

Abstract
The critical immortalizing activity of the human papillomavirus (HPV) type-16 E6 oncoprotein is to induce expression of hTERT, the catalytic and rate-limiting subunit of telomerase. Additionally, E6 binds to a cellular protein called E6-associated protein (E6-AP) to form an E3 ubiquitin ligase that targets p53 for proteasome-dependent degradation. Although telomerase induction and p53 degradation are separable and distinct functions of E6, binding of E6 to E6-AP strongly correlated with the induction of hTERT. Here, we demonstrate using shRNAs to reduce E6-AP expression that E6-AP is required for E6-mediated telomerase induction. A yeast two-hybrid screen to find new targets of the E6/E6-AP E3 ubiquitin ligase complex identified NFX1. Two isoforms of NFX1 were found: NFX1-123, which coactivated with c-Myc at the hTERT promoter, and NFX1-91, which repressed the hTERT promoter. NFX1-91 was highly ubiquitinated and destabilized in epithelial cells expressing E6. Furthermore, knockdown of NFX1-91 by shRNA resulted in derepression of the endogenous hTERT promoter and elevated levels of telomerase activity. We propose that the induction of telomerase by the HPV-16 E6/E6-AP complex involves targeting of NFX1-91, a newly identified repressor of telomerase, for ubiquitination and degradation.
Keywords: Telomerase, HPV, transcriptional repressor, ubiquitin, E6, E6-AP
Immortalization is a critical step in the process of transformation (Hahn et al. 1999a; Elenbaas et al. 2001). Several recent investigations have determined that the combined effects of disruption of the Rb/p16 pathway and induction of telomerase activity are sufficient to immortalize human epithelial cells (Kiyono et al. 1998; Garbe et al. 1999; Lundberg et al. 2002). The human papillomavirus (HPV) type-16 oncoproteins, E6 and E7, target these pathways to efficiently immortalize primary epithelial cells (Kiyono et al. 1998; Fehrmann and Laimins 2003). Although the E7 oncoprotein abrogates the Rb/p16 pathway by disrupting the Rb/E2F interaction and targeting Rb for degradation (Boyer et al. 1996; Helt and Galloway 2001), the E6 oncoprotein promotes the degradation of p53 through its interaction with the cellular E6-associated protein, E6-AP, an E3 ubiquitin ligase (Scheffner et al. 1993). In addition, E6 induces telomerase activity, thereby contributing to the immortalization of epithelial cells by maintaining telomere length (Klingelhutz et al. 1996). Studies of mutated E6 proteins reveal that induction of telomerase is an independent function of E6 separable from p53 degradation and that telomerase activation rather than p53 degradation is the critical step for immortalization of epithelial cells (Klingelhutz et al. 1996; Kiyono et al. 1998).
Several groups, including ours, have found that activation of telomerase by E6 is due to its ability to induce expression of hTERT, the catalytic and rate-limiting subunit of telomerase (Gewin and Galloway 2001; Oh et al. 2001; Veldman et al. 2001). Given that the majority of tumor cells have induced expression of hTERT, the mechanisms regulating the hTERT promoter are the subject of extensive investigation. Although the hTERT promoter region lies within a CpG island, promoter methylation does not appear to be the primary mode of silencing hTERT expression in telomerase negative cells (Devereux et al. 1999; Dessain et al. 2000). Chromatin conformation, on the other hand, does seem to play a key role in hTERT activity. Several studies have demonstrated that inhibition of histone deacetylases by trichostatin A in certain telomerase-negative cells can alter the chromatin structure and induce telomerase expression (Cong and Bacchetti 2000; Takakura et al. 2001; Hou et al. 2002). Interestingly, induction of telomerase by E6 also seems to involve an epigenetic mechanism that regulates the extent of hTERT induction. Different clones of human foreskin keratinocytes (HFKs) transduced with E6 express varying levels of hTERT that increase as cells are passaged in culture despite no accompanying increase in E6 expression levels (Klingelhutz et al. 1996; Kiyono et al. 1998; Baege et al. 2002).
The hTERT promoter contains many known transcription factor-binding sites, including several E boxes and Sp1 sites (Horikawa et al. 1999; Takakura et al. 1999; Wick et al. 1999). Although c-Myc is able to bind the E boxes in the hTERT promoter and can induce hTERT expression in many cell types (Wang et al. 1998; Greenberg et al. 1999; Wu et al. 1999), we and others have found no correlation between c-Myc expression and the ability of E6 to induce hTERT (Gewin and Galloway 2001; Oh et al. 2001; Veldman et al. 2001). We have, however, determined that the promoter region including the proximal E box is important for E6-mediated hTERT induction (Gewin and Galloway 2001; Veldman et al. 2001).
In an effort to elucidate the mechanism by which E6 induces hTERT, we began to characterize the ability of E6 and mutated versions of E6 to induce hTERT, to target p53 for degradation, and to bind to E6-AP. A strong correlation between E6-AP binding and hTERT induction prompted the search for possible new targets of the E6/E6-AP complex by a yeast two-hybrid screen. In addition to several known targets of E6/E6-AP, the screen identified a transcriptional repressor known as NFX1. This work describes both a requirement for E6-AP and a role for NFX1 in E6-mediated hTERT induction.
Results
E6-AP is required for telomerase activation of HPV-16 E6
The initial discovery that E6 induces expression of hTERT immediately prompted speculation that this function might be linked to the well-established p53 degradation function of E6. Although overexpression of p53 has been shown to repress hTERT expression in some tumor cell lines (Kanaya et al. 2000; Xu et al. 2000), inactivation of p53 is insufficient to induce telomerase (Hahn et al. 1999a; Opitz et al. 2001). Furthermore, hTERT induction and p53 degradation are separable and distinct functions of E6 as indicated by E6 mutants such as F2V, 8S/9A/10T, and Y54H that can activate telomerase but do not target p53 for degradation (Table 1; Klingelhutz et al. 1996; Liu et al. 1999). Yet, review of several published studies characterizing mutants of E6 revealed that mutants that retain the ability to target p53 for degradation could also induce hTERT expression (Table 1; Foster et al. 1994; Dalal et al. 1996; Zimmermann et al. 1999). As the E6/E6-AP E3 ubiquitin ligase complex targets p53 for degradation, binding of E6 to E6-AP may also be required for hTERT induction. Additional in vitro E6/E6-AP binding studies (data not shown), as well as published data (Table 1), revealed a strong correlation between E6 binding to E6-AP and telomerase activation. All of the mutated E6 proteins that could bind E6-AP induced hTERT expression or immortalized post-M0-mammary epithelial cells.
Table 1.
Telomerase activation and E6-AP binding of E6 proteins
| HPV 16E6 protein | TRAP activity | E6-AP binding | p53 degradation | References |
|---|---|---|---|---|
| 16E6 wt | + | + | + | Klingelhutz et al. 1996; Liu et al. 1999 |
| 16E6 F2V | + | + | − | Liu et al. 1999; data not shown |
| 16E6 8S/9A/10T | + | + | − | Foster et al. 1994; Klingelhutz et al. 1996; Gewin and Galloway 2001 |
| 16E6Δ9-13 | − | − | − | Foster et al. 1994; Gewin and Galloway 2001 |
| 16E6 L50G | − | − | − | Zimmermann et al. 1999; data not shown |
| 16E6 Y54H | +a | + | − | Liu et al. 1999 |
| 16E6 Δ118-122b | −/+a | −/+ | −/+ | Foster et al. 1994; Dalal et al. 1996; Kiyono et al. 1998 |
| 16E6 Δ123-127 | − | − | − | Foster et al. 1994; Klingelhutz et al. 1996; Liu et al. 1999 |
| 16E6 Δ146-151 | + | NT | + | Foster et al. 1994; Klingelhutz et al. 1996; Kiyono et al. 1998 |
(NT) Not tested.
TRAP activity is inferred by the ability of these mutants to immmortalize mammary epithelial cells (MECs).
16E6 Δ118-122 appears to have low-level activity in all of these functions.
To further support the hypothesis that E6-AP binding is required for E6 to induce hTERT expression, we developed short-hairpin RNA (shRNA) retroviral constructs to reduce E6-AP expression in primary HFKs. These experiments were performed using constructs to stably express the shRNAs driven by an RNA polymerase III promoter, either U6 or H1 (Paddison et al. 2002; Grandori et al. 2003; Smith et al. 2003). Three different 26- to 29-nt regions of the E6-AP gene were targeted to identify a construct that would give the greatest decrease in E6-AP expression. Figure 1A shows shRNA1 (esh1) was the most effective of the three, reducing expression of E6-AP RNA to ∼29% of that in the vector-control infected cells. The other two hairpins (esh2 and esh3) exhibited intermediate levels of effectiveness.
Figure 1.

