colon cancer? Biomarkers in medicine 6, 297–300 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altucci L., Leibowitz M. D., Ogilvie K. M., De Lera A. R. & Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nature Reviews Drug Discovery 6, 793–810 (2007). [DOI] [PubMed] [Google Scholar]
- Altucci L. & Gronemeyer H. The promise of retinoids to fight against cancer. Nature Reviews Cancer 1, 181–193 (2001). [DOI] [PubMed] [Google Scholar]
- Filippova G. N. Genetics and epigenetics of the multifunctional protein CTCF. Current topics in developmental biology 80, 337–360 (2007). [DOI] [PubMed] [Google Scholar]
- Liao D. Emerging roles of the EBF family of transcription factors in tumor suppression. Molecular Cancer Research 7, 1893–1901 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badve S. et al. FOXA1 expression in breast cancer—correlation with luminal subtype A and survival. Clinical Cancer Research 13, 4415–4421 (2007). [DOI] [PubMed] [Google Scholar]
- Uhlen M. et al. Tissue-based map of the human proteome. Science 347, 394, doi: 10.1126/science.1260419 (2015). [DOI] [PubMed] [Google Scholar]
- Kaplan E. & Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association 53, 457–481, doi: 10.1080/01621459.1958.10501452 (1958). [DOI] [Google Scholar]
- Lamb J. A., Ventura J.-J., Hess P., Flavell R. A. & Davis R. J. JunD mediates survival signaling by the JNK signal transduction pathway. Molecular cell 11, 1479–1489 (2003). [DOI] [PubMed] [Google Scholar]
- Rasko J. E. et al. Cell growth inhibition by the multifunctional multivalent zinc-finger factor CTCF. Cancer research 61, 6002–6007 (2001). [PubMed] [Google Scholar]
- Aran D., Sirota M. & Butte A. J. Systematic pan-cancer analysis of tumour purity. Nature communications 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICGC Data Portal, https://dcc.icgc.org/ (2015).
- TCGA Data Portal, https://tcga-data.nci.nih.gov/tcga/ (2015).
- Robinson M. D. & Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25, doi: 10.1186/gb-2010-11-3-r25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res In press (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. Linear Models for Microarray Data, https://bioconductor.org/packages/release/bioc/html/limma.html (2015).
- Law C. W., Chen Y., Shi W. & Smyth G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29, doi: 10.1186/gb-2014-15-2-r29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Protein Atlas http://www.proteinatlas.org/ (2015).
- Ensembl. Datasets and Data Processing, regulation sources, http://www.ensembl.org/info/genome/funcgen/regulation_sources.html (2015).
- Cunningham F. et al. Ensembl 2015. Nucleic Acids Res 43, D662–669, doi: 10.1093/nar/gku1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne J. C. et al. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res 36, D102–106, doi: 10.1093/nar/gkm955 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensembl regulatory elements, http://www.ensembl.org/info/genome/funcgen/regulatory_build.html#tfbs (2016).
- Bleda M. et al. CellBase, a comprehensive collection of RESTful web services for retrieving relevant biological information from heterogeneous sources. Nucleic Acids Res 40, W609–614, doi: 10.1093/nar/gks575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina I. The CellBase database, http://wwwdev.ebi.ac.uk/cellbase/webservices/ (2015).
- Sartor M. A., Leikauf G. D. & Medvedovic M. LRpath: A logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics 25, 211–217 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner D. & Dopazo J. Multidimensional gene set analysis of genomic data. PLoS ONE 5, e10348, doi: 10.1371/journal.pone.0010348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. & Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B 57, 289–300 (1995). [Google Scholar]
- Therneau T. Survival Analysis, https://cran.r-project.org/web/packages/survival/ (2015).
- Cox D. Regression models and life-tables. Journal of the Royal Statistical Society. Series B (Methodological) 34, 187–220 (1972). [Google Scholar]
- Ripley B. Choose a model by AIC in a Stepwise Algorithm, https://stat.ethz.ch/R-manual/R-devel/library/stats/html/step.html (2015).
- Venables W. & Ripley B. Modern Applied Statistics with S. (Springer, 2002). [Google Scholar]
- Yoshihara K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature communications 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.