


Abstract
The RAG1/RAG2 endonuclease initiates V(D)J recombination at antigen receptor loci but also binds to thousands of places outside of these loci. RAG2 localizes directly to lysine 4 trimethylated histone 3 (H3K4me3) through a plant homeodomain (PHD) finger. The relative contribution of RAG2-dependent and RAG1-intrinsic mechanisms in determining RAG1 binding patterns is not known. Through analysis of deep RAG1 ChIP-seq data, we provide a quantitative description of the forces underlying genome-wide targeting of RAG1. Surprisingly, sequence-specific DNA binding contributes minimally to RAG1 targeting outside of antigen receptor loci. Instead, RAG1 binding is driven by two distinct modes of interaction with chromatin: the first is driven by H3K4me3, promoter-focused and dependent on the RAG2 PHD, and the second is defined by H3K27Ac, enhancer-focused and dependent on ‘non-core’ portions of RAG1. Based on this and additional chromatin and genomic features, we formulated a predictive model of RAG1 targeting to the genome. RAG1 binding sites predicted by our model correlate well with observed patterns of RAG1-mediated breaks in human pro-B acute lymphoblastic leukemia. Overall, this study provides an integrative model for RAG1 genome-wide binding and off-target activity and reveals a novel role for the RAG1 non-core region in RAG1 targeting.
INTRODUCTION
V(D)J recombination occurs during early B- and T-lymphocyte development. During this process, antigen receptor genes are assembled from arrays of V, D and J gene segments. The reaction is initiated by the Recombination Activating Gene (RAG) endonuclease, which introduces double-strand breaks at recombination signal sequences (RSSs) flanking the V, D and J gene segments. RAG is comprised of a catalytic subunit (RAG1) and an essential cofactor (RAG2). The ‘core’ domains of RAG1 and RAG2 have been defined as the minimal portions required for RAG activity in vitro. The RAG1 core contains the active site residues responsible for DNA cleavage and makes extensive sequence-specific and non-specific contacts with the RSS and flanking DNA (1–3). RAG2 contributes no catalytic activity, but is an essential accessory factor. Its core domain strongly promotes DNA cleavage by RAG1 but makes only a small number of non-specific DNA contacts with the gene segment (1–4). The ‘non-core’ domains of the RAG proteins provide important regulatory functions (5). The RAG1 N-terminal region enhances V(D)J recombination activity (6,7), but the mechanism by which it does so is not well understood. This region contains a RING domain, which homodimerizes, harbors E3 ubiquitin ligase activity and interacts with and ubiquitylates histone H3 (8–11).
The RAG2 ‘non-core’ domain contains a plant homeodomain (PHD) that binds to H3K4me3. PHDs are widely distributed in phylogeny, exist in many chromatin-associated proteins (12–14) and have been shown to interact with methylated histone H3, particularly H3K36me3 and H3K4me3 (14–17). The RAG2–PHD–H3K4me3 interaction enhances the catalytic activity of the RAG complex (18–20) and recruits RAG2 to regions of transcriptionally active chromatin (21,22).
The substrate for RAG cleavage, the RSS, consists of a conserved heptamer (consensus 5′-CACAGTG) and nonamer (consensus 5′-ACAAAAACC) separated by a non-conserved spacer of either 12 or 23 base pairs (bp). Efficient cleavage requires a pair of RSSs, one containing a 12-bp spacer and one containing a 23-bp spacer (a requirement known as the ‘12/23 rule’). The heptamer specifies the site of cleavage, while the nonamer provides an important binding site for RAG1 (3). RSSs exhibit a high level of sequence variability, with only the first three nucleotides of the heptamer (5′-CAC) being perfectly conserved (2). As a consequence of the intrinsic degeneracy of the RSS, a large number cryptic RSSs (cRSSs) exist outside of the antigen receptor genes. Most of these consist of heptamer or heptamer-like sequences, such as CA repeats and CAC trinucleotides (23–26). The prediction of active cRSSs therefore presents significant computational challenges. Position weight matrices (PWMs), which calculate the similarity of a sequence to a reference motif (i.e. bona fide RSSs from antigen receptor loci (bRSS)) by summing up the similarity at each position, have been used to predict potential cRSSs (26). A more sophisticated computational approach—RSS information content (RIC)—like PWMs, relies on sequence similarity of the cRSS to bRSS, but also takes into account the dependence between different positions and assesses RSS quality by the product of joint probabilities of dependent positions, drawn from bRSS sequences. RIC scores of bRSSs have been shown to correlate with measured recombination efficiencies (27).