E6-AP expression is required for telomerase induction by HPV 16E6. (A) Northern blot. Three shRNA constructs targeting E6-AP (esh1,2,3) were transduced into HFKs and RNA was harvested to examine levels of E6-AP message. 36B4 is a loading control. Relative expression levels are presented normalized to 36B4 and the pB empty vector control. (B) Western blot. HFKs expressing the E6-AP shRNA constructs were subsequently transduced with LXSN empty vector (-) or LXSN-16E6 (+). p53 levels were examined by Western blot. Actin is a loading control. (C) TRAP assay. Extracts from the same cells shown in B were assayed for telomerase activity. HeLa cells are a positive control lysate. CHAPs is a lysis buffer negative control. TSR8 is a synthetic template of eight telomeric repeats used as a PCR positive control. (D) RT-PCR. Expression of hTERT RNA was examined by RT-PCR of RNA extracts. 36B4 is a loading control.
HFK cell lines stably expressing the E6-AP shRNAs were subsequently infected with empty vector or E6 retroviral constructs. Figure 1B demonstrates that knockdown of E6-AP abrogated the ability of E6 to target p53 for degradation, with esh1 again proving to be the most effective of the three shRNA-expressing cell lines. In addition, assays for telomerase activation revealed that E6-AP depletion disrupted E6-mediated hTERT induction in HFKs (Fig. 1C). TRAP activity in these cells directly correlated with the level of hTERT RNA detected by RT-PCR (Fig. 1D). These experiments demonstrated that E6-AP expression was required for hTERT induction by HPV-16 E6.
A new target of E6/E6-AP
Our studies revealed that E6 must bind to E6-AP but not necessarily to p53 in order to induce hTERT. A survey of previously identified cellular targets of E6/E6-AP did not reveal any logical contenders for hTERT transcriptional regulators; therefore, we began to search for new targets of E6/E6-AP by a yeast two-hybrid screen. The bait construct consisted of a catalytically defective E6-AP (C833A) fused to the Gal4 DNA-binding domain. To ensure equal dosage of both E6 and E6-AP, the plasmid also encoded the E6 oncoprotein. In a screen of both fetal brain and HeLa cell cDNA libraries, several known E6/E6-AP interactors were identified, including p53, HHR23A (human homolog of rad23), and a homolog of the E6TP1 gene (Unigene Hs. 406879; Scheffner et al. 1993; Gao et al. 1999; Kumar et al. 1999). In addition, a new E6/E6-AP target protein identified in the screen was NFX1 (nuclear factor binds to the X1 box), a transcriptional repressor of MHC class II genes (Song et al. 1994). NFX1 was originally described and cloned in a screen for proteins that bound to the X-box region of MHC class II genes and is thought to be involved in a feedback loop to limit the immune response following infection (Song et al. 1994).
Recent advancements in the annotation of the human genome revealed that NFX1 has two splice variants encoding isoforms with identical N termini and variant C termini (Fig. 2A, Unigene Hs. 413074). We will specify the longer 1120-amino acid isoform here as NFX1-123, as it is ∼123 kDa, and the shorter 833-amino acid isoform as NFX1-91 (∼91 kDa). Both isoforms have a RING finger/PHD finger domain; found in many E3 ubiquitin ligases, this domain has been shown to confer autoubiquitination activity to NFX1 in in vitro assays (Lorick et al. 1999). The RING finger domain is followed by several cysteine/histidine-rich sequences identified as NFX1-type zinc fingers by the Pfam database (http://www.sanger.ac.uk/cgi-bin/Pfam/getacc?PF01422). Identified as the DNA-binding domain of NFX1 (Song et al. 1994), these zinc fingers [C-X(1-6)-H-X-C-X3-C-(H/C)-X(3-4)(H/C)-X(1-10)-C] are highly homologous to one another but do not exhibit the typical spacing of traditional zinc fingers. NFX1-91 contains the first six zinc fingers followed by a unique lysine-rich stretch of 25 amino acids, whereas NFX1-123 contains eight zinc fingers as well as a region known as an R3H domain believed to be involved in single-stranded nucleic acid binding (Fig. 2A). The NFX1 gene is highly conserved among eukaryotic species, though little is known about the functions of its homologs.
Figure 2.

NFX1 isoforms have differential repressive and coactivating functions in hTERT reporter assays. (A) Schematic of NFX1 isoforms. The black box represents a PHD/RING finger domain. Multiple hatched boxes indicate NFX1-type zinc-finger domains. The dotted box represents an R3H domain present only in NFX1-123. The cross-hatched box indicates the unique lysine-rich C-terminal domain of NFX1-91. (B) Schematic of hTERT promoter and the regions included in the luciferase reporter constructs. Several potential SP1-binding sites and E boxes are indicated. Potential NFX1-binding sites are indicated by Xs. (C) hTERT reporter assay using the 710 hTERT construct in transient transfections in HFKS. The indicated genes were cotransfected with CMV, CMV-NFX1-123, or CMV-NFX1-91. The experiment was done in triplicate. (D) hTERT reporter assay with two different regions of the promoter in attempt to map the activity of the two NFX1 isoforms. The indicated DNAs were cotransfected with either the 219 hTERT or 710 hTERT reporter construct in HFKs. The data are a representative experiment of three trials done in duplicate.
Differential repressor and activator functions of the NFX1 isoforms on the hTERT promoter
Activity in hTERT luciferase reporter assays was used as a functional screen for the hits from the yeast two-hybrid screen. Although the hTERT promoter is inactive in primary HFKs, expression of either c-Myc or E6 can induce an hTERT luciferase reporter construct (Fig. 2B). Transfection of the NFX1 isoforms alone with the reporter construct had subtle effects, with NFX1-91 reducing activity to ∼60% of background levels, whereas NFX1-123 had virtually no effect (Fig. 2C). To investigate the activities of the NFX1 isoforms in the context of an activated promoter, we performed cotransfection experiments with c-myc (Fig. 2C). NFX1-123 strongly coactivated the hTERT promoter when cotransfected with c-myc, whereas NFX1-91 robustly repressed induction by c-myc. Interestingly, cotransfection of c-myc and E6 cooperatively increased hTERT promoter activity, with even higher activity on NFX1-123 cotransfection. Coexpression of NFX1-91 reduced hTERT activity. These assays indicated that NFX1-91 functioned as a transcriptional repressor, whereas NFX1-123 appeared to be a coactivator of transcription.
As NFX1 was originally identified in a screen for proteins that bind the X box of MHC class II genes, the presence of similar sequences in the hTERT promoter was investigated. The MHC class II X-box sequence is CCTAGCAACAGATG (highly conserved residues are underlined; Song et al. 1994). Sequence scanning of the promoter found two possible X-box-like sequences within the hTERT proximal promoter (Fig. 2B). One of these X-box-like sequences (CGTGGGAAGCCCTG) overlapped with the proximal E box and the other (CCTGGGAACAGGTG) lay in the reverse orientation ∼400 bp upstream of the transcription start site. In an attempt to map the regions of the hTERT promoter bound by the NFX1 isoforms, luciferase assays were performed with a truncated version of the hTERT promoter, lacking the upstream putative X box. Activity of the NFX1 isoforms on an ∼800-bp region (710 hTERT) of the hTERT promoter was compared with that on a ∼300-bp region (219 hTERT; Fig. 2B,D). Interestingly, whereas the coactivation function of NFX1-123 was clearly apparent with the longer promoter construct, it was greatly diminished with the minimal 219 hTERT promoter. In contrast, the repressive effects of NFX1-91 could be demonstrated using both promoter constructs. This suggests that these two isoforms may have differential binding affinities for specific elements within the hTERT promoter or NFX1-123 may require the presence of additional cofactors bound upstream. It should be noted that c-Myc induction of the 219 hTERT construct was significantly lower than that seen with the longer promoter region and this may impact the coactivation ability of NFX1-123.
In summary, luciferase reporter assays indicated that NFX1-91 was a repressor of the hTERT promoter, whereas NFX1-123 could coactivate the hTERT promoter dependent on sequences located upstream of the minimal 219 promoter fragment.
NFX1-91 binds a putative X box in the proximal promoter of hTERT
To address whether the NFX1 isoforms directly bind the proximal putative X box within the hTERT promoter, we generated recombinant His-tagged NFX1 proteins and purified them for use in electrophoretic mobility shift assays (EMSA). The recombinant proteins were truncated to include only the RING finger domain and the NFX1-type zinc fingers, as this is the proposed DNA-binding region (Fig. 3A; Song et al. 1994). Using titrations of recombinant protein, we found that His-NFX1-91 bound and shifted a 48-bp region surrounding the proximal E box and overlapping putative X box much more strongly than similar quantities of His-NFX1-123 (Fig. 3A). The ability of both proteins to bind this region was slightly diminished on mutation of five residues within the X box. Thus, NFX1-91 appeared to have a higher affinity than NFX1-123 for the hTERT proximal promoter that appeared to be specific for the X-box region.
Figure 3.
