

Abstract
The recurrence of renal disease after renal transplantation is becoming one of the main causes of graft loss after kidney transplantation. This principally concerns some of the original diseases as the atypical hemolytic uremic syndrome (HUS), the membranoproliferative glomerulonephritis (MPGN), in particular the MPGN now called C3 glomerulopathy. Both this groups of renal diseases are characterized by congenital (genetic) or acquired (auto-antibodies) modifications of the alternative pathway of complement. These abnormalities often remain after transplantation because they are constitutional and poorly influenced by the immunosuppression. This fact justifies the high recurrence rate of these diseases. Early diagnosis of recurrence is essential for an optimal therapeutically approach, whenever possible. Patients affected by end stage renal disease due to C3 glomerulopathies or to atypical HUS, may be transplanted with extreme caution. Living donor donation from relatives is not recommended because members of the same family may be affected by the same gene mutation. Different therapeutically approaches have been attempted either for recurrence prevention and treatment. The most promising approach is represented by complement inhibitors. Eculizumab, a monoclonal antibody against C5 convertase is the most promising drug, even if to date is not known how long the therapy should be continued and which are the best dosing. These facts face the high costs of the treatment. Eculizumab resistant patients have been described. They could benefit by a C3 convertase inhibitor, but this class of drugs is by now the object of randomized controlled trials.
Keywords: Kidney disease recurrence, Complement dysregulation, Atypical hemolytic uremic syndrome, C3 glomerulopathies, Dense deposit disease, Plasma therapy, Eculizumab, C3 glomerulonephritis
Core tip: Complement cascade is an important pathway of several kidney diseases. A distinction should be made between kidney diseases with complement overactivation and those with complement dysregulation. The latter are related to congenital or acquired abnormalities of complement factors. These diseases are linked to constitutional abnormalities of the patients, have high recurrence rate after renal transplantation and represent an important cause of graft loss. Diagnosis and treatment are not easy to be made. Just in the last decade a growing knowledge in the field of genetic and biology allowed the complement inhibitors to be the first class drug in the treatment.
INTRODUCTION
The aim of this review is to highlight the relevance of recurrent diseases after kidney transplantation and, in particular, to discuss the frequency and severity of recurrence of two groups of renal diseases strictly related to each other: C3 glomerulopathy (C3G) and thrombotic microangiopathy (TMA).
Along with the improved control of acute rejections and infections, the recurrence of primary nephropathy has become the most important cause of graft loss principally for patients who have glomerulonephritis (GN) as the primary disease[1,2]. In some series, recurrence of the original disease was reported to be the principal cause of graft loss more than one year after transplantation[2] (Table 1). Some renal diseases have a higher risk of recurrence and recurrence-related graft loss. Hariharan et al[3] observed, in a total of 4913 renal transplants, that the greatest relative risk (RR) for graft failure was related to the recurrence of hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP) (5.36), membranoproliferative glomerulonephritis (MPGN) (2.77) and focal segmental glomerulosclerosis (FSGS) (2.25). Interestingly, HUS/TTP and many cases of MPGN were related to complement cascade dysregulation and were ascribed to genetic abnormalities or to acquired abnormalities of components of the complement system[4].
Table 1.
Causes of graft loss (living kidney transplantation)
|
< 1 yr |
> 1 yr |
|||
| Non identical | Identical | Non identical | Identical | |
| Acute rejection | 5 (41.7%) | 0 (0%) | ||
| CAN with/without CR | 2 (16.7%) | 0 (0%) | 16 (31.4%) | 5 (23.8%) |
| CNI nephrotoxicity | 0 (0%) | 0 (0%) | 2 (3.9%) | 1 (4.8%) |
| Recurrence of original disease | 1 (8.3%) | 0 (0%) | 10 (19.6%) | 6 (28.6%) |
| Death with functioning graft | 2 (16.7%) | 1 (50%) | 19 (37.3%) | 7 (33.3%) |
| Discontinuation of immunosuppressant | 0 (0%) | 1 (50%) | 4 (7.8%) | 1 (4.8%) |
| Non-compliance | 1 (8.3%) | 0 (0%) | 0 (0%) | 1 (4.8%) |
| Others | 1 (8.3%) | 0 (0%) | 0 (0%) | 0 (0%) |
| P | 0.2002 | 0.6158 | ||
CAN: Chronic allograft nephropathy; CR: Chronic rejection; CNI: Calcineurin inhibitor.
ROLE OF COMPLEMENT IN KIDNEY DISEASES
The complement proteins may be seen in the biopsies of all forms of GN, and all three activation pathways have been documented in different kidney diseases and may be activated by different triggers[5] (Figure 1). Indeed, the complement system is activated by three pathways (the alternative, classical and lectin pathways) that generate a common terminal pathway. The classical (CP) and lectin pathways (LP) are triggered by the recognition of pathogens or damaged cell surfaces by antibodies and recognition molecules[6]. Many glomerular diseases, such as membranous nephropathy, IgA nephropathy and lupus nephropathy, involve these pathways. The activation of the alternative pathway (AP) is relatively complex. The AP undergoes continuous low-grade activation in the fluid phase by spontaneous C3 hydrolysis that is responsible for the deposition of a low amount of C3b onto cell surfaces (Figure 2). Self-surfaces are protected from complement damage by several regulators that are either membrane-anchored or in the fluid phase. Perturbation of the balance between complement activators and regulators provides the basis for aHUS and MPGN/C3G[7].
Figure 1.
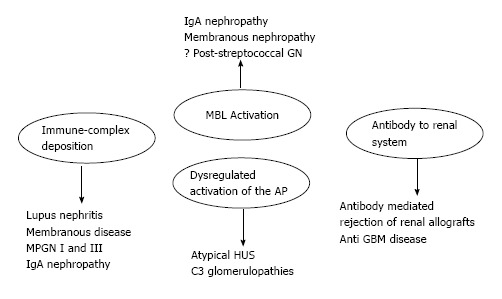
Mechanisms of complement activation in kidney disease. HUS: Hemolytic uremic syndrome; MBL: Mannose binding lectin; IgA: Immunoglobulin A; MPGN: Membranoproliferative glomerulonephritis; GBM: Glomerular basement membrane; AP: Alternative pathway.
Figure 2.
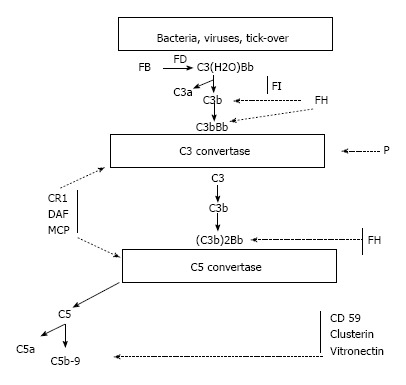
The C3 complement alternative pathway. C3(H2O) Bb: Alternative pathway initiation convertase; FB: Complement factor B; FD: Complement factor D; FH: Complement factor H; FI: Complement factor I; CR1: Complement receptor 1; DAF: Decay accelerating factor; MCP: Membrane cofactor protein; P: Properdin.
As mentioned above, the complement system is involved in the vast majority of kidney diseases. Two broad categories of kidney diseases should be distinguished. The first category is associated with complement over-activation and characterizes diseases such as lupus nephritis, membranous nephropathy, immune complex-associated MPGN and IgA nephropathy. The second category is related to complement dysregulation and characterizes diseases such as aHUS and C3G. In the former category, complement is activated by other factors, including immune-complex formation and deposition. After transplantation, the original disease may recur but is also more easily controlled by the immunosuppression needed to support the transplanted kidney. In the latter disease, complement activation may occur spontaneously and is often related to abnormalities of complement regulating factors. These nephropathies often recur after renal transplantation because the diseases are related to a constitutional and often genetically determined abnormality of the complement proteins. These abnormalities are not corrected either by the transplant itself or by the immunosuppressive therapy.
Tremendous advances are being made in our understanding of both aHUS/TMA and C3G. With the improvement of our understanding of genetics and biology, it has become increasingly clear that different disease mechanisms may cause the disease formerly called TTP/HUS. Furthermore, these mechanisms may deeply influence the recurrence rate after transplantation[8].
Similarly, the role of complement in C3G has been better defined[9], thus allowing us to move from a histologically based classification of the MPGNs to a new classification based on pathophysiology[10,11].
To date, the term aHUS applies to a heterogeneous group of diseases that have in common a TMA associated with some degree of renal failure. Frequently, aHUS patients have a complement abnormality (a genetic mutation or an autoantibody to complement factors) as the primary etiology. As a consequence, they are affected by a complement mediated TMA (Figure 3)[8].
Figure 3.
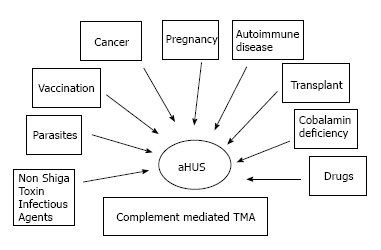
Heterogeneity of atypical hemolytic uremic syndrome. TMA: Thrombotic microangiopathy; HUS: Hemolytic uremic syndrome.