The described approaches predict thousands of potential cRSSs distributed fairly uniformly throughout the genome, some of which strongly resemble bRSSs. Though sites of RAG-mediated genomic instability tend to be enriched in cRSSs, the genome-wide distribution of off-target RAG activity is neither as frequent nor as uniform as the frequency and distribution of cRSSs would predict. Instead, illegitimate RAG-mediated events associated with leukemias and lymphomas are focused in active promoters and enhancers (25,26). Hence, prediction of RAG off-target activity requires an understanding of the mechanism by which RAG1 is targeted to specific places in chromatin, rather than merely predicting the location of cRSSs.
In a recent study, we showed that genome-wide RAG1 and RAG2 binding patterns overlap with sites marked by H3K4me3. A strong linear correlation was observed between RAG2 binding intensity and H3K4me3 density. RAG1 was found to occupy a subset of the RAG2(+) H3K4me3(+) sites in the genome. However, the majority of H3K4me3 peaks showed no evidence of RAG1 binding, and strikingly, RAG1 binding intensity did not linearly correlate with H3K4me3 density. This suggested that genome-wide RAG1 binding patterns cannot be fully explained by co-recruitment to H3K4me3 through RAG2, and that RAG2-independent mechanisms contribute to the targeting of RAG1 to chromatin (22).
One potential RAG2-independent recruitment mechanism is the direct interaction of RAG1 with histones (Figure 1A). The N-terminal RING domain of RAG1 can directly bind to and ubiquitylate histone 3 (H3) in vitro, and this has been suggested to play a role in regulation of V(D)J recombination (10,11,28). However, to date, there is no direct evidence that the RAG1 N-terminal region directs RAG1 localization in vivo.
Figure 1.

RAG1 targeting genome-wide is multifactorial. (A) Four possible modes of RAG1–chromatin interactions are considered in this study: (i) sequence-specific DNA interactions, (ii) sequence non-specific DNA interactions, (iii) RAG2-dependent histone interactions via H3K4me3 and (iv) RAG1 interactions with chromatin proteins, perhaps a histone. (B) Schematic diagram of the RAG variants used in the study: WT, R1-D708A (active site mutant), R2ΔC and cR1. (C) H3K4me3 versus RAG1 levels of H3K4me3 peaks. Most H3K4me3 peaks do not overlap with a RAG1 peak (orange; N = 15 645) while those that do (blue; N = 4738) are concentrated in the region with higher levels of H3K4me3. (D) H3K4me3 versus RAG2 levels of H3K4me3 peaks. Unlike RAG1, RAG2 linearly correlates with H3K4me3 and virtually all H3K4me3 high peaks are RAG2(+) (green; N = 16 242).
Another means by which RAG1 can be targeted is through direct interaction with DNA (Figure 1A). The RAG1 nonamer binding domain (NBD) mediates a well-characterized interaction with the nonamer, providing an important platform for sequence-specific recognition of the RSS, and other portions of the RAG1 core make numerous additional DNA contacts (1,29). Though the presence of paired RSSs is clearly important for stabilizing and maintaining RAG1 binding (30), it remains unclear whether an RSS is sufficient for the initial recruitment of RAG1 to chromatin.
RAG1 also exhibits non-specific DNA-binding activity in vitro (3) and the RAG1 NBD has been implicated in sequence-independent DNA binding in vivo (22). This intrinsic, non-specific affinity for DNA is partially masked in the presence of RAG2 in vitro (31) (Figure 1A). In addition, RAG can also recognize and cleave non-B-form DNA structures, exemplified by an off-target RAG cleavage site in BCL2, which underlies a recurrent translocation in follicular lymphoma (32).
Thus, the genome-wide RAG1 binding patterns could reflect at least four distinct modes of substrate recognition (Figure 1A): (i) indirect recruitment via RAG2 to H3K4me3-rich chromatin, (ii) direct binding of RAG1 to histones, (iii) sequence-specific binding to RSSs or RSS-like sequences and (iv) non-specific binding to DNA. The degree to which each of these modes contributes to RAG1 binding in vivo is poorly understood. To address this question, we constructed a regression model for RAG1 recruitment in vivo using previously published RAG1 ChIP-seq datasets (22), along with a new, deeply-sequenced dataset from mouse thymocytes. The model, based on features of chromatin state and DNA sequence, revealed two distinct modes for widespread RAG1 binding that are defined primarily by the histone marks H3K4me3 and H3K27Ac, and are dependent on the non-core regions of RAG2 and RAG1, respectively, with specific DNA binding making little contribution. The utility of the model is revealed by its ability to predict illegitimate RAG-mediated recombination events in human leukemia cells, establishing a correlation between off-target RAG1 binding and off-target activity.
MATERIALS AND METHODS
Data
The sources of all the data used in this study are listed in Supplementary Information (SI) appendix.