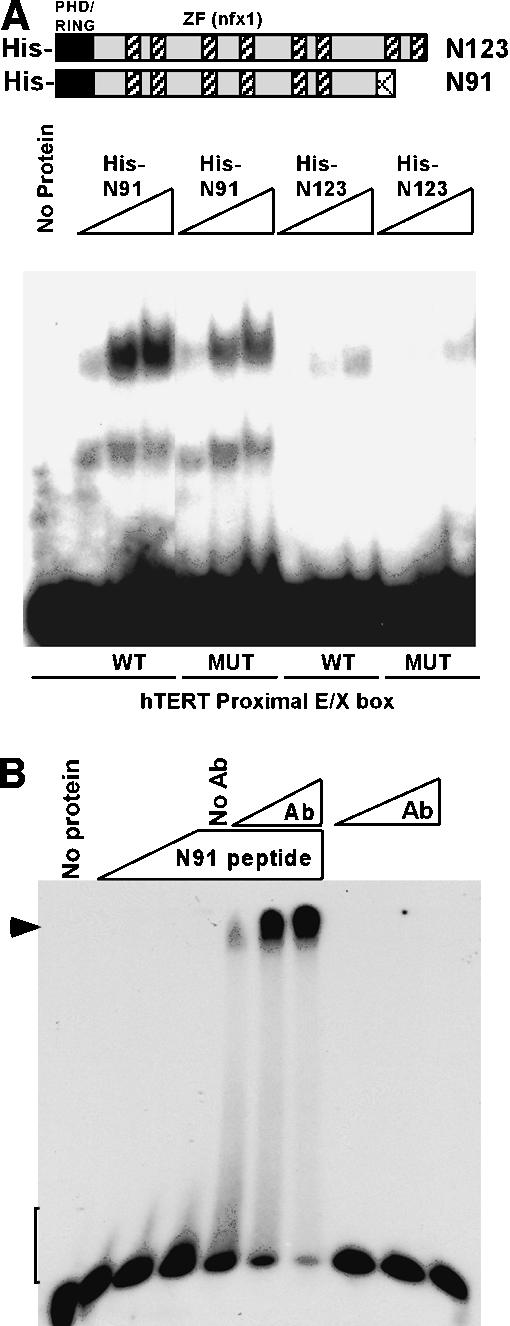
NFX1-91 binds to the proximal putative X box in the hTERT promoter. EMSA assays. (A) His-tagged recombinant NFX1 protein fragments, schematically represented at the top of the figure, were generated and purified from E. coli. Titrations of His-tagged NFX1-91 (His-N91) were able to shift a wild-type (WT) probe of 48 bp surrounding the proximal E box and including the overlapping putative X box. This shift was slightly diminished on mutation of five of the nucleotides within the putative X-box region (MUT). His-tagged NFX1-123 (His-N123) was much less efficient at binding and shifting the same probes. (B) A C-terminal peptide of NFX1-91 was sufficient to induce a small shifting (brackets) of the wild-type (WT) X-box DNA probe that could be dramatically supershifted (arrowhead) with titrations of a rabbit polyclonal antibody (Ab) raised to this peptide. The last three lanes demonstrate that antibody alone is unable to induce a similar shift.
Furthermore, the increased affinity of NFX1-91 for binding to the X box appears to reside within the unique C-terminal end of the protein. In another EMSA experiment, a C-terminal peptide (Ac-CASTQKKRSHYMK KIPAH-amide) was sufficient to induce a small shift of the probe DNA that could be dramatically supershifted with a rabbit polyclonal antibody raised to this peptide. Importantly, antibody alone did not induce a similar shift. These data suggest that the unique C terminus of NFX1-91 plays a significant role in the affinity of the protein for DNA.
16E6 preferentially binds and destabilizes NFX1-91 rather than NFX1-123
Given the repressive effects of NFX1-91 on the hTERT promoter in luciferase assays and its ability to bind the proximal promoter of hTERT, an attractive hypothesis emerged that E6/E6-AP might target NFX1-91 for degradation, thereby relieving repression at the hTERT promoter. Analysis of the RNA expression levels of the NFX1 isoforms in HFKs expressing E6 or a vector control (LXSN), HeLa (HPV 18-positive cervical carcinoma), C33A (HPV-negative cervical carcinoma), and irrelevant U2OS (osteosarcoma) cells by RT-PCR revealed that the level of expression of the two isoforms did not vary greatly between cell types (Supplementary Fig. S1). As the E6/E6-AP complex reduces expression of its targets by ubiquitin-mediated protein degradation, the effect of E6 expression on NFX1 protein levels was determined. A time course of cells treated with cycloheximide revealed substantial differences in the expression level and half-life of NFX1-91 in HFK/E6 cells compared with HFK/LXSN cells. Although NFX1-91 had an apparently short half-life in the absence of E6, its stability was further decreased in the presence of E6 (Fig. 4A). In contrast, NFX1-123 seemed to be an abundant, stable protein that was largely unaffected by E6 expression (Fig. 4A). To more accurately measure the half-life of NFX1-91 in the presence and absence of E6 expression, we performed pulse-chase labeling of HFKs expressing E6 wild-type and mutant proteins followed by immunoprecipitation with an antibody specific for NFX1-91. As shown in Figure 4B, E6 expression reduced the half-life of NFX1-91 ∼46%, from 2.5 h in vector control cells to 1.3 h in E6-expressing cells. Expression of two E6 mutants, F2V and 8S/9A/10T, which induce telomerase but do not target p53 for degradation despite their ability to bind E6-AP, caused reductions in NFX1-91 half-life similar to that found with E6 wild type (Fig. 4B). The reduction of NFX1-91 protein levels in E6-expressing HFKs was proteasome dependent, as treatment with the proteasome inhibitor MG-132 restored NFX1-91 protein levels to that found in vector-transduced HFKs (Fig. 4C). Although E6 interacted with NFX1-123 in the yeast two-hybrid screen and in in vitro binding assays (data not shown), E6 preferentially bound to NFX1-91 and not to NFX1-123 in coimmunoprecipitations from 293Ts transiently transfected with AU1-tagged E6 (Fig. 4D). Although we were unable to confirm the interaction with the reciprocal coimmunoprecipitation using NFX1 rabbit polyclonal antibody, the sum of the other protein expression data and binding data strongly suggested that NFX1-91 was a novel target of the E6/E6-AP E3 ubiquitin ligase complex.
Figure 4.

NFX1-91 rather than NFX1-123 is targeted by the E6/E6-AP complex. (A) Western blot. HFK/LXSN and HFK/E6 cells were treated with 25 μg/mL cycloheximide for the indicated time points. Lysates were assayed for NFX1 expression levels using an affinity-purified NFX1 antibody. c-Myc is shown as a positive control for a short-lived protein. Nucleolin is a stable protein used as a loading control. (B) Half-life of NFX1-91. A pulse-chase experiment to calculate the half-life of NFX1-91 was performed using metabolically labeled HFKs expressing the indicated E6 construct or LXSN vector control. The data presented are the average of two or three independent experiments for each cell line. Error bars represent the standard error of the mean. (C) Decreased expression of NFX1-91 seen in E6-expressing HFKs was proteasome dependent. Proteasome inhibition with MG-132 restored NFX1-91 protein levels. NFX1 protein was detected using an IgG-purified NFX1 antibody. The asterisk indicates a nonspecific background band used as a loading control. p53 expression is shown as a control for proteasome inhibition. (D) Endogenous NFX1-91 coimmunoprecipitated with AU1-tagged 16E6 in transiently transfected 293T cells. A longer exposure of the NFX1 blot shows the presence of a ubiquitin ladder. The reciprocal IP to precipitate AU1-16E6 with NFX1 antibody did not work.
NFX1-91 is highly ubiquitinated in the presence of E6
As E6 appeared to preferentially bind and destabilize NFX1-91 in a proteasome-dependent manner, we examined the ubiquitination status of NFX1-91 versus NFX1-123 in cell lysates. For these experiments, Flag-tagged versions of the NFX1 isoforms were transiently transfected into 293T cells in order to specifically immunoprecipitate the two different isoforms. Although expression of both isoforms was driven by the CMV promoter, NFX1-123 was much more highly expressed than NFX1-91 on transfection (Fig. 5A), consistent with the differences in protein stability previously observed (Fig. 4). In addition, Western blots with a ubiquitin antibody revealed a strong ladder signal only in the Flag-tagged NFX1-91 immunoprecipitations (Fig. 5B), indicating NFX1-91 was highly ubiquitinated whereas NFX1-123 was not, thus explaining their different half-lives. As this experiment was conducted in cells lacking E6 expression, it indicated that NFX1-91 protein levels, much like p53 protein levels, were regulated in a proteasome-dependent manner even in the absence of E6. To determine if E6 expression could influence the degree of NFX1-91 ubiquitination, we repeated the experiment in HFK/LXSN and HFK/E6 cells with endogenous NFX1 protein. We developed specific antibodies to separately immunoprecipitate each isoform of NFX1. As seen in Figure 5C, ubiquitinated NFX1-91 was much more prevalent in HFK/E6 cells than in HFK/LXSN cells, and NFX1-123 did not appear to be ubiquitinated. A longer exposure of the ubiquitin Western blot shown in Figure 5C indicated that there is some ubiquitinated NFX1-91 in HFK/LXSN cells (data not shown). Therefore, as we saw in Figure 5B with the 293T cells, an E6-independent means of ubiquitinating NFX1-91 seems to exist. Significantly, the higher levels of ubiquitinated NFX1-91 detected in HFK/E6 cells directly correlated with the decreased stability of NFX1-91 protein in the presence of E6 (Fig. 4), strongly supporting the hypothesis that NFX1-91 is a target of E6/E6-AP-mediated degradation.