Similarly, after reclassification, the MPGNs are distinguished into immune-complex- mediated MPGNs and C3Gs. The latter have clear signs of C3 staining with little or no immunoglobulin deposition evident on renal biopsy. C3Gs are further divided into dense deposit diseases (DDD) and the recently recognized entity C3GN[11].
EPIDEMIOLOGY
GNs that occur in the transplanted kidney may be caused either by recurrent or de novo disease. In clinical and in the epidemiological studies is necessary to distinguish between these conditions. True recurrence occurs when: (1) post-transplant proteinuria or hematuria or elevated serum creatinine is found after transplantation; (2) biopsy-proven kidney disease is diagnosed in the native kidneys; or (3) the same disease is proven by biopsy in the transplanted kidney[12]. Challenges to the diagnosis of recurrent diseases are manifold. They include: (1) misdiagnosis or mislabeling of native kidney disease; (2) lack of a unified approach to using diagnostic tools for the diagnosis of recurrent disease; and (3) difficulties in differentiating recurrent disease from other causes of renal damage such as drug toxicity and chronic rejection[3,13].
There are still other potential biases occurring among registries dealing with recurrences of renal diseases mediated by complement dysregulation. For example Shiga toxin-related HUS combined with aHUS in many registries. Additionally, the vast majority of registries or networks report data by using the classification of MPGNs, which precedes the results of the consensus report on C3G[14] and the recent consensus report and reclassification of GNs[10,11].
Because of the above-mentioned factors, the data reported by different registries as the North American Pediatric Renal Transplant Collaborative Study (NAPRTCS), the Australia New Zealand Dialysis Transplant Data System (ANZDATA), the Renal Allograft Disease Registry (RADR) and the United States Renal Data System (USRDS) differ significantly in reporting the prevalence of recurrent GNs after transplantation[3,12,15-18]. A study by Shimmura et al[2] on 266 living kidney transplants clearly documents that recurrence of the original disease is the third leading cause of graft loss after one year from transplantation (Table 1). The aforementioned study by Hariharan[3] documents the highest RR for graft failure for HUS/TTP and MPGN.
Two other studies on pediatric patients[19,20] report high rates of recurrence for aHUS and type I and II MPGN according to the old classification, although there is a wide range of rates among the studies.
Series related to the early 2000s indicated that the risk of post-transplant recurrence for aHUS was 20% in pediatric patients and 50% in adult patients[21]. Recently, in 280 patients with aHUS screened for CFH, IF or MCP mutations, post-transplant aHUS recurrence was reported in 33%[22], 37%[23] and 60%[24], respectively.
Fewer data are available regarding the epidemiology of MPGN recurrence according to the new classification. Indeed, many registries are still using the old classification. According to these data, MPGN type I recurs in 20%-30% of patients, whereas MPGN type II recurs in 80%-100% of patients[25].
More recently, Kasiske et al[26], observing 1574 MP GNs in 140109 transplant patients recorded in the USRDS an observation that the true recurrence rate of MPGN increased over time, with the most frequent recurrences of GN between 1995 and 2003.
After the reclassification[10,11], the most interesting and recent data on C3G recurrence are those reported by Zand et al[27]. According to these data, the recurrence rate of C3GN is 66.7%, and graft failure occurs in 50% of patients with recurrence.
PATHOPHYSIOLOGY OF TMA AND ITS RECURRENCE
As mentioned above, the complement AP is constitutively active. After the generation of C3b, it binds either to either pathogens or the host cells. This necessitates the prompt and tight control of its activity. In turn, C3b may generate new C3 convertases (C3bBb) that act as an auto-amplifier by creating new C3b molecules. The same enzymes may also generate the C5 convertases that activate C5, the anaphylatoxins C5a and C5b and activate the membrane attack complex (MAC) C5b-C9[28]. In normal conditions, the AP may be spontaneously activated by the process called tick over. Cell surfaces are protected from auto-activation by several factors both in the fluid phase and anchored to the cell membranes.
The principal inhibiting factor is complement factor H (CFH), which acts both in the fluid phase and on cell surfaces. Factor H also act as a co-factor to complement factor I (CFI)[29-31]. Cell surfaces are also protected by at least 4 specific membrane regulators: (1) complement receptor 1 (CR1/CD 35); (2) membrane cofactor protein (MCP/CD46); (3) decay accelerating factor (DAF/CD55); and (4) protectin (CD59), which blocks MAC formation (Figure 4)[32,33].
Figure 4.
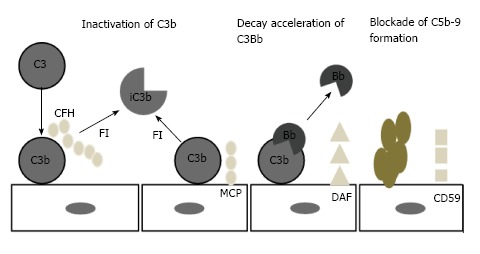
Regulation of complement on surfaces. CFH: Complement factor H; FI: Complement factor I; iC3b: Inactivated C3b; MCP: Membrane cofactor protein; DAF: Decay accelerating factor; CD59: MAC inhibitor protein.
The loss of this complex regulation results in complement activation, with consequent cell damage[34,35]. The role of complement dysregulation is increasingly recognized as the principal cause of TMAs. It may be caused by genetic mutations or by autoantibodies. Additionally, a triggering factor, often from the environment, is needed.
Figure 5 represents the whole spectrum of TMAs. In this figure, three different conditions are possible: (1) complement-driven TMA (i.e., aHUS), where there is an underlying complement defect; (2) complement-enhanced TMA, as in Shiga-toxin-producing Escherichia coli-HUS (STEC-HUS); and (3) TMA with coexisting disease (e.g., pregnancy, lupus). The most frequently mutated gene in aHUS is CFH. Mutations have been identified in 25% of sporadic cases and 40% of familial cases[6,36,37]. In addition to CFH, the CFH gene family includes five other complement factor H-related genes (CFHR1-5). Deletion, duplications and hybrid proteins occur[38]. As a result, the loss of cell surface protection may occur and aHUS may develop[36,39].
Figure 5.
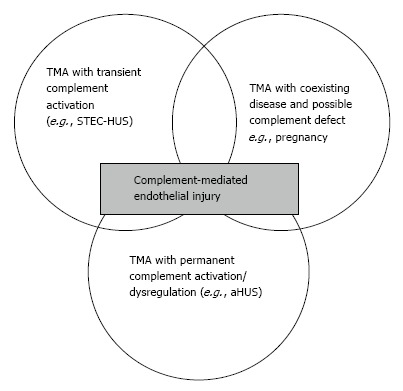
Spectrum of thrombotic microangiopathies. TMA: Thrombotic microangiopathies; STEC-HUS: Shiga toxin-producing Escherichia coli-hemolytic uremic syndrome.
The CFHR3-CFHR1 deletion is the most frequent deletion associated with aHUS. Recently a new role for the FHR protein was identified by Goicochea de Jorge et al[40]. According to these findings, FHRs could act as competitive antagonists to FH. MCP (CD46) mutations are found in 5%-9% of aHUS patients[36]. The protein is membrane-anchored and serves as a co-factor of FI. FI mutations lead to a lack of complement control accounting for 4%-8% of aHUS[36,37].
In addition to mutations in regulatory genes, gain of function mutations of effector proteins (C3 and factor B) has been reported. Factor B mutations are reported in < 4% cases of aHUS[41-43]. Factor B mutations enhance the C3bB formation. Gain of function mutations in C3 are reported in 2%-8% of aHUS[36,37]. These mutations impair MCP and confer resistance to cleavage by CFI[44,45].
Mutations in the thrombomodulin gene (THBD) have been reported in up to 5% of aHUS patients[36]. Few data have been reported on this mutation. It is probable that the THBD mutation may influence disease severity in association with other mutations[46].
Autoantibodies against different complement factors, independent of genetic mutations or in association with genetic abnormalities, may cause aHUS. Antibodies to anti-complement factor H (FHAA) account for 3%-8% of aHUS and are often found in association with FHR1 deficiency or deletion[36]. Recently, in India, an association of FHAA and aHUS was reported in approximately 60% of the pediatric population[47]. Auto antibodies to anti-factor I have been reported only in three patients[48]. Their role is not yet clear.
In many cases, the development of aHUS in patients who are affected by complement abnormalities needs a triggering factor. Among these well-known factors are pregnancy, drugs, and autoimmune disorders. Complement abnormalities were identified in 83% of patients with pregnancy associated aHUS[49]. CFH mutations have been reported in 4 patients with aHUS associated with ticlopidine[50]. Further 4% of patients affected by lupus disease documented an aHUS related to complement abnormalities[51,52]. Complement abnormalities strongly influence aHUS recurrence after renal transplantation. Indeed, aHUS recurrence was reported at rates from 20% to 50% in the era when genetic analysis of complement proteins was not available[53-56]. Recently, several studies have highlighted the risk of aHUS recurrence according to different genes abnormalities[53] (Table 2). Complement abnormalities have also been found in conditions that should not be affected by recurrence.
Table 2.