RAG1 enrichment at RSSs
A Poisson test was used to determine if RAG1 binding was enriched at RSSs/cRSSs in the genome. λ was defined as the mean value of the number of reads in the ±300 bp surrounding RSSs/cRSSs, using a sliding window of either 28 bp for 12RSS or 39 bp for 23RSS; and X was defined as the number of reads on the corresponding RSSs.
Footprint analysis
Assay for transposase-accessible chromatin using sequencing (ATAC-seq) data was processed as described in Teng et al. (22). The 30 bp surrounding each pre-B RAG1 or pro-B PU.1 ChIP-seq summit were defined as the ‘motif’ region and the surrounding ±70 bp were defined as the ‘flanking’ region. Summits were then grouped by their ChIP-seq level percentile (see Figure 2). For each group of summits, the number of ATAC-seq reads in each position was calculated for both strands and a binomial test was employed to calculate the depletion of the ATAC-seq signal at the ‘summit’ region compared to the upstream ‘flanking’ region for each strand, as described previously (33). To control for Tn5 transposase sequence biases (34), we used data from naked DNA and calculated the relative frequency of each trinucleotide at the Tn5 integration site. For each group of summits, we summed the frequencies of the ‘motif’ and ‘flanking’ region and calculated the expected motif/flanking read frequency, to be used in the binomial test. A Bonferroni correction was used to account for multiple testing. A summit group showing significance on both strands was considered to be a footprint.
Figure 2.

The role of DNA sequence in RAG1 targeting. (A) RAG1 D708A ChIP-seq profiles at RSSs and cryptic RSSs (cRSSs). Top, TCRα locus (black), middle, Igκ locus (brown) and bottom, a non-antigen receptor locus Cnot2 (red). Red and blue triangles, 12 and 23RSSs, respectively; c12RSS, red hatched triangles. Panels in the center show a magnified section of each locus. (B and C) D708A-RAG1 ChIP-seq signal plotted for the 500 bp surrounding RSS/cRSS in the indicated genotypes. (B) 12RSS or 12cRSS or (C) 23 23RSS or 23cRSSs. cRSSs were identified using the RIC algorithm (27) with stringent thresholds of RIC > −28 for 12cRSSs and RIC > −44 for 23cRSSs. All data derive from mouse thymus except for Jκ and Vκ plots, in which the data derive from mouse pre-B cells. (D and E) Footprinting analysis—cut-site probability was taken as the mean ATAC-seq signal for 200 bp surrounding (D) RAG1 or (E) PU.1 ChIP-seq summits. Footprint were analyzed as described in ‘Materials and Methods’ section. Pie charts indicate the distribution of motifs as a fraction of (D) RAG1 and(E) PU.1 summits; 3.4% of RAG1 summits (±10 bp) share a sequence motif (E-value (motif) = 4.1e-4), while 55% of PU.1 summits share at least one motif, with the dominant motif representing 30% of the peaks (E-value (motif) = 2.2e-1291). See Supplementary Figure S1 for motif sequences.
RAG1 targeting model
The model for RAG1 targeting was built using data from mouse thymocytes. H3K4me3 peaks were first called using MACS-2.1.0 as described previously (17,53), resulting in 20 383 H3K4me3 peaks. Then, the RAG1, H3K27Ac and DNAseI-HS RPKM were calculated for these peaks. The GC content, CpG value, CpA content and number of 12RSSs, 23RSSs, heptamers and nonamers were determined for the 2 kb surrounding peak summits (see SI appendix for details). For all features, a normalized value was obtained by subtracting the mean value and dividing by the standard deviation.
The set of features were then used to predict RAG1 levels using non-linear support vector regression (SVR) (35) implemented by the R package e1071 SVM function (https://cran.r-project.org/web/packages/e1071/vignettes/svmdoc.pdf) with radial basis function (rbf) kernel and default parameters. To select the set of features that best predicts RAG1 targeting, a random subset of 2000 peaks was iteratively taken from the full set of 20 383 H3K4me3 peaks. We randomized the values of three of the features in each iteration. We ran the model using 2-fold cross validation (learning on 1000 samples and testing on the remaining 1000), calculated the mean square error (MSE) with and without these features and took the difference in MSE (ΔMSE) as the measure of feature importance. A two-tailed t-test was used to compare the importance of each candidate feature to the importance of a random feature, generated by randomizing H3K4me3 levels (Figure 3A). We then looked for the optimal set of features by adding features one by one starting from the highest ranked feature and re-calculated the MSE upon addition of each feature. The subset of features that minimized the MSE was selected (Figure 3B). A cutoff of 1 normalized RAG1 RPKM was used to divide the H3K4me3 peaks set into RAG1hi and RAG1lo peaks (3134 and 17 249 peaks, respectively). A two tailed t-test was used to compare feature level in RAG1hi and RAG1lo total peaks, promoters and enhancers (Figures 3C and 4B). The performance of the model, build on mouse thymocytes data, was tested using independent ChIP-seq sets from mouse pre-B cells (Figure 3D) and human thymocytes (Supplementary Figure S2), as described in SI appendix.