Figure 5.

NFX1-91 is ubiquitinated by E6-independent and E6-dependent means. (A) Western blot. 293Ts were transiently transfected with Flag-tagged forms of NFX1. Cells were treated with MG-132 prior to lysis. Proteins were detected with rabbit anti-Flag antibody. (B) The lysates shown in A were immunoprecipitated with rabbit IgG (rIgG) or a rabbit anti-Flag antibody (α-Flag). Western blots with a mouse anti-ubiquitin antibody indicate that NFX1-91 was highly ubiquitinated, whereas NFX1-123 was not. (C) Endogenous NFX1 isoforms were immunoprecipitated from HFK/LXSN and HFK/E6 cells with antibodies specific for each isoform or with preimmune serum. Anti-NFX1 detects both NFX1 isoforms and anti-ubiquitin detects ubiquitinated proteins. Nucleolin is a loading control for the input lysates.
In vivo evidence for NFX1-91 as a repressor of hTERT
Thus far, luciferase reporter assays suggested NFX1-91 functions as a repressor at the hTERT promoter and protein expression data indicated that the E6/E6-AP complex could destabilize NFX1-91 protein, suggestive of a relief of repression mechanism for E6-mediated hTERT induction. To address directly whether NFX1-91 functions as a transcriptional repressor at the endogenous hTERT promoter, we reduced NFX1-91 expression using stable shRNA expression in HFKs. We constructed an shRNA construct targeted to the unique 3′ untranslated region (3′UTR) of NFX1-91. The NFX1-91 shRNA (n91sh) reduced NFX1-91 protein levels in HFKs while not affecting NFX1-123 protein levels (Fig. 6A). This was accompanied by derepression of the hTERT promoter even in cells lacking E6 expression (Fig. 6B). The previously characterized E6-AP sh1 (esh1) construct (Fig. 1) was included as a negative control. When the shRNA-expressing cell lines were subsequently transduced with empty vector or E6, the E6 cells with reduced NFX1-91 expression had a greater than twofold increased expression of hTERT as demonstrated by RT-PCR and TRAP assay (Fig. 6C,D). Furthermore, the lower telomerase activity resulting from decreasing E6-AP expression in HFK/E6 cells correlated with increased NFX1-91 protein levels, decreased detection of ubiquitinated NFX1-91, and an increased half-life of NFX1-91 from 1.2 to 1.7 h (Fig. 6E,F). These data indicated that NFX1-91 functions as a transcriptional repressor at the endogenous hTERT promoter in HFKs and that the activity of the hTERT promoter increased in response to decreased NFX1-91 protein expression levels, either by shRNA expression or by E6/E6-AP-mediated ubiquitination and degradation.
Figure 6.

Knockdown of NFX1-91 expression using shRNAs derepresses the endogenous hTERT gene in HFKs. (A) Western blot. NFX1-91 protein expression was reduced with expression of n91sh. (B) TRAP assay. Lysates from HFKs transduced with pB, esh1, or n91sh were analyzed for telomerase activity. Two micrograms and 0.5 μg of each lysate were used in the TRAP reactions. HeLa is a positive control lysate (0.2 μg). The percent of TRAP activity is presented relative to that in the HeLa lane. (C) TRAP assay. The cells shown in A and B were subsequently transduced with LXSN or LXSN-16E6. Lysates were examined for telomerase activity. The percent of TRAP activity is presented relative to that in the pB/LXSN-16E6 cells. (D) RT-PCR. Expression of hTERT and NFX1-91 was examined in RNA extracts from the cells in C. (E) Western blots. In vivo ubiquitination assays were performed as in Figure 4 with HFKs transduced with pB/LXSN, pB/E6, or esh1/E6. The blots on the left show protein expression in the lysates used for IP (5% input). The blots on the right show levels of ubiquitinated NFX1-91 immunoprecipitated from the cells. (F) NFX1-91 half-life. A pulse-chase analysis of NFX1-91 protein half-life in two independent lines of HFKs expressing E6 and empty vector (E6) or E6 and the esh1 shRNA (E6 + esh1) found an increase in protein half-life from 1.2 to 1.7 h. Error bars represent the standard deviation of two independent experiments.
To further validate the role of NFX1-91 in regulation of telomerase activity, we examined the impact of reduced NFX1-91 expression on the lifespan of cells in culture. The cells presented in Figure 6C were continually passaged, splitting 1:3 as needed for ∼2 mo (Fig. 7A). As cells divide, the telomeres of cells that do not express telomerase continually shorten and the telomere structure becomes disrupted (Hahn et al. 1999b; Masutomi et al. 2003). Cells respond to the chromosome ends as though they are a double-stranded DNA break and generally attempt to repair the exposed chromosome ends or initiate a senescent arrest (Espejel and Blasco 2002; d'Adda di Fagagna et al. 2003; Takai et al. 2003). Senescent cells can be identified by a change in cellular morphology and with a marker for senescent-associated (SA)-β-galactosidase activity (Dimri et al. 1995). As expected, there were fewer senescent cells present in the E6-expressing population compared with the vector control population (Fig. 7B). Furthermore, about half of the E6 cells with reduced E6-AP expression stained positive for SA-β-galactosidase and appeared large and flattened with many vacuoles (Fig. 7B,C). In contrast, the n91sh-expressing cells, even in the absence of E6 expression, were more rounded like rapidly dividing cells and displayed reduced SA-β-galactosidase staining. The percent of cells exhibiting SA-β-galactosidase activity (Fig. 7C) was inversely related to the telomerase activity detected at earlier passages (Fig. 6C,D). Additionally, the cells with increased telomerase activity had increased proliferative capacity in culture, as indicated by the number of population doublings each cell line achieved during the same number of days in culture (Fig. 7A,B). Therefore, reduction of NFX1-91 protein levels either via reduction of mRNA levels with shRNA or via E6/E6-AP-mediated ubiquitination and degradation was sufficient to induce hTERT expression and delay senescent growth arrest in primary human epithelial cells. Conversely, reduced E6-AP expression prohibits E6-mediated telomerase induction and lifespan extension through the stabilization of the hTERT transcriptional repressor, NFX1-91.
Figure 7.

Senescence-associated β-galactosidase activity in late-passage HFKs. (A) Growth curves. HFKs expressing the indicated shRNA and LXSN (closed symbols) or E6 (open symbols) were grown in culture for ∼2 mo. The pBabe vector control is indicated by black squares, the esh1 cells are represented by red circles, and the n91sh cells are indicated by green triangles. The time point at which cells were stained for SA-β-galactosidase is indicated. (B) Micrographs of SA-β-galactosidase staining of late-passage HFKs expressing the indicated shRNA and LXSN empty vector or LXSN-16E6. The blue staining indicates senescent cells. (C) Quantitation of SA-β-galactosidase staining seen in B. The data represent the average percent of blue cells counted in four different fields at 100× magnification. The error bars indicate the standard error of the mean.
Although delayed senescence correlated well with increased telomerase activity, we were unable to observe any significant differences in the telomere lengths of the cells presented in Figures 6 and 7 (data not shown). Telomere-length Southern blots indicated that telomeres continued to shorten regardless of the level of telomerase expression (data not shown). Previous studies of mortal cultures of E6-expressing epithelial cells have also observed a lack of telomere-length maintenance despite the induction of telomerase expression (Kiyono et al. 1998). This may indicate that telomerase is acting only at the shortest telomeres to maintain proper telomere structure rather than increasing telomere lengths (Hemann et al. 2001; Masutomi et al. 2003). We propose that the continued proliferation and lack of β-galactosidase-positive cells in the telomerase-expressing cells more accurately reflects the maintenance of proper telomere structure than the gross changes in telomere length observed by telomere-length Southern blot analysis.