Risk of atypical hemolytic uremic syndrome recurrence according to the implicated genetic abnormality
| Gene | Protein location | Functional impact | Mutation frequency in aHUS (%) | Recurrence frequency after transplantation (%) |
| Mutation | ||||
| CFH | Plasma | Loss | 20-30 | 75-90 |
| CFI | Plasma | Loss | 2-12 | 45-80 |
| CFB | Plasma | Gain | 1-2 | 100 |
| C3 | Plasma | Gain | 5-10 | 40-70 |
| MCP | Membrane | Loss | 10-15 | 15-20 |
| THBD | Membrane | Loss | 5 | 1 case |
| Genetic polymorphism (frequency in control population) | ||||
| Homozygous CFHR1del (3%-8%) | Circulating | Undetermined | 14-23 (> 90% in patients with anti-CFH antibodies | NA |
aHUS: Atypical hemolytic uremic syndrome; C3: Complement C3; CFH: Complement factor H; CFI: Complement factor I; CFB: Complement factor B; MCP: Membrane cofactor protein; THBD: Thrombomodulin; CFHR1: Complement factor H receptor 1; NA: Not available.
STEC-HUS of the native kidneys should be protected from recurrence. However, two patients with a history of STEC-HUS were recently diagnosed with post-transplant recurrence[57]. Both patients were recognized to be affected by complement abnormalities; one had a heterozygous CFI mutation, and the other had a heterozygous MCP mutation.
Patients affected by MCP mutations rarely have aHUS recurrence after transplantation[58]. Recently, however, transplant failures due to aHUS recurrence have been observed in patients affected by MCP mutations[59]. Almost all of these patients were affected by combined MCP and CFH or CFI mutations.
Patients affected by CFH mutations are at a high risk of recurrence. In two French case series, recurrences were observed in 80% of children[22] and 75% of adults[60]. The graft failure rates in the case of recurrence are approximately 86%.
Interestingly, the location of CFH mutation impacts the recurrence risk[61]. Indeed, mutations involving the C-terminal domain of CFH confer higher risk and have a worse prognosis[49]. This finding is consistent with the critical role of the CFH C-terminal domain in binding to the endothelium and exerting the protective role of the endothelial cells[62].
The majority of FHAA is directed against the C-terminal domain of factor H. As a consequence, a higher risk of recurrence should be expected due to FHAA. However, this does not seem to be the case, because aHUS recurrence due to FHAA is uncommon[63].
Nonetheless, the recurrence risk due to FHAA is not easily understood because 40% of patients with FHAA are also affected by mutations in the complement genes[64]. Additionally, a reduction of FHAA is achievable with the immunosuppressant therapy, thereby reducing the risk of aHUS recurrence.
In summary, the risk of recurrence is 4 times higher in patients with mutations in the CFH gene or carriers of the hybrid gene between CFH/CFHR1. In a recent study by Le Quintrec[65], patients with the hybrid gene lost their grafts due to early recurrence.
The relevance of CFI mutations on aHUS recurrence has discordant results and interpretations. The first studies to CFI mutations reported a high recurrence rate and graft loss[10,31,53,58,66,67]. A study by Bienaimé et al[68] in 2010 reported that patients with CFI mutations do not seem to carry a higher risk of recurrence. These data were more recently confirmed by the study mentioned above by Le Quintrec et al[65].
MCP mutations rarely affect aHUS recurrence because the endothelial cell surfaces of the transplanted kidney normally express MCP. Only three recurrences have been reported in the literature[69,70]. In these patients, recurrence might be ascribed to combined complement gene mutations[59] or microchimerism from the recipient endothelial cells[70].
Data on the role of THBD are scarce. aHUS recurrence due to THBD mutations should not occur because the molecule is membrane-anchored as MCP. Additionally, a small proportion of THBD is present in soluble form. Nonetheless, sporadic cases of recurrence due to THBD have been reported[71,72]. In one patient the recurrence occurred early post-transplantation during the ischemia-reperfusion phase. During this phase, the soluble form of THBD might be not adequate to protect from recurrence.
Patients affected by gain of function mutations (CFB, C3) are also exposed to the risk of recurrence. To date, four patient carriers of the CFB mutation have been reported to have aHUS recurrences and consequent graft loss[73,74]. Data on recurrence in patients affected by C3 mutations are discordant. Le Quintrec[65] reported a high recurrence rate, with 4 recurrences in 5 grafts. Previously, Noris et al[75] reported only two recurrences in 7 transplanted patients. In an attempt to explain the difference, Zuber et al[53] speculated that for some patients, the intra-graft production of normal C3 might occur and might be protective.
Several environmental triggers might act to damage the graft endothelium and to facilitate aHUS recurrence on already damaged cells in patients with genetic abnormalities.
Anti-HLA antibodies[76], ischemia-reperfusion events[77], immunosuppressant drugs[78] and viral infections[79], either isolated or in association, might play a relevant role and favor aHUS recurrence in genetically predisposed patients.
Le Quintrec et al[65] attempted to identify the risk factors for aHUS recurrence. Low C3 levels and the presence of a mutation were significant in the univariate analysis. In a multivariate analysis of mutations, a mammalian target of rapamycin (mTOR) inhibitor regimen and recipient age were significantly associated with increased aHUS recurrence rates.
PATHOPHYSIOLOGY AND RECURRENCE OF C3G
After the reclassification of MPGNs, as mentioned above[10,14], C3Gs included the MPGNs caused by complement dysregulation rather than the MPGN immune-complex-related disorders[80]. As a consequence, C3Gs include the GNs for which immunofluorescence microscopy is C3-positive and immunoglobulin-negative.
C3Gs may be sub-divided into DDD and C3GN based on electron microscopy, even if, in some cases the distinction is challenging[14,81]. Recently, advances toward an improved understanding of the characteristics of C3 deposits have been made through proteomic analysis and laser microdissection (LMD)[82]. Laser dissection and mass spectrometry of glomeruli from patients with C3G documented an accumulation of the AP and the terminal complement complex proteins, thus confirming that C3G results from abnormalities of the AP, which lead to glomerular damage[81].
The pathophysiology of AP pathway activation in C3GN and DDD is very similar, with fluid phase dysregulation due to gene mutations or autoantibodies occurring in both disorders. Indeed, as for aHUS, the complement abnormalities in C3Gs may occur on a genetic basis or as acquired factors as autoantibodies.
The most common acquired complement defect is represented by the presence of an antibody called the C3 nephritic factor (C3NeF), which blocks CFH-mediated decay and stabilizes C3 convertase[81,83]. In particular, C3NeF binds to C3 convertase and inhibits the action of factor H, CR1 and DAF, blocking the dissociation of the convertase. C3NeF enhances C3 convertase activity 10 fold[9,84,85]. The frequency of C3NeF is high in C3G, ranging from 50% to 80% of patients[83]. C3NeF may also be associated with genetic mutations. Recently, other auto antibodies have been found in C3Gs. These auto antibodies are directed against C3 convertase, factor B[86] or anti-factor H[87,88]. AP dysregulation in DDD is more frequently autoantibody-induced with respect to TMA. Genetic abnormalities also have been encountered. Few patients have been identified with genetic mutations of factor I, MCP, C3, factor B and factor H[83,89]. In an extensive study by Servais et al[83], only 5.3% of the patients affected by C3GN had CFI mutations, and 1.8% had MCP mutations.
In 2010, Martinez-Barricarte et al[90] identified a mutant C3 protein resistant to factor H inactivation in a patient affected by DDD. More recently, a different C3 mutation has been identified.
Mutations in factor H have been reported more frequently among patients affected by C3Gs. Mutations may result in a defective protein or a complete lack of protein H. Mutations may occur in a homozygous or heterozygous manner[91,92] and may be associated with C3NeF, thus documenting the association of different risk factors.
In recent years genetic mutations of the CFHR gene cluster have been reported among patients with C3G[93]. CFHR family gene mutations[94], deletions[95], duplication[96] and hybrid genes[97] have been reported in patients with C3Gs either in isolated patients or family groups.
For example, Gale et al[96] reported two Cypriot families whose members were affected by a CFHR5 mutation. The protein produced by the mutated gene was poorly effective in binding to C3b on cell surfaces and thus led to the deregulation of the fluid phase of the AP. The disease was called CFHR5 nephropathy.
Recently, Malik et al[98] reported patients from the same family affected by C3G due to abnormal copies in the CFHR3 and CFHR1 loci. The finding of familial cases of C3G highlights the genetic origin of several C3Gs and the related complement AP dysregulation.
In summary the specific cause of C3G is inadequate regulation of the complement system. The causes of complement dysregulation may be divided into genetic and acquired factors. Among the former are changes in many of the complement genes: Among the latter are specific antibodies called C3 nephritic factors or C3NeFs that impair normal regulation of the complement system. It appears that patients with DDD are more likely to have C3NeFs, while patients with C3GN are more likely to have abnormalities in a group of proteins called the “Complement Factor H-Related” proteins.
Additionally, genetic defects may represent the basis of either C3G or aHUS (Table 3). Indeed, in recent years, a large number of genetic studies have established a strong association between the factor H-related proteins and different diseases involving complement dysregulation. This association, together with the recent functional data on factor H-related proteins such as FH competitors and complement deregulators, has gained the attention of the complement scientific community[99].
Table 3.