Figure 3.

Construction of a RAG1 binding model. (A) Candidate features were ranked by their importance for the RAG1 targeting model as measured by Δmean square error (MSE) (see ‘Materials and Methods’ section). The benchmark for importance was determined using a random feature, created by scrambling of H3K4me3 values (red). (B) The regression model was applied using an increasing number of features added by the ranking of their importance. Red dashed line shows the subset that yielded the minimal MSE and was used in subsequent analyses. (C) Levels of selected features at RAG1hi versus RAG1lo H3K4me3 peaks. RAG1hi peaks were higher in H3K4me3 and H3K27Ac, closer to transcription start sites and depleted in CA dinucleotides compared to RAG1lo peaks (P = 0 for all features). (D) The RAG1 targeting model based on mouse thymocytes was used to predict the RAG1 distribution in mouse pre-B cells. Regression error characteristic (REC) curve, plotting the fraction of the peak set (y-axis) that was predicted with a certain maximal residual (x-axis), illustrate prediction quality of the full regression model (blue line), compared with regression using either H3K4me3 only (orange line), H3K27Ac only (purple line). Upper and lower limit curves were traced by calculating the residuals between ChIP-seq replicates (black dashed line) or a random feature (red dashed line), respectively.
Figure 4.

RAG1 peaks segregate into two clusters. (A) Hierarchical clustering of RAG1hi H3K4me3 peaks according to peak-specific feature importance. Each column represents a RAG1hi peak and each row represents a feature. Feature importance is color coded. Dendrogram above the heat map represents the hierarchical clustering of the peak set. (B) Boxplots of the levels of each feature in RAG1hi promoter (orange; N = 2368) and enhancer (purple; N = 307) clusters (filled boxes) compared with RAG1lo regions (empty boxes; 1407 promoters and 1454 enhancers) selected to be comparable for certain ‘fixed’ parameters, as indicated by red boxes. (C and D) Snapshots of representative RAG1hi versus RAG1lo (C) promoters and (D) enhancers illustrate the levels of WT RAG1, H3K4me3, H3K27Ac and DNaseI-HS. The numbers in parentheses indicate the maximal RPKM.
The performance of the model was evaluated using independent ChIP-seq sets from mouse pre-B cells (Figure 3D) and human thymocytes (Supplementary Figure S2). For this purpose, we used a regression error characteristic (REC) curve, plotting the cumulative distribution function of the prediction errors obtained by the model (e.g. fraction of the peak set (y-axis) that was predicted with a certain maximal residual (x-axis)) (36). The area under the resulting curve (AUC) can be used as a measure for the model performance (a perfect model, where all data points are predicted with 0 residual, will have AUC = 1). To account for the inherent variance between ChIP-seq experiments, the AUCs were then normalized by the AUC resulted from a comparison of two mouse thymus RAG1 ChIP-seq replicates.
Peaks clustering
RAG1hi H3K4me3 peaks were clustered according to the relative importance of each feature to each peak (Figure 4). For this purpose, we defined an importance matrix M such that
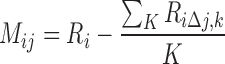 |
where 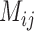 is the importance of feature j to sample i.
is the importance of feature j to sample i. 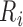 is the residual i using the SVR model and
is the residual i using the SVR model and 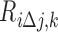 is the ith residual after replacing feature j with a random number, kεK, where k is a number within the range in which feature j is distributed—K, using intervals of 0.1. The higher the change of the residual, the more important the feature for the sample.
is the ith residual after replacing feature j with a random number, kεK, where k is a number within the range in which feature j is distributed—K, using intervals of 0.1. The higher the change of the residual, the more important the feature for the sample.
Next, we took the peaks that were most accurately predicted (MSE < 1; overall – 2675 peaks). These peaks were then clustered according to the above matrix by hierarchical clustering using pheatmap R (https://cran.rproject.org/web/packages/pheatmap/pheatmap.pdf) with Euclidian distance and complete linkage. A non-parametric Wilcoxon-test was used to compare the RAG1 levels in two clusters of peaks (Figure 5B).
Figure 5.