Discussion
Induction of telomerase appears to be a common and requisite event in the immortalization and transformation of many cell types. In our studies to determine how HPV-16 E6 induces telomerase in epithelial cells, we have identified NFX1-91 as a cellular repressor of the human hTERT promoter both in vivo and in vitro that is destabilized by the E6/E6-AP complex. Previous work by our group and others suggested a correlation between the ability of E6 to bind its cellular partner E6-AP and the induction of telomerase (Table 1). The E6/E6-AP complex is proposed to target many different cellular proteins for ubiquitination and degradation (Fehrmann and Laimins 2003; Scheffner and Whitaker 2003), including a large family of PDZ domain-containing proteins that are membrane-associated guanylate kinases, or MAGUKs (Thomas et al. 2001). The PDZ-binding domain of E6, encompassing the four C-terminal amino acids, has been shown to be required for transformation of established rodent cells by E6 (Kiyono et al. 1997) but is dispensable for immortalization by E6 (Kiyono et al. 1998). Therefore it is unlikely that any of the PDZ-containing proteins targeted by E6 are involved in telomerase activation. Here, we provide direct evidence of the requirement for E6-AP expression for E6-mediated hTERT induction (Fig. 1). These findings prompted the search for novel targets of the E6/E6-AP complex that might function as transcriptional repressors at the hTERT promoter.
Identified as an interactor of E6/E6-AP in a yeast two-hybrid screen, NFX1, a known transcriptional repressor, seemed an ideal candidate for a hypothesized telomerase repressor degraded on E6 expression in epithelial cells. This gene actually encoded two splice variants, NFX1-123 and NFX1-91, with opposing activities in hTERT reporter assays (Fig. 2). NFX1-123 strongly coactivated with c-Myc at the hTERT promoter, whereas NFX1-91 repressed the activity of the promoter. Coexpression of E6 with either isoform increased the activity of the promoter, suggesting that E6 could co-operatively activate hTERT with NFX1-123 and could decrease the repressive effects of NFX1-91. Furthermore, coimmunoprecipitation experiments indicated that the E6/E6-AP complex preferentially interacted with the NFX1-91 isoform and stimulated its ubiquitination and degradation (Figs. 4, 5). We propose that E6/E6-AP induces telomerase by destabilizing the hTERT transcriptional repressor, NFX1-91. In support of this hypothesis, decreased expression of NFX1-91 using shRNAs was sufficient to induce hTERT expression and telomerase activity in primary human epithelial cells (Fig. 6). Unfortunately, as seen in Figure 5A, NFX1-91 did not overexpress well despite repeated attempts (data not shown); therefore, we were unable to demonstrate repression of the hTERT promoter in response to increased expression of NFX1-91.
A recent study by Lin and Elledge (2003) found three tumor suppressor/oncogene pathways involved in hTERT repression; these were Mad1, menin, and SIP1 (Lin and Elledge 2003). Reduced expression of any one of these hTERT repressors was sufficient to induce hTERT expression in previously telomerase-negative cells. It is striking that these repressors do not seem redundant; oncogenic stimulation that abrogates any one of them is sufficient to relieve repression. Therefore, it is not implausible that, despite the identification of several hTERT repressor proteins, E6/E6AP specifically targets only one transcriptional repressor, NFX1-91, to induce hTERT expression. It is likely that future investigations will reveal different cell-type specificities for hTERT repressors and variability in the responsiveness of different cell types to perturbations of these repressors.
In reporter assays, E6 seems to require an intact proximal E box in the hTERT promoter for activity (Gewin and Galloway 2001; Veldman et al. 2003). Although c-Myc levels do not appear to change dramatically on E6 expression (Gewin and Galloway 2001; Veldman et al. 2001), two groups have recently found by chromatin immunoprecipitation that c-Myc is bound to the hTERT promoter in E6-expressing cells (Baege et al. 2002; Veldman et al. 2003). McMurray and McCance (2003) suggest that in E6-expressing cells, c-Myc replaces USF1 and USF2 at the hTERT promoter, and Veldman et al. (2003) find that c-Myc is bound to the hTERT promoter in both hTERT-negative E7-expressing cells and hTERT-positive E6-expressing cells. In unpublished data from our lab, we also found that c-Myc was bound at the hTERT promoter in both vector control and E6-expressing HFKs. Therefore, binding of c-Myc to the hTERT promoter was not sufficient for promoter activity. Because our data suggested that disruption of the NFX1-91 repressor of telomerase was involved in the activation of telomerase, it is interesting to note that a putative NFX1-binding site overlaps with the proximal E box. EMSA experiments indicate that a recombinant fragment of NFX1-91 binds to this region with significantly higher affinity than a largely similar fragment of NFX1-123. These data help to clarify how NFX1-91, an unstable protein expressed at much lower levels than NFX1-123, could repress the hTERT promoter in the presence of abundant NFX1-123 protein levels. Our future efforts will determine whether either of the two NFX1 isoforms is differentially bound to this site in vivo in E6-expressing cells.
Although reporter assays indicated that the NFX1-123 isoform could function as a coactivator of the hTERT promoter, this isoform is highly expressed, stable, and apparently unaffected by E6 expression. NFX1-123, with its two additional zinc fingers and R3H domain, most likely exists in a very different conformation than NFX1-91 that is not recognized by the E6/E6-AP complex. As structure relates to function, this likely influences the DNA-binding affinity, as seen by EMSA in Figure 3, and functional consequences of DNA binding for NFX1-123. Whether the coactivation function of NFX1-123 is an integral part of E6-mediated hTERT induction remains to be tested. Interestingly, the instability and repressive functions of NFX1-91 as well as its higher affinity for the X-box-like sequences in the hTERT promoter appeared to reside in the unique lysine-rich C terminus.
Although we propose that the targeting of NFX1-91 for ubiquitination and degradation by the E6/E6-AP complex is involved in the induction of telomerase in epithelial cells, this may be only one step in the process of robustly inducing hTERT expression by E6. Previous data have indicated that induction of hTERT by HPV-16 E6 may involve epigenetic phenomenon such as histone acetylation or chromatin remodeling (Klingelhutz et al. 1996; Kiyono et al. 1998; Baege et al. 2002). Many transcriptional regulators function by covalently modifying the histones associated with the nearby chromatin. Recently, it has been demonstrated that ubiquitination of histone H2B may influence the methylation status of histone H3 and thereby mark the chromatin as active for transcription (Muratani and Tansey 2003). Given that E6/E6-AP and the NFX1 RING finger possess E3 ubiquitin ligase activity, it will be interesting to investigate the ubiquitination status of histone H2B at the hTERT promoter in E6-expressing cells.
Ubiquitination and transcriptional activation may also be intimately linked in a developing model involving the ubiquitination and degradation of transcriptional activators bound at promoters (Muratani and Tansey 2003). The activators rely on E3 ligase activity found within coactivators to license their activity and link increased protein turnover with clearing of the promoter for subsequent rounds of transcription. In fact, the Skp2 ubiquitin ligase is a coactivator of c-Myc at the cyclin D2 promoter and loss of Skp2 can stabilize c-Myc protein levels and reduce transactivation of c-Myc-responsive promoters (Kim et al. 2003; von der Lehr et al. 2003). Although c-Myc steady-state levels do not change on E6 expression, it is possible that this is a result of balanced c-Myc induction (Kinoshita et al. 1997) and increased degradation (Gross-Mesilaty et al. 1998). E6/E6-AP has been demonstrated to ubiquitinate c-Myc in vitro and in vivo (Gross-Mesilaty et al. 1998). In fact, E6 has been found at the hTERT promoter and E6 can immunoprecipitate c-Myc from cell lysates (Veldman et al. 2003). Although we have been unable to demonstrate an interaction between c-Myc and E6, it remains possible that these proteins do interact in a complex at the hTERT promoter. An attractive synthesis of this model with our data is that E6/E6-AP may function at multiple levels to induce hTERT. First, the E6/E6-AP complex may target NFX1-91 for increased turnover to derepress the promoter; then, an E3 ligase may ubiquitinate and activate the c-Myc bound at the hTERT promoter. This theory and the possible role of the E3 ligase activity of the NFX1 isoforms or the E6/E6-AP complex to ubiquitinate either histone H2B or c-Myc remains to be tested.
In summary, we have identified NFX1-91 as a novel cellular repressor of the hTERT promoter in primary human epithelial cells. Significantly, interference with its expression is sufficient to induce telomerase expression and extend the lifespan of primary epithelial cells, thus making NFX1-91 an important new target of transformation mechanisms.
Materials and methods
Cell culture
Primary human keratinocytes (HFKs) were derived from neonatal foreskins and grown in EpiLife medium supplemented with calcium chloride (60 μM) and human keratinocyte growth supplement (Cascade Biologics). 293T, HeLa, C33A, and U20S cells were grown in Dulbecco's modified Eagle's medium (GIBCO-BRL) containing 10% fetal calf serum (FCS) and penicillin-streptomycin. SF9 cells were grown in SF 900 II serum-free medium (GIBCO-BRL) containing 5% FCS and gentamycin.