Overview of mutations in complement factor H related protein genes
| Genetic defect | Phenotypical expression |
| Duplication in the CFHR5 gene | C3 glomerulopathy (CFHR5 nephropathy) |
| Duplication in the CFHR1 gene | C3 glomerulopathy |
| Hybrid CFHR3/CFHR1 gene | C3 glomerulopathy |
| Hybrid CFHR2/CFHR5 gene | C3 glomerulopathy |
| Hybrid CFH/CFHR1 gene | aHUS |
| Hybrid CFH/CFHR3 gene | aHUS |
CFHR: Complement factor H related; aHUS: Atypical hemolytic uremic syndrome.
From the pathophysiological point of view, many cases of C3Gs and TMA are associated with defective control of the AP. The inevitable questions are whether C3G and TMA are the other sides of the same coin and which factors determine whether a patient develops one disease instead of the other[5].
Animal models highlight that C3G may be the consequence of prevalent dysregulation of fluid phase complement activation, whereas TMA is principally related to complement activation on the capillary wall. The same studies determined that an absolute deficiency of factor H favors fluid phase complement activation and C3G, whereas the absence or abnormality of the binding region of factor H favors TMA[100]. It has also been hypothesized that CFH and CFH/CFHR mutations induce aHUS to inhibit the CFH binding to most cell surfaces, whereas C3G-associated mutant CFHRs do not inhibit CFH binding to endothelial cell surfaces[6].
Concerning C3G recurrence after transplantation, the finding of familial cases of C3GN highlights the genetic origin and the related complement AP dysregulation of the vast majority of C3GN. These data form the basis of its recurrence after transplantation. However, fewer data are available on C3G recurrence compared to TMA. Indeed, C3G is a rare disease and principally, its pathogenesis and its complement-dependent nature have been recognized only recently. More data are available on DDD recurrence. Indeed, this disease was identified a long time ago based on its characteristic microscopic aspects. This finding occurred long before our understanding of its pathogenesis. In a retrospective analysis of 75 children, the 5-year graft survival rate was only 50%[101]. Almost all adult patients had recurrences after transplantation and up to 25% lost their graft[19].
In a large, retrospective cohort study of 80 adults and children affected by C3G, Medjeral-Thomas et al[102] reported a histological recurrence following renal transplantation in all 6 DDD patients. Recurrence was associated with graft loss in 50% of patients. Similarly, four of seven C3GN patients transplanted had histological recurrences. Graft loss occurred in 3 patients. A UNOS review reported a 10-year graft survival rate of 57.5% for patients affected by DDD recurrence[103]. In different studies, the reported rate of DDD recurrence is variable ranging from 18% to 100%[104,105].
Considering only those patients whose diagnosis was made by renal biopsy, the recurrence rate was over 70%[106,107]. Disease recurrence may occur suddenly after transplantation. However, cases of recurrence many years later are also described[107]. The risk factors for recurrence and graft loss for DDD are not well defined. No relationship with preTx disease presentation or C3 serum levels has been found. Additionally, the C3NeF levels do not correlate with the risk of recurrence[108]. The presence of heavy proteinuria seems to be the only risk factor related to recurrence.
The different genetic variants responsible of C3GN have been already described. Overall, C3GN recurs in two-thirds of transplanted patients and graft loss is common[27,81,83]. Histologically, it recurs with a membranoproliferative pattern. Risk factors for recurrence are still now debated. According to some studies[25], they include the severity of histological lesions in the native kidneys, HLA-B8 DR3, living related donors and previous graft loss for recurrence[109]. To date, our understanding of C3GN recurrence is only based on case reports. Furthermore, the broadest study on C3GN outcomes after recurrence by Zand et al[27] was unable to find any risk factor for recurrence. The multiple defects in complement regulatory proteins causing C3GN likely impair the establishment of any well-defined recurrence risk.
Eleven patients affected by CFHR5 nephropathy were successfully transplanted[110]; however protocol biopsies have documented recurrence[111]. The recurrence may be early after transplantation and demonstrates that renal-derived CFHR5 protein cannot prevent the development of CFHR5 graft nephropathy. Very recently Wong et al[112] described a high recurrence rate in 5 patients affected by hybrid CFHR3 1 gene-associated C3GN.
DIAGNOSIS OF RECURRENCE
Diagnosis of recurrence may be easy if the clinical history of the recipient is known and the diagnosis of C3G/aHUS of the native kidneys has been made after an etiological workup and a kidney biopsy. Unfortunately, the clinical history of the recipient and a renal biopsy of the native kidneys are often not available.
In such patients, if the graft is not doing well, a renal biopsy should be promptly performed and examined by light microscopy, immunofluorescence and electron microscopy. When the diagnosis of C3G/aHUS is suspected, a complete workup should be undertaken. The diagnostic approach should include a comprehensive biochemical, genetic and pathologic analysis of the complement AP. This approach should include complement factors and complement regulatory protein levels, measurement of MCP on peripheral blood leukocytes as well as screening for anti-CFH antibodies and C3NeFs. Additionally, the genetic investigation should include mutation screening of CFH, CFI, MCP, C3 and CFB. The screening requires an extensive sequencing of all coding exons. Additionally, a study of recombination in the CFHR region should be made[113]. The genetic studies are not easy to perform because the spectrum of genes currently known to be involved is rapidly expanding[114]. Nonetheless, such studies are vital because the importance of genetic mutation screening to determine the outcome of retransplantation following a failed kidney allograft from a patient with recurrent aHUS has recently been documented[115]. In other words, not all mutations have the similar detrimental effects. The absence of a more severe genotype could facilitate the successful treatment of the recurrence.
RECCOMENDATIONS, PREVENTION ND TREATMENT OF POST-TRANSPLANT aHUS AND C3Gs RECURRENCE
The vast majority of data are available for aHUS because C3G has been only recently defined and data on prevention and treatment rely more on case reports than on evidence-based medicine.
Recommendations
Patients with aHUS as a primary disease and patients with suspected aHUS and with STEC-HUS should be screened for all complement factors and regulating proteins. Additionally, a genotyping for CFH, CFHR, CFI, MCP CFB and C3 should be performed[114].
Patients with a suspected diagnosis of C3G should also be screened for C3NeF and for other autoantibodies that are known to be involved in this disease.
Living donor renal transplantation, even in the eculizumab era, is not indicated for patients with mutations in CFH, CFI, C3 and CFB. In patients with aHUS due to a mutation in MCP, donation may be safe after exclusion of other mutations often associated with MCP mutation. However, increased evidence for a polygenic pattern for aHUS and C3G and the still-unknown polymorphisms should always consider a living donation with extreme caution[53].
Patients affected by aHUS but with no identified mutations should be recommended to proceed with transplantation combined with intensive plasma exchange (PE)[21].
Prevention
To date, there is limited evidence for preventing C3G recurrence after transplantation. The more validated experience refers to the use of eculizumab to prevent aHUS recurrence[80]. Whether these strategies may be recommended to prevent C3G will be subject to future research.
More data are available concerning aHUS prevention. The avoidance of any possible endothelial insult has been highlighted[113]. Post-transplant conditions that may cause endothelial insult include ischemia-reperfusion injury, infections, and immunosuppressive drugs. All of these factors could act as triggers to activate the AP in predisposed patients.
An association between calcineurin inhibitors (CNIs) and aHUS recurrence has been hypothesized[7]. Other studies do not confirm this association and note that mTOR inhibitors are frequently used to avoid CNIs and may, per se, induce aHUS[116,117]. PE has been used to prevent aHUS recurrence[53]. However, PE has several drawbacks.
First, in some cases, PE fails to prevent aHUS[118]. Second, there is a risk of recurrence when PE is interrupted. Third, the evidence of subclinical recurrent aHUS in patients still under treatment indicates that in some cases, PE does not control complement activation[118].
Pre-transplant rituximab administration has been effective for patients with anti-CFH antibodies[40,119,120]. In these patients, the association of PE may improve the treatment efficacy. The anti-C5 monoclonal antibody (eculizumab) has been used to prevent post-transplant aHUS in several patients. Among the reported patients, nine had either CFH mutations or a CFH/CFHR1 hybrid gene. Another patient had a C3 mutation[118,121-124]. All of these patients had a complement genetic abnormality with a risk of aHUS recurrence greater than 80%. Only one patient lost the graft due to an arterial thrombosis. All other patients had a successful recurrence-free post-transplant course, even if, to our knowledge, they are still undergoing eculizumab treatment[116].
Treatment
In a retrospective study, Zand et al[27] reviewed the outcomes of 14 patients diagnosed with a C3G recurrence after transplantation. Ten patients did not receive any additional treatment. Three patients received rituximab treatment, but the overall outcome was poor.
Another study reported the beneficial effect of plasma infusions (PI) in patients with a genetic mutation in factor H[125]. Case reports documented the efficacy of eculizumab in patients with DDD recurrence[106] and patients with C3GN recurrence[126], although the patient with C3GN repeat allograft biopsies showed progression of the disease. Other studies[127,128] reported eculizumab efficacy for the treatment of recurrent DDD and C3GN. A randomized clinical trial to evaluate the efficacy of eculizumab in patients with C3G is ongoing[129].