RAG1hi clusters represent two modes of interaction mediated by different RAG1 and RAG2 domains. (A) Schematic representation of the expected RAG1 targeting interactions in wt, cR1 and R2ΔC genotypes. (B) Comparison of RAG1 levels in Cluster 1 (H3K4me3-driven) and Cluster 2 (H3K27Ac-driven) in WT, cR1 and R2ΔC genotypes. In both WT and cR1, RAG1 levels are higher in Cluster 1 than Cluster 2, but the differences are much greater in cR1 (t-test; P < 1e-5 and P < 1e-35, respectively). In R2ΔC, RAG1 levels are higher in cluster 2 (P < 1e-9). (C) Correlation between the levels of RAG1 in the indicated genotypes and H3K4me3, H3K27Ac or DNaseI-HS. Spearman correlation coefficients are indicated on each plot in the bottom right corner and the MSE is indicated by background color.
RESULTS
In a recent study, we performed RAG1 and RAG2 ChIP-seq in developing lymphocytes from wild-type (WT) or RAG1 catalytic mutant mice (22). The latter are deficient in endogenous RAG1, but harbor a bacterial artificial chromosome expressing a RAG1 active site mutant (D708A) that retains its DNA binding properties but not catalytic activity. This mutant allowed us to assay for RAG1 binding to chromatin in the absence of RAG-mediated genome rearrangements. We also analyzed cells from RAG1-/- and RAG2-/- mice (‘R1-/-’ and ‘R2-/-‚’ respectively), core RAG1 (‘cR1’) mice (expressing the catalytic core of RAG1, aa 384–1008 of 1040 aa) and ‘R2ΔC’ mice (expressing a C-terminally deleted RAG2 lacking the PHD, aa 1–352 of 527 aa) (Figure 1B) (22). Both lymphocyte development and V(D)J recombination occur with reduced efficiencies in cR1 and R2ΔC mice (6,37). Our prior study revealed that RAG1 binds thousands of places in the mouse genome, co-localizes with H3K4me3 and overlaps almost exclusively with active promoters and enhancers. We herein use a newly generated RAG1 ChIP-seq dataset from WT thymocytes, along with the published data described above, to show that factors distinct from H3K4me3 and RAG2 contribute to the chromatin targeting pattern of RAG1.
H3K4me3 density does not fully explain the variance in RAG1 binding
Mouse thymocytes yielded 20 383 H3K4me3 peaks, 4738 of which overlapped with RAG1 peaks. H3K4me3 levels showed a bi-Gaussian distribution of H3K4me3(hi) and H3K4me3(lo) peaks with means of ∼5 and ∼20 H3K4me3 RPKM, respectively (Figure 1C). These two populations differed in their RAG1 distribution: while virtually all H3K4me3(lo) peaks showed little or no RAG1 binding, H3K4me3(hi) peaks exhibited a wide range of RAG1 levels (Figure 1C; blue indicates H3K4me3 peaks that overlap with significant RAG1 peaks). In contrast, RAG2 levels correlated linearly with H3K4me3 (Figure 1D). The non-linear correlation between RAG1 and H3K4me3 suggests that other factors likely contribute to determining the strength of RAG1 binding (Figure 1A).
Neither the RSS nor any other specific DNA motif plays a major role in RAG1 targeting outside of antigen receptor genes
We first sought to evaluate the extent to which DNA sequence dictates RAG1 binding patterns. While the RSS is vital for RAG1 cleavage activity, its in vivo role in recruiting RAG1 to chromatin is not well understood. We reasoned that if the RSS is a dominant attractor of RAG1, we should expect an RSS-centric RAG1 ChIP-seq signal, as observed for classical transcription factors and their corresponding binding sites (38).
To establish a benchmark for RSS-centric RAG1 binding, we examined the RAG1 signal surrounding bRSSs in antigen receptor loci using ChIP-seq data obtained from mouse thymocytes or pre-B cells that express the D708A RAG1 catalytic mutant (Figure 1B). In antigen receptor loci, binding in two distinct types of regions should be distinguished. The first is recombination centers, which are thought to represent the initial site of RAG recruitment (39). Recombination centers focus on J and J-proximal D gene segments and are characterized by high levels of transcription and a strong signature of H3K4me3, RAG2 and RAG1 (Figure 2A; Jα and Jκ (39)). The second is the V portion of the locus, where RAG binding might be a consequence of interaction with the recombination center (Figure 2A; Vα and Vκ). A clear enrichment of RAG1 was observed at Tcrα and Igκ bRSSs in both types of regions, albeit with distinct characteristics (Figure 2B, C, black and brown traces, respectively, Poisson test; P-value < 1e-7). In particular, while in recombination centers the RSS peak rose above the strong signal seen across the entire recombination center, the RAG1 binding peak at the corresponding V region RSSs was much sharper—albeit much weaker. This difference is addressed in the discussion. Our ability here to detect RAG1 binding at groups of Igκ and Tcrα V RSSs extends the findings of our previous studies, where analysis of individual V regions did not reveal a clear RAG1 binding signal (21,22).