Plasmids
The E6-AP and NFX1-91 shRNA constructs were generated by previously described methods (Paddison et al. 2002; Grandori et al. 2003; Smith et al. 2003). Briefly, oligos containing the 26- to 29-nt shRNA sequence were used in PCR reactions to clone the U6 or H1 RNA polymerase III promoter upstream into a pBabepuro-based vector. The E6-AP shRNA targeted sequences were as follows: esh1 5′-CTAATAGAACGCTACTACCACCAGTTAAC-3′, esh2 5′-AGAGATTGTTGAAGGCCATCACGTATGCC-3′, and esh3 5′-ACAATGAAGAAGATGATGAAGAGCCCATC-3′. The n91sh construct was targeted to the 3′UTR region (5′-TGTGGAACCAGCCCAACTGCCCATCAGTCAA-3′). The NFX1-123 isoform was PCR cloned from a HeLa cell cDNA library and the NFX1-91 isoform was cloned from a fetal brain cDNA library. These genes (with and without a Flag tag) were subsequently inserted by restriction digest or via the GATEWAY recombination-based system (Invitrogen) into a CMV-based vector for transient transfections. The AU1 tag (DTYRYI) was fused to the N terminus of 16E6 by PCR and subsequently cloned into a CMV-based vector using the GATEWAY system (Invitrogen). The pBabe-c-myc vector was obtained from Carla Grandori. The pGL3-based hTERT luciferase reporter constructs have been previously described (Gewin and Galloway 2001). The His-tagged NFX1 isoforms were generated by PCR cloning the indicated fragments into the BamHI site of the pET16b vector (Novagen) in frame with multiple histidine residues.
Retroviral infections
Retroviruses were produced either in established viral producer cell lines (PA317 or PG13) or in 293Ts by a transient VSV-G-pseudotyped virus production protocol as previously described (Bartz and Vodicka 1997). Cells were infected at ∼60% confluence in 6-cm tissue culture plates. Twenty-four hours after infection, cells were expanded to 10-cm plates and allowed to adhere to the plate for 4-24 h before adding selective media. HFKs were selected in 0.5 μg/mL puromycin or 50 μg/mL G418. Cells expressing shRNAs were maintained in puromycin-containing media.
Northern blotting
Total cellular RNA was isolated using the RNeasy kit (QIAGEN). From 20 to 60 μg of total RNA was electrophoresed on 1% agarose-formaldehyde gels, transferred to Hybond-N membranes (Amersham), and hybridized to 32P-labeled probes. The E6-AP probe was generated by random primer labeling (Roche) a 440-bp XhoI fragment of the E6-AP gene. The 36B4 loading control probe has been described previously (Kiyono et al. 1998).
Yeast two-hybrid screen
The yeast two-hybrid screen was performed using the Match-maker GAL4 two-hybrid system (Clontech). A catalytically defective E6-AP mutant (C833A) was fused to the GAL4 DNA-binding domain in the pGBT9 plasmid. The 16E6 gene was also cloned into this plasmid under the control of the ADH2 promoter. Both a HeLa cell cDNA library and a fetal brain cDNA library were fused to the GAL4 activation domain in the pGAD and pACT2 plasmids, respectively. Clones that grew on selective media were subjected to a secondary screen for β-galactosidase activity. Full-length clones were obtained by 5′ RACE (rapid amplification of cDNA ends) using the Marathon cDNA amplification kit (Clontech).
Luciferase assays
Luciferase assays were performed as previously described (Gewin and Galloway 2001). Briefly, HFKs were grown to 50%-60% confluence in six-well plates and transfected with a pGL3-based hTERT reporter plasmid (710 hTERT or 219 hTERT) and CMV- or pBabe-based expression constructs. A total of 2 μg of DNA was transfected into each well using a 1:3 DNA:FuGENE (Roche) ratio. Cells were incubated for 24 h after transfection, rinsed in phosphate-buffered saline (PBS), and lysed in the well by freeze-thawing in 100 μL of reporter lysis buffer (Promega). Cell debris was removed by centrifugation. Luminescence was quantitated in 10 μL of each lysate on mixing with luciferase assay buffer (Promega) in a Monolight 2010 luminometer. Each experiment was done several times in duplicate and normalized for total protein concentration.
Recombinant protein purification
pET16b constructs expressing His-tagged NFX1 isoforms were transformed into Codon (+) DE3 cells and grown in 100-mL cultures to an O.D.600 of ∼0.6. Protein expression was induced for 2 h at room temperature with 0.4 mM isopropyl-β-D-thio-galactopyranoside (IPTG). Proteins were extracted from inclusion bodies using denaturing conditions (8 M urea, 10 mM Tris-Cl, 100 mM NaH2PO4, 20 mM β-mercaptoethanol, 10% glycerol, 1% NP-40, 300 mM sodium chloride, 20 μM zinc chloride adjusted to pH 8) and purified on Ni-NTA beads (Qiagen). After washing with lysis buffer adjusted to pH 6.3, the beads and bound recombinant proteins were dialyzed stepwise to remove urea and NP-40 prior to elution with increasing concentrations of imidazole (100-250 mM).
EMSA
Probes were generated by radiolabeling, annealing, and gel purifying complementary oligonucleotides containing sequences from the hTERT promoter. Sequences correspond to 15 bp upstream of the proximal E box (CACGTG) and extend 27 bp downstream. Five base pairs were changed (GGAAGCCCTG to TGGGGCCCGA) to generate the X-box mutant probe. Titrations of recombinant proteins (1-6 μL) were incubated with 5000 cpm of radiolabeled probes in 40-μL binding reactions containing 25 mM HEPES (pH 7.5), 5% glycerol, 5 μM zinc chloride, 5 mM magnesium chloride, 50 mM potassium chloride, and 0.1 mg of bovine serum albumin per milliliter. The peptide shifts were performed identically using titrations (20-100 ng) of peptide and supershifted with titrations (1-4 μg) of affinity-purified rabbit polyclonal antibody raised to this peptide. Reactions were run on 4.5% polyacrylamide gels in 25 mM HEPES (pH 7.5).
RT-PCR
RNA was isolated from cells using the RNeasy kit (QIAGEN). cDNA was synthesized using random hexamers and the Superscript II reverse transcriptase system (Invitrogen). RNase H (2 units) was added to the reactions and incubated for 20 min at 37°C. Expression of hTERT, NFX1-123, NFX1-91, and 36B4 was detected by PCR using either ethidium bromide staining or [32P]α-dCTP incorporation and autoradiography. Sequences of hTERT and 36B4 primers are previously described (Gewin and Galloway 2001). NFX1-123 primers are (F) 5′-TCCCTCCCAT GAACAGAGAC-3′ and (R) 5′-TTTCAAGCACACCTGTCAG C-3′. NFX1-91 primers are (F) 5′-TTACCCTCCAGTTCCCT GTG-3′ and (R) 5′-CATGCGTGTGCAGGTATCTT-3′.
Telomerase activity
Telomerase activity was detected using the radioisotopic detection method of the TRAPeze telomerase detection kit (Serologicals Corporation, Chemicon International).
Generation of rabbit polyclonal antibodies
Rabbit polyclonal antibodies were generated to recognize both NFX1 isoforms (α-NFX1) and each isoform, NFX1-123 (α-NFX1-123) and NFX1-91 (α-NFX1-91), individually. Recombinant full-length NFX1-123 was generated in SF9 cells using a baculovirus system. Briefly, NFX1-123 was cloned into the pVIC1 plasmid in which NFX1 was fused to a chitin-binding domain with an intervening intein sequence as described previously (Chong et al. 1997; Pradhan et al. 1999). This construct was transfected into SF9 cells using the BaculoGold system (Pharmingen) to generate infectious baculovirus. SF9s infected with baculovirus were harvested and lysed in buffer M (50 mM Tris-HCl at pH 7.4, 1 mM EDTA, 500 mM sodium chloride, and COMPLETE protease inhibitor tablet). The SF9 protein lysate was precleared on cellulose resin and subsequently purified with chitin beads. Full-length NFX1-123 was cleaved from the chitin-binding domain in two volumes of buffer M plus 0.5% Tween-20, 4 mM dithiothreitol, and 5% glycerol incubated at 16°C overnight and used to generate a rabbit polyclonal antibody to recognize both NFX1 isoforms. The NFX1-123-specific antibody was raised to a recombinant His-tagged C-terminal fragment of NFX1-123 (amino acids 932-1120) expressed in and purified from Escherichia coli. The NFX1-91-specific antibody was raised to a C-terminal peptide (Ac-CASTQKKRSHYMKKIPAH-amide) generated by BIOSOURCE.
Western blotting
Whole-cell lysates were prepared for Western blotting by trypsinizing cells, washing with PBS, and resuspending in WE16th lysis buffer (50 mM Tris-HCl at pH 7.5, 250 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate [SDS], 20% glycerol, 10 μM zinc chloride, 2 mM dithiothreitol, 80 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, and a COMPLETE protease inhibitor tablet [Roche]). Lysates were then sonicated and clarified by centrifugation. Protein concentrations were determined by using the DC protein assay (Bio-Rad). Protein lysates were electrophoresed on SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore). Western blots were performed with goat anti-actin (Santa Cruz Biotechnology, I-19), mouse anti-p53 (Oncogene Science, Ab6), mouse anti-c-myc (Santa Cruz Biotechnology, C-33), mouse anti-nucleolin (Santa Cruz Biotechnology, C-23), rabbit anti-AU1 (Bethyl), mouse anti-Flag (Sigma, M2), and mouse anti-ubiquitin (Covance, P4G7). Rabbit polyclonal NFX1, NFX1-123, and NFX1-91 antibodies are described earlier.