An exciting new approach to C3G treatment is the soluble complement receptor 1 (CR1), which promotes the breakdown of active C3b. The infusion of soluble CR1 was reported to improve C3 and serum MAC levels in a patient with DDD recurrence[97].
Before the eculizumab era, patients affected by aHUS recurrence were extensively treated with PE. In the French survey, the outcomes of aHUS recurrence were not different among patients, regardless of treatment with PE[116]. PE combined with belatacept was effective for one patient, as reported by Midvedt et al[130]. Eculizumab has been reported to be effective in a recent study by Matar et al[131], regardless of concomitant PE treatment. The largest experience in treating recurrent aHUS with eculizumab was reported by Zuber et al[118].
According to their findings, eculizumab was efficient in treating aHUS recurrence after transplantation. The treatment should be started as early as possible, and the treatment tolerance is excellent. Interestingly, two patients who received a single dose regimen experienced a delayed relapse[132]. Two attempts of eculizumab discontinuation were followed by new relapses[133].
Overall, these experiences suggest that a high risk of relapse may persist after a first recurrence. This fact suggests caution in withdrawing eculizumab in this setting.
Additionally, active HUS lesions have been observed in patients with a documented C5 blockade receiving eculizumab regularly[118]. Whether a C3 convertase blocker could more efficiently treat these patients is currently unknown.
Two additional studies have documented eculizumab efficacy in plasma therapy resistant or dependent patients with recurrent aHUS[134,135]. More than 80% of the patients achieved TMA-free status.
The efficacy of eculizumab has changed our approach to aHUS and C3G recurrence after transplantation. However several questions remain to be answered, including: (1) Do complement investigations impact therapeutic decisions? (2) For how long should patients with recurrent aHUS or C3G be given eculizumab? and (3) Does eculizumab change our indications for renal transplant for patients on dialysis for aHUS or C3G?[136].
It is crucial to explore the most appropriate dose, dosing intervals and duration of treatment to reduce the enormous financial burden of eculizumab therapy[137].
CONCLUSION
Recurrence of primary disease after renal transplantation is currently one of the most important causes of graft loss.
Recurrence is principally common for those diseases, often glomerulonephritis, caused by constitutional abnormalities of the patient, not kidney related. Among these abnormalities are diseases caused by complement dysregulation such as aHUS and C3Gs. To date, aHUS and C3Gs often represent a contraindication to renal transplantation due to the frequency and severity of recurrent disease. The clinical use of the anti-C5 inhibitor, eculizumab, seems to overcome the limitations to kidney transplantation for selected patients. However, we have highlighted the drawbacks of this therapy, principally represented by the high costs of lifelong therapy. The main perspectives in the field of renal transplantation of avoiding or treating recurrences are either diagnostic and therapeutic. An improved understanding of genetics and biology will allow an improved knowledge of gene mutations and the possibility of opening new methods in the field of living donor transplantation; Future therapeutic approaches are represented by the availability of purified deficient gene products and the availability of C3 convertase inhibitors. In addition to CR1 as mentioned above, the current targets of research include the compstatin analog Cp40, which can block C3b[138]. Similarly, another research target is a monoclonal antibody able to inhibit the C3 convertase induced by C3NeF[139].
Footnotes
Conflict-of-interest statement: No conflict of interest for both authors as in the enclosed declaration.
Manuscript source: Invited manuscript
Specialty type: Transplantation
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: September 13, 2016
First decision: October 21, 2016
Article in press: November 16, 2016
P- Reviewer: Artunc F, Duan SB, Sun CK S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ. Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med. 2002;347:103–109. doi: 10.1056/NEJMoa013036. [DOI] [PubMed] [Google Scholar]
- 2.Shimmura H, Tanabe K, Ishida H, Miyamoto N, Tokumoto T, Ishikawa N, Toma H. Long-term results of living kidney transplantation from HLA-identical sibling donors under calcineurin inhibitor immunosuppression. Int J Urol. 2006;13:502–508. doi: 10.1111/j.1442-2042.2006.01350.x. [DOI] [PubMed] [Google Scholar]
- 3.Hariharan S, Adams MB, Brennan DC, Davis CL, First MR, Johnson CP, Ouseph R, Peddi VR, Pelz CJ, Roza AM, et al. Recurrent and de novo glomerular disease after renal transplantation: a report from Renal Allograft Disease Registry (RADR) Transplantation. 1999;68:635–641. doi: 10.1097/00007890-199909150-00007. [DOI] [PubMed] [Google Scholar]
- 4.McCaughan JA, O’Rourke DM, Courtney AE. The complement cascade in kidney disease: from sideline to center stage. Am J Kidney Dis. 2013;62:604–614. doi: 10.1053/j.ajkd.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 5.Thurman JM. Complement in kidney disease: core curriculum 2015. Am J Kidney Dis. 2015;65:156–168. doi: 10.1053/j.ajkd.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noris M, Remuzzi G. Glomerular Diseases Dependent on Complement Activation, Including Atypical Hemolytic Uremic Syndrome, Membranoproliferative Glomerulonephritis, and C3 Glomerulopathy: Core Curriculum 2015. Am J Kidney Dis. 2015;66:359–375. doi: 10.1053/j.ajkd.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, Kavanagh D, Noris M, Pickering M, Sanchez-Corral P, et al. Atypical aHUS: State of the art. Mol Immunol. 2015;67:31–42. doi: 10.1016/j.molimm.2015.03.246. [DOI] [PubMed] [Google Scholar]
- 9.Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, Fremeaux-Bacchi V, Nester C, de Córdoba SR, Noris M, et al. The role of complement in C3 glomerulopathy. Mol Immunol. 2015;67:21–30. doi: 10.1016/j.molimm.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Sethi S, Haas M, Markowitz GS, D’Agati VD, Rennke HG, Jennette JC, Bajema IM, Alpers CE, Chang A, Cornell LD, et al. Mayo Clinic/Renal Pathology Society Consensus Report on Pathologic Classification, Diagnosis, and Reporting of GN. J Am Soc Nephrol. 2016;27:1278–1287. doi: 10.1681/ASN.2015060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salvadori M, Rosso G. Reclassification of membranoproliferative glomerulonephritis: Identification of a new GN: C3GN. World J Nephrol. 2016;5:308–320. doi: 10.5527/wjn.v5.i4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Golgert WA, Appel GB, Hariharan S. Recurrent glomerulonephritis after renal transplantation: an unsolved problem. Clin J Am Soc Nephrol. 2008;3:800–807. doi: 10.2215/CJN.04050907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, McIntosh MJ, Stablein D. Improved graft survival after renal transplantation in the United States, 1988 to 1996. N Engl J Med. 2000;342:605–612. doi: 10.1056/NEJM200003023420901. [DOI] [PubMed] [Google Scholar]
- 14.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.North American Pediatric Renal Trials and Collaborative Studies 2006 Annual Report, 2006. Available from: https://www.emmes.com/study/ped.
- 16.United States Renal Data Syspem: Annual Data Report, 2007. Available from: http://www.usrds.org/adr.htm.
- 17.Australia and New Zealand Dialysis and Transplant Registry: the 29th Annual Report, 2006. Available from: http://www.anzdata.org.au.