We then determined whether the genome-wide sites of RAG1 binding in mouse thymocytes and pre-B cells were centered on cRSSs, using the above RSS-centric RAG1 signal in Tcrα and Igκ as a comparator. We identified cRSSs in the mouse genome using the RIC algorithm, which seeks to predict the functionality of a cRSS by assessing its similarity to bRSSs (27). A stringent RIC cutoff of −28 and −44 for 12RSSs and 23RSSs, respectively (blue lines in Supplementary Figure S1A and B), captures the vast majority of the bRSSs that flank antigen receptor gene segments and yields 3877 c12RSSs (cryptic 12RSSs) and 1866 c23RSSs (cryptic 23RSSs) distributed throughout the genome. In contrast to the strong RSS-centric RAG1 binding profile in Tcrα and Igκ, no significant enrichment of RAG1 binding was observed at high quality cRSSs (Figure 2B, C, red traces; Poisson test; P-value > 0.4 for cRSSs).
We next analyzed RAG1 ChIP-seq data from R2-/- and R2ΔC thymocytes in which the absence of RAG2 or its PHD finger removes the potential for indirect coupling of RAG1 to H3K4me3 via RAG2. We previously observed that RAG1 binding spreads to additional sites in the genomes of R2-/- and R2ΔC thymocytes relative to thymocytes expressing WT RAG2 (22). We reasoned that this expanded binding repertoire might allow RAG1 to interact more broadly with cRSSs scattered in the genome. However, no RAG1 binding signal was observed over c12RSSs in these genotypes (Figure 2B, blue and green traces; Poisson test; P-value > 0.1). A statistically significant enrichment of RAG1 binding was observed at c23RSSs, but the signal was far lower than seen at bRSSs (Figure 2C; Poisson test; P-value = 0.02 for R2ΔC and 0.008 for R2-/-). Together, these data indicate that high scoring cRSSs in the genome have little or no ability to recruit RAG1 above background levels, in contrast to bRSS, where sequence-targeted RAG1 recruitment is readily observed.
While the above analysis rules out a major role for cRSSs in RAG1 targeting, it does not address the possibility that RAG1 binds to DNA sequences that do not fit the classical criteria for cRSSs. A powerful, unbiased approach to identify direct interactions with DNA is provided by the analysis of digital footprints in DNase-I-seq/ATAC-seq data. This approach takes advantage of the fact that proteins that bind directly to DNA provide local protection against endonuclease or transposase activity within hypersensitive hotspots (34). Aggregating the signal from multiple binding sites results in a pattern characterized by a depletion of DNase-seq signal at a candidate-binding site compared to the flanking DNA (33). We therefore scanned a mouse pre-B cell ATAC-seq dataset (40) for RAG1 footprints. Typically, the reference positions for footprint scanning, around which the signal is searched, are conserved transcription factor binding sites. However, because RAG1 does not have a known binding site distinct from the RSS, we took advantage of our mouse pre-B cell RAG1 ChIP-seq data and set the reference points as the summits of RAG1 peaks (overall 3386 peaks; see ‘Materials and Methods’ section for details). The summit ±20 bp was set as the ‘binding site’ and was extended for 100 bp on either side.
For each summit region, a Binomial test was used to evaluate the depletion of ATAC-seq reads on the summit (±15 bp) compared to its flanks. To control for possible Tn5 transposase sequence biases we used ATAC-seq data derived from naked DNA (Supplementary Figure S1C and D) as a background model, as described in ‘Materials and Methods’ section. No digital footprint could be discerned at the RAG1 summit regions in pre-B cells, regardless of the ChIP-seq signal strength (Figure 2D; Binomial test P = 0.19 for the strongest RAG1 binding sites), indicating again that RAG1 binding patterns throughout the genome are not driven by sequence-specific interactions. To ensure that this approach was capable of detecting a footprint for a classical DNA binding protein using this ATAC-seq dataset, we repeated the same analysis for the transcription factor PU.1 using ChIP-seq data derived from pro-B cells (21 657 peaks) (41). A clear footprint is observed for PU.1 among the strongest PU.1 binding sites (Figure 2E; Binomial test; P < 1e-190).