Proteasome inhibition and cycloheximide treatment
In proteasome inhibition experiments, cells were treated with 10-20 μM MG-132 (Calbiochem) or an equal volume of dimethyl sulfoxide (solvent control) for 4 h at 37°C. For half-life analysis, HFKs were treated with 25 μM cycloheximide (Calbiochem) and harvested in WE16th lysis buffer at the indicated time points.
Pulse-chase labeling of cells and protein half-life calculations
Keratinocytes were metabolically labeled with Express Label [35S]cysteine-[35S]methionine (DuPont NEN) in 154XP medium (Cascade Biologics) for 1 h. Cells were chased for various periods of time with EpiLife keratinocyte medium, washed two times in cold PBS, and lysed on ice for 30 min in RIPA buffer + 1 mM EDTA. Following a brief sonication, the lysates were frozen in liquid nitrogen and stored at -80°C until completion of the time course. Lysates were thawed quickly and insoluble debris was pelleted. Lysates were subsequently precleared with protein A agarose prior to immunoprecipitation with preimmune or anti-NFX1-91 rabbit serum. After incubation with protein A agarose beads, immunocomplexes were washed once with RIPA buffer, twice with high-salt buffer (500 mM sodium chloride, 50 mM Tris at pH 8, 1% NP-40), and once more with RIPA buffer. The beads were boiled in sample buffer and electrophoresed on a 6% SDS-polyacrylamide gel. The gels were dried and subjected to phosphorimaging (Molecular Dynamics). Band intensities were measured using ImageQuant.
Immunoprecipitations
293Ts at 60% confluence in 15-cm plates were transfected with 20 μg of CMV-AU1-16E6 with FuGENE (Roche). Twenty-four hours later, cells were treated with 10 μM MG-132 (Calbiochem) for 2 h prior to harvest. 293Ts were harvested for immunoprecipitation by rinsing with cold PBS. Cells were pelleted and resuspended in an NP-40 lysis buffer (1× PBS, 0.5% NP-40, 10% glycerol, 10 μM zinc chloride, 2 mM dithiothreitol, 80 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, and a COMPLETE protease inhibitor tablet [Roche]). Cells were lysed by quick freezing in liquid nitrogen and thawing in a room-temperature water bath. Cell debris was pelleted at 14,000 rpm for 15 min and lysates were precleared by rotating at 4°C with 50 μL of protein A agarose (Roche). After centrifugation to remove the beads, lysates were incubated with the appropriate antibody for 1-2 h at 4°C and purified by adding protein A agarose and rotating for another hour at 4°C. Immunocomplexes were washed three times with lysis buffer and eluted by heating for 10 min at 70°C in 2× sample buffer. Elutions were electrophoresed on NuPAGE 4%-12% Tris-Bis gradient gels (Invitrogen) to resolve both AU1-tagged 16E6 and NFX1 isoforms and Western blotted as described earlier.
In vivo ubiquitination assay
Cells were treated with 20 μM MG-132 for 2 h prior to harvest. Lysates were prepared by trypsinizing cells and washing with PBS. Cell pellets were resuspended in 2% SDS in PBS and boiled for 10 min, then diluted in five to six volumes of 1% Triton X-100 in PBS. The lysates were sonicated on ice and clarified by centrifugation followed by preclearing with protein A agarose for 30 min at 4°C. The lysate was subsequently divided for individual immunoprecipitations with the appropriate antibody and incubated at 4°C for 1-2 h. Preimmune rabbit serum and normal rabbit IgG (Santa Cruz Biotechnology) were used as negative controls. To precipitate bound proteins, we added protein A agarose to each immunoprecipitation and rotated it for 1 h at 4°C. Bound proteins were washed three times with PBS and eluted by boiling for 5 min in 2× sample buffer. Elutions were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting as described earlier.
SA-β-galactosidase staining
Cells were washed three times in PBS containing 1 mM magnesium chloride and fixed in 3% formaldehyde in PBS for 5 min at room temperature. After three washes in PBS (pH 6.0), cells were stained in PBS (pH 6.0) containing 1 mM magnesium chloride, 0.12 mM potassium ferricyanide, 0.12 mM potassium ferrocyanide, and 1 mM X-GAL at 37°C overnight.
Acknowledgments
We thank Carla Grandori for critical reading of this manuscript and members of the Galloway and Eisenman labs for advice, reagents, and technical assistance. This work was supported by grant CA64795 to D.A.G. L.G. was supported in part by the Viral Oncology Training Grant (PHS 5 T32 CA 09229-23) and the STD/AIDS Research Training Grant (T32 AI07140).
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1214704.
References
- Baege A.C., Berger, A., Schlegel, R., Veldman, T., and Schlegel, R. 2002. Cervical epithelial cells transduced with the papillomavirus E6/E7 oncogenes maintain stable levels of oncoprotein expression but exhibit progressive, major increases in hTERT gene expression and telomerase activity. Am. J. Pathol. 160: 1251-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz S.R. and Vodicka, M.A. 1997. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods 12: 337-342. [DOI] [PubMed] [Google Scholar]
- Boyer S.N., Wazer, D.E., and Band, V. 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56: 4620-4624. [PubMed] [Google Scholar]
- Chong S.R., Mersha, F.B., Comb, D.G., Scott, M.E., Landry, D., Vence, L.M., Perler, F.B., Benner, J., Kucera, R.B., Hirvonen, C.A., et al. 1997. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene 192: 271-281. [DOI] [PubMed] [Google Scholar]
- Cong Y.S. and Bacchetti, S. 2000. Histone deacetylation is involved in the transcriptional repression of hTERT in normal human cells. J. Biol. Chem. 275: 35665-35668. [DOI] [PubMed] [Google Scholar]
- Dalal S., Gao, Q.S., Androphy, E.J., and Band, V. 1996. Mutational analysis of human papillomavirus type 16 E6 demonstrates that p53 degradation is necessary for immortalization of mammary epithelial cells. J. Virol. 70: 683-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessain S.K., Yu, H.Y., Reddel, R.R., Beijersbergen, R.L., and Weinberg, R.A. 2000. Methylation of the human telomerase gene CpG island. Cancer Res. 60: 537-541. [PubMed] [Google Scholar]
- Devereux T.R., Horikawa, I., Anna, C.H., Annab, L.A., Afshari, C.A., and Barrett, J.C. 1999. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 59: 6087-6090. [PubMed] [Google Scholar]
- d'Adda di Fagagna F., Reaper, P.M., Clay-Farrace, L., Fiegler, H., Carr, P., von Zglinicki, T., Saretzki, G., Carter, N.P., and Jackson, S.P. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194-198. [DOI] [PubMed] [Google Scholar]
- Dimri G.P., Lee, X.H., Basile, G., Acosta, M., Scott, C., Roskelley, C., Medrano, E.E., Linskens, M., Rubelj, I., Pereirasmith, O., et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. 92: 9363-9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B., Spirio, L., Koerner, F., Fleming, M.D., Zimonjic, D.B., Donaher, J.L., Popescu, N.C., Hahn, W.C., and Weinberg, R.A. 2001. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes & Dev. 15: 50-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel S. and Blasco, M.A. 2002. Identification of telomere-dependent `senescence-like' arrest in mouse embryonic fibroblasts. Exp. Cell Res. 276: 242-248. [DOI] [PubMed] [Google Scholar]
- Fehrmann F. and Laimins, L.A. 2003. Human papillomaviruses: Targeting differentiating epithelial cells for malignant transformation. Oncogene 22: 5201-5207. [DOI] [PubMed] [Google Scholar]
- Foster S.A., Demers, G.W., Etscheid, B.G., and Galloway, D.A. 1994. The ability of human papillomavirus E6 proteins to target p53 for degradation in vivo correlates with their ability to abrogate actinomycin D-induced growth arrest. J. Virol. 68: 5698-5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Srinivasan, S., Boyer, S.N., Wazer, D.E., and Band, V. 1999. The E6 oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for degradation. Mol. Cell. Biol. 19: 733-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe J., Wong, M., Wigington, D., Yaswen, P., and Stampfer, M.R. 1999. Viral oncogenes accelerate conversion to immortality of cultured conditionally immortal human mammary epithelial cells. Oncogene 18: 2169-2180. [DOI] [PubMed] [Google Scholar]
- Gewin L. and Galloway, D.A. 2001. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J. Virol. 75: 7198-7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori C., Wu, K.J., Fernandez, P., Ngouenet, C., Grim, J., Clurman, B.E., Moser, M.J., Oshima, J., Russell, D.W., Swisshelm, K., et al. 2003. Werner syndrome protein limits MYC-induced cellular senescence. Genes & Dev. 17: 1569-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg R.A., O'Hagan, R.C., Deng, H., Xiao, Q., Hann, S.R., Adams, R.R., Lichtsteiner, S., Chin, L., Morin, G.B., and DePinho, R.A. 1999. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene 18: 1219-1226. [DOI] [PubMed] [Google Scholar]