- 18.Hariharan S, Peddi VR, Savin VJ, Johnson CP, First MR, Roza AM, Adams MB. Recurrent and de novo renal diseases after renal transplantation: a report from the renal allograft disease registry. Am J Kidney Dis. 1998;31:928–931. doi: 10.1053/ajkd.1998.v31.pm9631835. [DOI] [PubMed] [Google Scholar]
- 19.Cochat P, Fargue S, Mestrallet G, Jungraithmayr T, Koch-Nogueira P, Ranchin B, Zimmerhackl LB. Disease recurrence in paediatric renal transplantation. Pediatr Nephrol. 2009;24:2097–2108. doi: 10.1007/s00467-009-1137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Stralen KJ, Verrina E, Belingheri M, Dudley J, Dusek J, Grenda R, Macher MA, Puretic Z, Rubic J, Rudaitis S, et al. Impact of graft loss among kidney diseases with a high risk of post-transplant recurrence in the paediatric population. Nephrol Dial Transplant. 2013;28:1031–1038. doi: 10.1093/ndt/gfs549. [DOI] [PubMed] [Google Scholar]
- 21.Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transplant. 2008;12:619–629. doi: 10.1111/j.1399-3046.2008.00910.x. [DOI] [PubMed] [Google Scholar]
- 22.Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudailliez B, Bouissou F, Deschenes G, Gie S, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392–2400. doi: 10.1681/ASN.2006080811. [DOI] [PubMed] [Google Scholar]
- 23.Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, Goodship TH, Remuzzi G. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1:88–99. doi: 10.2215/CJN.00050505. [DOI] [PubMed] [Google Scholar]
- 25.Andresdottir MB, Assmann KJ, Hoitsma AJ, Koene RA, Wetzels JF. Recurrence of type I membranoproliferative glomerulonephritis after renal transplantation: analysis of the incidence, risk factors, and impact on graft survival. Transplantation. 1997;63:1628–1633. doi: 10.1097/00007890-199706150-00016. [DOI] [PubMed] [Google Scholar]
- 26.Kasiske BL, Snyder JJ, Peng Yi, Connaire JJ. Changes in outcomes for kidney transplant patients with type I membranoproliferative glomerulonephritis. Am J Transplant. 2006;6 S2:654. [Google Scholar]
- 27.Zand L, Lorenz EC, Cosio FG, Fervenza FC, Nasr SH, Gandhi MJ, Smith RJ, Sethi S. Clinical findings, pathology, and outcomes of C3GN after kidney transplantation. J Am Soc Nephrol. 2014;25:1110–1117. doi: 10.1681/ASN.2013070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teoh CW, Riedl M, Licht C. The alternative pathway of complement and the thrombotic microangiopathies. Transfus Apher Sci. 2016;54:220–231. doi: 10.1016/j.transci.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt CQ, Herbert AP, Hocking HG, Uhrín D, Barlow PN. Translational mini-review series on complement factor H: structural and functional correlations for factor H. Clin Exp Immunol. 2008;151:14–24. doi: 10.1111/j.1365-2249.2007.03553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makou E, Herbert AP, Barlow PN. Functional anatomy of complement factor H. Biochemistry. 2013;52:3949–3962. doi: 10.1021/bi4003452. [DOI] [PubMed] [Google Scholar]
- 31.Bettoni S, Bresin E, Remuzzi G, Noris M, Donadelli R. Insights into the Effects of Complement Factor H on the Assembly and Decay of the Alternative Pathway C3 Proconvertase and C3 Convertase. J Biol Chem. 2016;291:8214–8230. doi: 10.1074/jbc.M115.693119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 33.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122:282–292. doi: 10.1182/blood-2013-03-489245. [DOI] [PubMed] [Google Scholar]
- 35.Camous L, Roumenina L, Bigot S, Brachemi S, Frémeaux-Bacchi V, Lesavre P, Halbwachs-Mecarelli L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 36.Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, Moulin B, Servais A, Provot F, Rostaing L, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554–562. doi: 10.2215/CJN.04760512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant. 2011;26:739–741. doi: 10.1093/ndt/gfq658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3:e41. doi: 10.1371/journal.pgen.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, Bresin E, Remuzzi G, Rodriguez de Cordoba S, Noris M. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol. 2015;26:209–219. doi: 10.1681/ASN.2013121339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, Hakobyan S, Morgan BP, Harris CL, Pickering MC, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci USA. 2013;110:4685–4690. doi: 10.1073/pnas.1219260110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Vriese AS, Sethi S, Van Praet J, Nath KA, Fervenza FC. Kidney Disease Caused by Dysregulation of the Complement Alternative Pathway: An Etiologic Approach. J Am Soc Nephrol. 2015;26:2917–2929. doi: 10.1681/ASN.2015020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Funato M, Uemura O, Ushijima K, Ohnishi H, Orii K, Kato Z, Yamakawa S, Nagai T, Ohara O, Kaneko H, et al. A complement factor B mutation in a large kindred with atypical hemolytic uremic syndrome. J Clin Immunol. 2014;34:691–695. doi: 10.1007/s10875-014-0058-8. [DOI] [PubMed] [Google Scholar]
- 43.Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, Cayla M, Tabarin F, Jablonski M, Hue C, et al. Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol. 2014;25:2053–2065. doi: 10.1681/ASN.2013070796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frémeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, Moghal N, Kaplan BS, Weiss RA, Lhotta K, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–4952. doi: 10.1182/blood-2008-01-133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lhotta K, Janecke AR, Scheiring J, Petzlberger B, Giner T, Fally V, Würzner R, Zimmerhackl LB, Mayer G, Fremeaux-Bacchi V. A large family with a gain-of-function mutation of complement C3 predisposing to atypical hemolytic uremic syndrome, microhematuria, hypertension and chronic renal failure. Clin J Am Soc Nephrol. 2009;4:1356–1362. doi: 10.2215/CJN.06281208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sánchez Chinchilla D, Pinto S, Hoppe B, Adragna M, Lopez L, Justa Roldan ML, Peña A, Lopez Trascasa M, Sánchez-Corral P, Rodríguez de Córdoba S. Complement mutations in diacylglycerol kinase-ε-associated atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2014;9:1611–1619. doi: 10.2215/CJN.01640214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, Saini H, Kotresh ST, Ali U, Bhatia D, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85:1151–1160. doi: 10.1038/ki.2013.373. [DOI] [PubMed] [Google Scholar]
- 48.Kavanagh D, Pappworth IY, Anderson H, Hayes CM, Moore I, Hunze EM, Bennaceur K, Roversi P, Lea S, Strain L, et al. Factor I autoantibodies in patients with atypical hemolytic uremic syndrome: disease-associated or an epiphenomenon? Clin J Am Soc Nephrol. 2012;7:417–426. doi: 10.2215/CJN.05750611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fakhouri F, Roumenina L, Provot F, Sallée M, Caillard S, Couzi L, Essig M, Ribes D, Dragon-Durey MA, Bridoux F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21:859–867. doi: 10.1681/ASN.2009070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chapin J, Eyler S, Smith R, Tsai HM, Laurence J. Complement factor H mutations are present in ADAMTS13-deficient, ticlopidine-associated thrombotic microangiopathies. Blood. 2013;121:4012–4013. doi: 10.1182/blood-2013-03-487694. [DOI] [PubMed] [Google Scholar]
- 51.Song D, Wu LH, Wang FM, Yang XW, Zhu D, Chen M, Yu F, Liu G, Zhao MH. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther. 2013;15:R12. doi: 10.1186/ar4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen MH, Chen MH, Chen WS, Mu-Hsin Chang P, Lee HT, Lin HY, Huang DF. Thrombotic microangiopathy in systemic lupus erythematosus: a cohort study in North Taiwan. Rheumatology (Oxford) 2011;50:768–775. doi: 10.1093/rheumatology/keq311. [DOI] [PubMed] [Google Scholar]
- 53.Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C. New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol. 2011;7:23–35. doi: 10.1038/nrneph.2010.155. [DOI] [PubMed] [Google Scholar]
- 54.Loirat C, Niaudet P. The risk of recurrence of hemolytic uremic syndrome after renal transplantation in children. Pediatr Nephrol. 2003;18:1095–1101. doi: 10.1007/s00467-003-1289-8. [DOI] [PubMed] [Google Scholar]
- 55.Artz MA, Steenbergen EJ, Hoitsma AJ, Monnens LA, Wetzels JF. Renal transplantation in patients with hemolytic uremic syndrome: high rate of recurrence and increased incidence of acute rejections. Transplantation. 2003;76:821–826. doi: 10.1097/01.TP.0000085083.74065.1B. [DOI] [PubMed] [Google Scholar]
- 56.Quan A, Sullivan EK, Alexander SR. Recurrence of hemolytic uremic syndrome after renal transplantation in children: a report of the North American Pediatric Renal Transplant Cooperative Study. Transplantation. 2001;72:742–745. doi: 10.1097/00007890-200108270-00033. [DOI] [PubMed] [Google Scholar]
- 57.Alberti M, Valoti E, Piras R, Bresin E, Galbusera M, Tripodo C, Thaiss F, Remuzzi G, Noris M. Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am J Transplant. 2013;13:2201–2206. doi: 10.1111/ajt.12297. [DOI] [PubMed] [Google Scholar]
- 58.Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant. 2010;10:1517–1523. doi: 10.1111/j.1600-6143.2010.03156.x. [DOI] [PubMed] [Google Scholar]
- 59.Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, Pinto S, Goodship TH, Alberti M, Ribes D, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24:475–486. doi: 10.1681/ASN.2012090884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jablonski M. Kidney transplant outcome in atypical hemolytic uremic syndrome: The French experience. Am Soc Nephrol. 2009 [Google Scholar]