Direct binding of a factor to DNA can also be revealed by the presence of a shared sequence motif coincident with ChIP-seq peaks. To investigate this, we searched pre-B cell RAG1 (and for comparison, PU.1) ChIP-seq data for a conserved motif in the summit ±10 bp using DREME (42). For PU.1, we found eight motifs (Supplementary Figure S1D) representing ∼55% of the peaks, with 30% of the peaks containing the most significant motif GGAAVT (Figure 2E and Supplementary Figure S1D; E-value = 2.2e-1291). Notably, this motif contains the consensus core PU.1 binding site GGAA (43). No such predominant sequence motif was present in RAG1 peak summits. One weak motif, CMGGAA, was identified in only 3.4% of the RAG1 peaks (Figure 2D and Supplementary Figure S1D) and its statistical significance (E-value = 4.1e-4) was much lower than that of the most prevalent PU.1 motif. Importantly, a higher percentage of H3K4me3 peaks that do not feature RAG1 binding also contain a related motif, RGGAAR, (9%; data not shown), indicating that motifs containing GGAA are common in active promoters of developing B cells (perhaps because many contain PU.1 binding sites), and are unlikely to be a specific feature of RAG1 peaks.
Finally, we examined evolutionary conservation of the DNA sequences surrounding RAG1 and PU.1 peaks, expecting greater conservation surrounding the summit than in the flanking DNA if the peaks derive from a functional binding site (44,45). This was clearly observed for PU.1 but not for RAG1 (Supplementary Figure S1F). Conservation over the entire interval analyzed was higher for RAG1 than for PU.1, likely reflecting the high percentage of RAG1 peaks that correspond to promoters and enhancers (Supplementary Figure S1G) (22), which are evolutionarily conserved elements (45).
Taken together, these four distinct analyses argue strongly that RAG1 genome-wide binding is not targeted to a specific DNA sequence, but instead must be driven primarily by other chromatin features. Satisfyingly, these findings complement our previous observation that off-target RAG1 binding sites are preferentially depleted of RAG1 cleavage targets (cRSS and heptamer-like motifs), which may serve as one means of protecting the genome from inappropriate RAG cleavage (22).
RAG1 targeting model—feature selection and performance
We next sought to define the features that, together with H3K4me3, are determinants of RAG1 targeting. We analyzed features associated with promoters, enhancers and RSSs (Supplementary Table S1) because of their likelihood of being informative for predicting RAG1 binding levels. RAG1 binding is highly restricted to promoter and enhancer regions and occurs in regions of the genome that are relatively depleted of potential cleavage sites (such as cRSSs and heptamers), but enriched in RAG1 binding motifs (nonamers) (22,26). We restricted our search to the H3K4me3 peak set, since more than 99% of RAG1 peaks overlap with H3K4me3 peaks (22). Using data from mouse thymocytes, the H3K4me3 peak set was characterized with respect to the above features and was then used to train a regression model for RAG1 binding level.
We used SVR, which can capture non-linear relationships, as the learning algorithm (see ‘Materials and Methods’ section). First, we evaluated the extent to which each of the selected features in Supplementary Table S1 contributed to the prediction of RAG1 level by calculating the MSE of the SVR model (the mean, over the set of peaks, of the squared differences between the real and predicted RAG1 binding levels). We then defined ‘importance’ of each feature as its differential MSE (ΔMSE)—the difference between the MSE of the SVR using the whole set of features and the MSE after randomizing the value of that feature. The higher the ΔMSE, the more important the feature to the prediction of RAG1 levels (Figure 3A). To filter out uninformative features, we ranked the features by their importance and iteratively trained the SVR model with an increasing number of features, starting with the most important feature. We then added features according to their importance ranking, calculating the MSE for each iteration, until the addition of features no longer resulted in a decreased MSE (Figure 3B).
This approach revealed five features that were informative for RAG1 binding (Figure 3A and B). H3K4me3 was the most important feature (t-test against random feature; P < 1e-42) and H3K27Ac also stood out in its importance (P < 1e-20). High DNaseI-HS (indicative of chromatin openness), %CA content and close proximity to a transcription start site (TSS) were also informative (P < 0.05 for each feature). Interestingly, the density of RSS-like elements was not significantly more important to the model than a random feature (P > 0.5). This emphasizes that selection against the presence of RAG1 cleavage motifs in off-target binding sites is focused on the heptamer, as reflected in the importance of %CA (22). To characterize the regions that attract RAG1 with respect to these informative features, we split the H3K4me3 peak set into RAG1hiand RAG1lo peaks, as described in ‘Materials and Methods’ section. Compared to RAG1lo peaks, RAG1hi peaks were typified by high H3K4me3, high H3K27Ac, chromatin openness (high DNaseI-HS), close proximity to TSSs and depletion of CA dinucleotides (Figure 3C; t-test; P = 0 for all features). We then evaluated the ability of the RAG1 targeting model (built on mouse thymocyte data) to predict RAG1 binding levels in mouse pre-B cells and human thymocytes. The RAG1 binding sites predicted by the RAG1 targeting model were then compared to the actual RAG1 binding patterns identified in our previously-generated RAG1 ChIP-seq datasets (22), to obtain a residual for each peak. The fraction of peaks as a function of residual cut-off was calculated and the area under curve (AUC) was used to evaluate performance. To trace the theoretical maximal AUC, we calculated the absolute differences between two RAG1 replicates taken from the actual RAG1 ChIP-seq data, with performance quantified as normalized AUC (AUC divided by the maximal AUC). The full model was able to predict RAG1 levels better than a linear regression model based on either H3K4me3 alone or H3K27Ac alone for mouse pre-B cells (0.96, 0.92 and 0.88 normalized AUC, respectively; Figure 3D). Prediction for human thymocytes gave similar results (0.91, 0.86 and 0.87, for full model, H3K4me3 alone and H3K27Ac alone, respectively; Supplementary Figure S2). The performance of a negative control in which the model was trained using permuted RAG1 levels was substantially poorer (0.75 normalized AUC for both mouse pre-B cells and human thymocytes). Therefore, the RAG1 binding pattern can be explained by similar chromatin features in different RAG1-expressing cell-types.