- Gross-Mesilaty S., Reinstein, E., Bercovich, B., Tobias, K.E., Schwartz, A.L., Kahana, C., and Ciechanover, A. 1998. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc. Natl. Acad. Sci. 95: 8058-8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn W.C., Counter, C.M., Lundberg, A.S., Beijersbergen, R.L., Brooks, M.W., and Weinberg, R.A. 1999a. Creation of human tumour cells with defined genetic elements. Nature 400: 464-468. [DOI] [PubMed] [Google Scholar]
- Hahn W.C., Stewart, S.A., Brooks, M.W., York, S.G., Eaton, E., Kurachi, A., Beijersbergen, R.L., Knoll, J.H., Meyerson, M., and Weinberg, R.A. 1999b. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 5: 1164-1170. [DOI] [PubMed] [Google Scholar]
- Helt A.M. and Galloway, D.A. 2001. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 75: 6737-6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemann M.T., Strong, M.A., Hao, L.Y., and Greider, C.W. 2001. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107: 67-77. [DOI] [PubMed] [Google Scholar]
- Horikawa I., Cable, P.L., Afshari, C., and Barrett, J.C. 1999. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 59: 826-830. [PubMed] [Google Scholar]
- Hou M., Wang, X.B., Popov, N., Zhang, A.J., Zhao, X.Y., Zhou, R., Zetterberg, A., Bjorkholm, M., Henriksson, M., Gruber, A., et al. 2002. The histone deacetylase inhibitor trichostatin a derepresses the telomerase reverse transcriptase (hTERT) gene in human cells. Exp. Cell Res. 274: 25-34. [DOI] [PubMed] [Google Scholar]
- Kanaya T., Kyo, S., Hamada, K., Takakura, M., Kitagawa, Y., Harada, H., and Inoue, M. 2000. Adenoviral expression of p53 represses telomerase activity through down-regulation of human telomerase reverse transcriptase transcription. Clin. Cancer Res. 6: 1239-1247. [PubMed] [Google Scholar]
- Kim S.Y., Herbst, A., Tworkowski, K.A., Salghetti, S.E., and Tansey, W.P. 2003. Skp2 regulates Myc protein stability and activity. Mol. Cell 11: 1177-1188. [DOI] [PubMed] [Google Scholar]
- Kinoshita T., Shirasawa, H., Shino, Y., Moriya, H., Desbarats, L., Eilers, M., and Simizu, B. 1997. Transactivation of prothymosin α and c-myc promoters by human papillomavirus type 16 E6 protein. Virology 232: 53-61. [DOI] [PubMed] [Google Scholar]
- Kiyono T., Hiraiwa, A., Fujita, M., Hayashi, Y., Akiyama, T., and Ishibashi, M. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. 94: 11612-11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyono T., Foster, S.A., Koop, J.I., McDougall, J.K., Galloway, D.A., and Klingelhutz, A.J. 1998. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396: 84-88. [DOI] [PubMed] [Google Scholar]
- Klingelhutz A.J., Foster, S.A., and McDougall, J.K. 1996. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 380: 79-82. [DOI] [PubMed] [Google Scholar]
- Kumar S., Talis, A.L., and Howley, P.M. 1999. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274: 18785-18792. [DOI] [PubMed] [Google Scholar]
- Lin S.Y. and Elledge, S.J. 2003. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 113: 881-889. [DOI] [PubMed] [Google Scholar]
- Liu Y., Chen, J.J., Gao, Q., Dalal, S., Hong, Y., Mansur, C.P., Band, V., and Androphy, E.J. 1999. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J. Virol. 73: 7297-7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorick K.L., Jensen, J.P., Fang, S., Ong, A.M., Hatakeyama, S., and Weissman, A.M. 1999. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. 96: 11364-11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg A.S., Randell, S.H., Stewart, S.A., Elenbaas, B., Hartwell, K.A., Brooks, M.W., Fleming, M.D., Olsen, J.C., Miller, S.W., Weinberg, R.A., et al. 2002. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 21: 4577-4586. [DOI] [PubMed] [Google Scholar]
- Masutomi K., Yu, E.Y., Khurts, S., Ben Porath, I., Currier, J.L., Metz, G.B., Brooks, M.W., Kaneko, S., Murakami, S., DeCaprio, J.A., et al. 2003. Telomerase maintains telomere structure in normal human cells. Cell 114: 241-253. [DOI] [PubMed] [Google Scholar]
- McMurray H.R. and McCance, D.J. 2003. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol. 77: 9852-9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M. and Tansey, W.R. 2003. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4: 192-201. [DOI] [PubMed] [Google Scholar]
- Oh S.T., Kyo, S., and Laimins, L.A. 2001. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 75: 5559-5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz O.G., Suliman, Y., Hahn, W.C., Harada, H., Blum, H.E., and Rustgi, A.K. 2001. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J. Clin. Invest. 108: 725-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison P.J., Caudy, A.A., Bernstein, E., Hannon, G.J., and Conklin, D.S. 2002. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes & Dev. 16: 948-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan S., Bacolla, A., Wells, R.D., and Roberts, R.J. 1999. Recombinant human DNA (cytosine-5) methyltransferase I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 274: 33002-33010. [DOI] [PubMed] [Google Scholar]
- Scheffner M. and Whitaker, N.J. 2003. Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system. Semin. Cancer Biol. 13: 59-67. [DOI] [PubMed] [Google Scholar]
- Scheffner M., Huibregtse, J.M., Vierstra, R.D., and Howley, P.M. 1993. The Hpv-16 E6 and E6-Ap complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75: 495-505. [DOI] [PubMed] [Google Scholar]
- Smith L.L., Coller, H.A., and Roberts, J.M. 2003. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell Biol. 5: 474-479. [DOI] [PubMed] [Google Scholar]
- Song Z., Krishna, S., Thanos, D., Strominger, J.L., and Ono, S.J. 1994. A novel cysteine-rich sequence-specific DNA-binding protein interacts with the conserved X-box motif of the human major histocompatibility complex class II genes via a repeated Cys-His domain and functions as a transcriptional repressor. J. Exp. Med. 180: 1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H., Smogorzewska, A., and de Lange, T. 2003. DNA damage foci at dysfunctional telomeres. Curr. Biol. 13: 1549-1556. [DOI] [PubMed] [Google Scholar]
- Takakura M., Kyo, S., Kanaya, T., Hirano, H., Takeda, J., Yutsudo, M., and Inoue, M. 1999. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 59: 551-557. [PubMed] [Google Scholar]
- Takakura M., Kyo, S., Sowa, Y., Wang, Z., Yatabe, N., Maida, Y., Tanaka, M., and Inoue, M. 2001. Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res. 29: 3006-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M., Glaunsinger, B., Pim, D., Javier, R., and Banks, L. 2001. HPV E6 and MAGUK protein interactions: Determination of the molecular basis for specific protein recognition and degradation. Oncogene 20: 5431-5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman T., Horikawa, I., Barrett, J.C., and Schlegel, R. 2001. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J. Virol. 75: 4467-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman T., Liu, X., Yuan, H., and Schlegel, R. 2003. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. 100: 8211-8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N., Johansson, S., Wu, S.Q., Bahram, F., Castell, A., Cetinkaya, C., Hydbring, P., Weidung, I., Nakayama, K., Nakayama, K.I., et al. 2003. The F-Box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 11: 1189-1200. [DOI] [PubMed] [Google Scholar]
- Wang J., Xie, L.Y., Allan, S., Beach, D., and Hannon, G.J. 1998. Myc activates telomerase. Genes & Dev. 12: 1769-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick M., Zubov, D., and Hagen, G. 1999. Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT). Gene 232: 97-106. [DOI] [PubMed] [Google Scholar]
- Wu K.J., Grandori, C., Amacker, M., Simon-Vermot, N., Polack, A., Lingner, J., and Dalla-Favera, R. 1999. Direct activation of TERT transcription by c-MYC. Nat. Genet. 21: 220-224. [DOI] [PubMed] [Google Scholar]
- Xu D.W., Wang, Q., Gruber, A., Bjorkholm, M., Chen, Z.G., Zaid, A., Selivanova, G., Peterson, C., Wiman, K.G., and Pisa, P. 2000. Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene 19: 5123-5133. [DOI] [PubMed] [Google Scholar]
- Zimmermann H., Degenkolbe, R., Bernard, H.U., and O'Connor, M.J. 1999. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 73: 6209-6219. [DOI] [PMC free article] [PubMed] [Google Scholar]