- 61.Saland JM, Ruggenenti P, Remuzzi G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2009;20:940–949. doi: 10.1681/ASN.2008080906. [DOI] [PubMed] [Google Scholar]
- 62.Pickering MC, de Jorge EG, Martinez-Barricarte R, Recalde S, Garcia-Layana A, Rose KL, Moss J, Walport MJ, Cook HT, de Córdoba SR, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249–1256. doi: 10.1084/jem.20070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le Quintrec M, Zuber J, Noel LH, Thervet E, Frémeaux-Bacchi V, Niaudet P, Fridman WH, Legendre C, Dragon-Durey MA. Anti-Factor H autoantibodies in a fifth renal transplant recipient with atypical hemolytic and uremic syndrome. Am J Transplant. 2009;9:1223–1229. doi: 10.1111/j.1600-6143.2009.02586.x. [DOI] [PubMed] [Google Scholar]
- 64.Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, Schmidt CQ, Staniforth SJ, Holmes LV, Ward R, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379–387. doi: 10.1182/blood-2009-05-221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, Chatelet V, Mousson C, Mourad G, Bridoux F, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013;13:663–675. doi: 10.1111/ajt.12077. [DOI] [PubMed] [Google Scholar]
- 66.Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, Loirat C, Rondeau E, Fridman WH. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan MR, Thomas CP, Torrealba JR, Djamali A, Fernandez LA, Nishimura CJ, Smith RJ, Samaniego MD. Recurrent atypical hemolytic uremic syndrome associated with factor I mutation in a living related renal transplant recipient. Am J Kidney Dis. 2009;53:321–326. doi: 10.1053/j.ajkd.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bienaime F, Dragon-Durey MA, Regnier CH, Nilsson SC, Kwan WH, Blouin J, Jablonski M, Renault N, Rameix-Welti MA, Loirat C, et al. Mutations in components of complement influence the outcome of Factor I-associated atypical hemolytic uremic syndrome. Kidney Int. 2010;77:339–349. doi: 10.1038/ki.2009.472. [DOI] [PubMed] [Google Scholar]
- 69.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, Dragon-Durey MA, Blouin J, Caudy A, Arzouk N, Cleper R, Francois M, Guest G, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–2025. doi: 10.1681/ASN.2005101051. [DOI] [PubMed] [Google Scholar]
- 70.Frémeaux-Bacchi V, Arzouk N, Ferlicot S, Charpentier B, Snanoudj R, Dürrbach A. Recurrence of HUS due to CD46/MCP mutation after renal transplantation: a role for endothelial microchimerism. Am J Transplant. 2007;7:2047–2051. doi: 10.1111/j.1600-6143.2007.01888.x. [DOI] [PubMed] [Google Scholar]
- 71.Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, Del-Favero J, Plaisance S, Claes B, Lambrechts D, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–357. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sinibaldi S, Guzzo I, Piras R, Bresin E, Emma F, Dello Strologo L. Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant. 2013;17:E177–E181. doi: 10.1111/petr.12151. [DOI] [PubMed] [Google Scholar]
- 73.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, López-Trascasa M, Sánchez-Corral P, Morgan BP, Rodríguez de Córdoba S. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–245. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roumenina LT, Jablonski M, Hue C, Blouin J, Dimitrov JD, Dragon-Durey MA, Cayla M, Fridman WH, Macher MA, Ribes D, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114:2837–2845. doi: 10.1182/blood-2009-01-197640. [DOI] [PubMed] [Google Scholar]
- 75.Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, Daina E, Fenili C, Castelletti F, Sorosina A, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–1859. doi: 10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Satoskar AA, Pelletier R, Adams P, Nadasdy GM, Brodsky S, Pesavento T, Henry M, Nadasdy T. De novo thrombotic microangiopathy in renal allograft biopsies-role of antibody-mediated rejection. Am J Transplant. 2010;10:1804–1811. doi: 10.1111/j.1600-6143.2010.03178.x. [DOI] [PubMed] [Google Scholar]
- 77.Kwon O, Hong SM, Sutton TA, Temm CJ. Preservation of peritubular capillary endothelial integrity and increasing pericytes may be critical to recovery from postischemic acute kidney injury. Am J Physiol Renal Physiol. 2008;295:F351–F359. doi: 10.1152/ajprenal.90276.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruggenenti P. Post-transplant hemolytic-uremic syndrome. Kidney Int. 2002;62:1093–1104. doi: 10.1046/j.1523-1755.2002.00543.x. [DOI] [PubMed] [Google Scholar]
- 79.Olie KH, Goodship TH, Verlaak R, Florquin S, Groothoff JW, Strain L, Weening JJ, Davin JC. Posttransplantation cytomegalovirus-induced recurrence of atypical hemolytic uremic syndrome associated with a factor H mutation: successful treatment with intensive plasma exchanges and ganciclovir. Am J Kidney Dis. 2005;45:e12–e15. doi: 10.1053/j.ajkd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 80.Barbour S, Gill JS. Advances in the understanding of complement mediated glomerular disease: implications for recurrence in the transplant setting. Am J Transplant. 2015;15:312–319. doi: 10.1111/ajt.13042. [DOI] [PubMed] [Google Scholar]
- 81.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, Theis JD, Dogan A, Smith RJ. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465–473. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sethi S, Vrana JA, Fervenza FC, Theis JD, Sethi A, Kurtin PJ, Zhang Y, Smith RJ. Characterization of C3 in C3 glomerulopathy. Nephrol Dial Transplant. 2016;pii:gfw290. doi: 10.1093/ndt/gfw290. [DOI] [PubMed] [Google Scholar]
- 83.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 84.Dragon-Durey MA, Blanc C, Marinozzi MC, van Schaarenburg RA, Trouw LA. Autoantibodies against complement components and functional consequences. Mol Immunol. 2013;56:213–221. doi: 10.1016/j.molimm.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 85.Smith RJ, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol. 2011;48:1604–1610. doi: 10.1016/j.molimm.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Q, Müller D, Rudolph B, Hartmann A, Kuwertz-Bröking E, Wu K, Kirschfink M, Skerka C, Zipfel PF. Combined C3b and factor B autoantibodies and MPGN type II. N Engl J Med. 2011;365:2340–2342. doi: 10.1056/NEJMc1107484. [DOI] [PubMed] [Google Scholar]
- 87.Goodship TH, Pappworth IY, Toth T, Denton M, Houlberg K, McCormick F, Warland D, Moore I, Hunze EM, Staniforth SJ, et al. Factor H autoantibodies in membranoproliferative glomerulonephritis. Mol Immunol. 2012;52:200–206. doi: 10.1016/j.molimm.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 88.Lorcy N, Rioux-Leclercq N, Lombard ML, Le Pogamp P, Vigneau C. Three kidneys, two diseases, one antibody? Nephrol Dial Transplant. 2011;26:3811–3813. doi: 10.1093/ndt/gfr436. [DOI] [PubMed] [Google Scholar]
- 89.Masani N, Jhaveri KD, Fishbane S. Update on membranoproliferative GN. Clin J Am Soc Nephrol. 2014;9:600–608. doi: 10.2215/CJN.06410613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martínez-Barricarte R, Heurich M, Valdes-Cañedo F, Vazquez-Martul E, Torreira E, Montes T, Tortajada A, Pinto S, Lopez-Trascasa M, Morgan BP, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120:3702–3712. doi: 10.1172/JCI43343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dragon-Durey MA, Frémeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, Deschenes G, Coppo P, Herman Fridman W, Weiss L. Heterozygous and homozygous factor h deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. 2004;15:787–795. doi: 10.1097/01.asn.0000115702.28859.a7. [DOI] [PubMed] [Google Scholar]
- 92.Servais A, Noël LH, Dragon-Durey MA, Gübler MC, Rémy P, Buob D, Cordonnier C, Makdassi R, Jaber W, Boulanger E, et al. Heterogeneous pattern of renal disease associated with homozygous factor H deficiency. Hum Pathol. 2011;42:1305–1311. doi: 10.1016/j.humpath.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 93.Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs) Mol Immunol. 2013;56:170–180. doi: 10.1016/j.molimm.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 94.Besbas N, Gulhan B, Gucer S, Korkmaz E, Ozaltin F. A novel CFHR5 mutation associated with C3 glomerulonephritis in a Turkish girl. J Nephrol. 2014;27:457–460. doi: 10.1007/s40620-013-0008-1. [DOI] [PubMed] [Google Scholar]
- 95.Chen Q, Manzke M, Hartmann A, Büttner M, Amann K, Pauly D, Wiesener M, Skerka C, Zipfel PF. Complement Factor H-Related 5-Hybrid Proteins Anchor Properdin and Activate Complement at Self-Surfaces. J Am Soc Nephrol. 2016;27:1413–1425. doi: 10.1681/ASN.2015020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, Pusey CD, Pierides A, Kyriacou K, Athanasiou Y, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, Meyer NC, Hunsicker LG, Sethi S, Smith RJ. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–1829. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malik TH, Lavin PJ, Goicoechea de Jorge E, Vernon KA, Rose KL, Patel MP, de Leeuw M, Neary JJ, Conlon PJ, Winn MP, et al. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol. 2012;23:1155–1160. doi: 10.1681/ASN.2012020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tortajada A, Garcìa SP, Gastoldi S, Fernandez JG, Martin Merinero H, Arjona E, Noris M, Rodriguez de Cordoba S. Prevalenr FHR1 mutant protein generated by gene conversion reveals crucial role of factor H polymorhisms in atypical Hemolytic Uremic Syndrome. Immunobiology. 2016;221:1199. [Google Scholar]
- 100.Goicoechea de Jorge E, Pickering MC. Atypical hemolytic uremic syndrome: telling the difference between H and Y. Kidney Int. 2010;78:721–723. doi: 10.1038/ki.2010.222. [DOI] [PubMed] [Google Scholar]
- 101.Braun MC, Stablein DM, Hamiwka LA, Bell L, Bartosh SM, Strife CF. Recurrence of membranoproliferative glomerulonephritis type II in renal allografts: The North American Pediatric Renal Transplant Cooperative Study experience. J Am Soc Nephrol. 2005;16:2225–2233. doi: 10.1681/ASN.2005020175. [DOI] [PubMed] [Google Scholar]
- 102.Medjeral-Thomas NR, O’Shaughnessy MM, O’Regan JA, Traynor C, Flanagan M, Wong L, Teoh CW, Awan A, Waldron M, Cairns T, et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol. 2014;9:46–53. doi: 10.2215/CJN.04700513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Angelo JR, Bell CS, Braun MC. Allograft failure in kidney transplant recipients with membranoproliferative glomerulonephritis. Am J Kidney Dis. 2011;57:291–299. doi: 10.1053/j.ajkd.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 104.Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, de Córdoba SR, Hageman GS, Jokiranta TS, Kimberling WJ, et al. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol. 2007;18:2447–2456. doi: 10.1681/ASN.2007030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol. 2005;16:1392–1403. doi: 10.1681/ASN.2005010078. [DOI] [PubMed] [Google Scholar]
- 106.McCaughan JA, O’Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant. 2012;12:1046–1051. doi: 10.1111/j.1600-6143.2011.03923.x. [DOI] [PubMed] [Google Scholar]
- 107.Andresdottir MB, Assmann KJ, Hoitsma AJ, Koene RA, Wetzels JF. Renal transplantation in patients with dense deposit disease: morphological characteristics of recurrent disease and clinical outcome. Nephrol Dial Transplant. 1999;14:1723–1731. doi: 10.1093/ndt/14.7.1723. [DOI] [PubMed] [Google Scholar]
- 108.Ponticelli C, Glassock RJ. Posttransplant recurrence of primary glomerulonephritis. Clin J Am Soc Nephrol. 2010;5:2363–2372. doi: 10.2215/CJN.06720810. [DOI] [PubMed] [Google Scholar]
- 109.Little MA, Dupont P, Campbell E, Dorman A, Walshe JJ. Severity of primary MPGN, rather than MPGN type, determines renal survival and post-transplantation recurrence risk. Kidney Int. 2006;69:504–511. doi: 10.1038/sj.ki.5000084. [DOI] [PubMed] [Google Scholar]
- 110.Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, Zavros M, Maxwell PH, Cook HT, Demosthenous P, Hadjisavvas A, et al. Familial C3 glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol. 2011;6:1436–1446. doi: 10.2215/CJN.09541010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vernon KA, Gale DP, de Jorge EG, McLean AG, Galliford J, Pierides A, Maxwell PH, Taube D, Pickering MC, Cook HT. Recurrence of complement factor H-related protein 5 nephropathy in a renal transplant. Am J Transplant. 2011;11:152–155. doi: 10.1111/j.1600-6143.2010.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wong L, Moran S, Lavin PJ, Dorman AM, Conlon PJ. Kidney transplant outcomes in familial C3 glomerulopathy. Clin Kidney J. 2016;9:403–407. doi: 10.1093/ckj/sfw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zuber J, Le Quintrec M, Morris H, Frémeaux-Bacchi V, Loirat C, Legendre C. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev (Orlando) 2013;27:117–125. doi: 10.1016/j.trre.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 114.Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, Van Praet J, Pani A, De Vriese AS. Diagnosis of complement alternative pathway disorders. Kidney Int. 2016;89:278–288. doi: 10.1016/j.kint.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 115.Chua S, Wong G, Lim WH. The importance of genetic mutation screening to determine retransplantation following failed kidney allograft from recurrent atypical haemolytic ureamic syndrome. BMJ Case Rep. 2014;2014:pii: bcr2013202875. doi: 10.1136/bcr-2013-202875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Noris M, Remuzzi G. Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens. 2013;22:704–712. doi: 10.1097/MNH.0b013e328365b3fe. [DOI] [PubMed] [Google Scholar]
- 117.Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, Birembaut P, Chanard J, Rieu P. Sirolimus-induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. Am J Transplant. 2005;5:2441–2447. doi: 10.1111/j.1600-6143.2005.01047.x. [DOI] [PubMed] [Google Scholar]
- 118.Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, Heyne N, Ardissino G, Chatelet V, Noël LH, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12:3337–3354. doi: 10.1111/j.1600-6143.2012.04252.x. [DOI] [PubMed] [Google Scholar]
- 119.Kwon T, Dragon-Durey MA, Macher MA, Baudouin V, Maisin A, Peuchmaur M, Fremeaux-Bacchi V, Loirat C. Successful pre-transplant management of a patient with anti-factor H autoantibodies-associated haemolytic uraemic syndrome. Nephrol Dial Transplant. 2008;23:2088–2090. doi: 10.1093/ndt/gfn063. [DOI] [PubMed] [Google Scholar]
- 120.Waters AM, Pappworth I, Marchbank K, Bockenhauer D, Tullus K, Pickering MC, Strain L, Sebire N, Shroff R, Marks SD, et al. Successful renal transplantation in factor H autoantibody associated HUS with CFHR1 and 3 deficiency and CFH variant G2850T. Am J Transplant. 2010;10:168–172. doi: 10.1111/j.1600-6143.2009.02870.x. [DOI] [PubMed] [Google Scholar]
- 121.Zimmerhackl LB, Hofer J, Cortina G, Mark W, Würzner R, Jungraithmayr TC, Khursigara G, Kliche KO, Radauer W. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N Engl J Med. 2010;362:1746–1748. doi: 10.1056/NEJMc1001060. [DOI] [PubMed] [Google Scholar]
- 122.Román-Ortiz E, Mendizabal Oteiza S, Pinto S, López-Trascasa M, Sánchez-Corral P, Rodríguez de Cordoba S. Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr Nephrol. 2014;29:149–153. doi: 10.1007/s00467-013-2591-8. [DOI] [PubMed] [Google Scholar]
- 123.Nester C, Stewart Z, Myers D, Jetton J, Nair R, Reed A, Thomas C, Smith R, Brophy P. Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2011;6:1488–1494. doi: 10.2215/CJN.10181110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krid S, Roumenina LT, Beury D, Charbit M, Boyer O, Frémeaux-Bacchi V, Niaudet P. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant. 2012;12:1938–1944. doi: 10.1111/j.1600-6143.2012.04051.x. [DOI] [PubMed] [Google Scholar]
- 125.Habbig S, Mihatsch MJ, Heinen S, Beck B, Emmel M, Skerka C, Kirschfink M, Hoppe B, Zipfel PF, Licht C. C3 deposition glomerulopathy due to a functional factor H defect. Kidney Int. 2009;75:1230–1234. doi: 10.1038/ki.2008.354. [DOI] [PubMed] [Google Scholar]
- 126.Gurkan S, Fyfe B, Weiss L, Xiao X, Zhang Y, Smith RJ. Eculizumab and recurrent C3 glomerulonephritis. Pediatr Nephrol. 2013;28:1975–1981. doi: 10.1007/s00467-013-2503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D’Agati VD, Canetta PA, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7:748–756. doi: 10.2215/CJN.12901211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, Appel GB, D’Agati VD. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol. 2012;23:1229–1237. doi: 10.1681/ASN.2011121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mario Negri Institute for Pharmacological Research. Eculizumab in Primary MPGN (EAGLE). In: ClinicalTrials.gov [Internet]. Bethesda(MD): National Library of Medicine (US) Available from: https://www.clinicaltrials.gov/ct2/show/NCT02093533 NLM Identifier: NCT02093533.
- 130.Midtvedt K, Bitter J, Dørje C, Bjørneklett R, Holdaas H. Belatacept as immunosuppression in patient with recurrence of hemolytic uremic syndrome after renal transplantation. Transplantation. 2009;87:1901–1903. doi: 10.1097/TP.0b013e3181a991ca. [DOI] [PubMed] [Google Scholar]
- 131.Matar D, Naqvi F, Racusen LC, Carter-Monroe N, Montgomery RA, Alachkar N. Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation. 2014;98:1205–1212. doi: 10.1097/TP.0000000000000200. [DOI] [PubMed] [Google Scholar]
- 132.Larrea CF, Cofan F, Oppenheimer F, Campistol JM, Escolar G, Lozano M. Efficacy of eculizumab in the treatment of recurrent atypical hemolytic-uremic syndrome after renal transplantation. Transplantation. 2010;89:903–904. doi: 10.1097/TP.0b013e3181ccd80d. [DOI] [PubMed] [Google Scholar]
- 133.Alachkar N, Bagnasco SM, Montgomery RA. Eculizumab for the treatment of two recurrences of atypical hemolytic uremic syndrome in a kidney allograft. Transpl Int. 2012;25:e93–e95. doi: 10.1111/j.1432-2277.2012.01497.x. [DOI] [PubMed] [Google Scholar]
- 134.Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A. A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS) BMJ Open. 2013;3:e003573. doi: 10.1136/bmjopen-2013-003573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alexion Pharmaceuticals. Open Label controlled trial of Eculizumab in Adult Patients with Plasma Therapy-sensitive Atypical Hemolytic Uremic Syndrome (aHUS). In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US) Available from: https://www.clinicaltrials.gov/ct2/show/NCT00838513 NLM Identifier: NCT00838513.
- 136.Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–657. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 137.Tanimoto T, Oshima Y, Kami M. Eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;369:1378–1379. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 138.Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, Smith RJ. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220:993–998. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Paixão-Cavalcante D, Torreira E, Lindorfer MA, Rodriguez de Cordoba S, Morgan BP, Taylor RP, Llorca O, Harris CL. A humanized antibody that regulates the alternative pathway convertase: potential for therapy of renal disease associated with nephritic factors. J Immunol. 2014;192:4844–4851. doi: 10.4049/jimmunol.1303131. [DOI] [PMC free article] [PubMed] [Google Scholar]