RAG1 peaks are divided into two clusters—one H3K4me3-driven and one H3K27Ac-driven
While ΔMSE of a feature, defined above, measures the overall importance of a feature to the prediction of the total peak set, it does not give information about the prediction of individual peaks. To identify which of the above features are important for the prediction of each peak, we first calculated the residual of each peak in the regression model (the absolute difference between the real and predicted RAG1 levels of a peak). We defined the differential residual (Δresidual) for feature i and peak j as the absolute difference of peak j residual in the regression model using all the five selected features, and the residual of the same peaks after randomizing feature i (see ‘Materials and Methods’ section). We then performed hierarchical clustering of the RAG1hi peaks by their relative feature importance, thereby arranging RAG1hipeaks, each represented by a vector of its feature importance, along a dendrogram based on the Euclidian distance between them. The dendrogram can then be divided into clusters.
Two clusters of RAG1 peaks were identified: a large cluster (Cluster 1) in which H3K4me3 is particularly important for predicting RAG1 binding levels (2368 out of the 2675 peaks) and a smaller cluster (Cluster 2) in which H3K27Ac is of particular importance (307 peaks) (Figure 4A and Supplementary Table S2). Cluster 1 predominantly represents promoters (high H3K4me3, low H3K27Ac, TSS proximal; Figure 4B; filled orange boxes) and Cluster 2 is composed primarily of enhancers (H3K4me3 low, H3K27Ac high, TSS distal; filled purple boxes). The two clusters showed similar levels of DNaseI-HS.
We next determined the differences between the two RAG1hi clusters and the corresponding RAG1lo regions. We identified a set of RAG1lo promoters as H3K4me3 peaks that were similar to RAG1hi promoters in their H3K4me3 levels and distance from TSS, but showed RAG1 normalized levels lower than 1 (Figure 4B, open orange boxes; an example of RAG1hi and RAG1lo promoters is shown in Figure 4C). We found that RAG1hi promoters had higher levels of H3K27Ac and lower CA dinucleotide percentage compared to RAG1lo promoters (t-test; P < 1e-95 and P < 1e-100, respectively). Similarly, RAG1lo enhancers were defined as peaks that were significantly similar to RAG1hi enhancers in their H3K27Ac levels and distance from TSS (Figure 4B, open purple boxes; an example of RAG1hi and RAG1lo enhancers is shown in Figure 4D). As in promoters, RAG1hi enhancers had higher levels of H3K4me3 and lower CA dinucleotide percentage than RAG1lo enhancers (t-test; P < 1e-113 and P < 1e-17, respectively). DNaseI-HS of both RAG1hi promoters and enhancers was similar to their RAG1lo counterparts (P = 0.22 and P = 0.6 for promoters and enhancers, respectively).
RAG1 clusters represent different modes of chromatin interaction
We then asked whether the two RAG1 binding clusters represent distinct modes by which RAG1 interacts with chromatin. For Cluster 1, driven by H3K4me3, we hypothesized that recruitment of RAG1 would depend strongly on its interaction with RAG2 and the ability of RAG2 to bind H3K4me3 via its PHD finger. This hypothesis was testable using RAG1 ChIP-seq data from R2ΔC thymocytes (Figure 1B). For Cluster 2, driven by H3K27Ac, we presumed that H3K4me3- and RAG2 PHD-independent mechanisms would play a significant role (see schematic in Figure 5A). Given recent findings implicating the N-terminal RAG1 non-core region in binding and ubiquitylation of histones (10,11,28), we used RAG1 ChIP-seq data from cR1 thymocytes (Figure 1B) as a starting point to probe for a role for RAG1 non-core regions in establishing Cluster 2. Comparisons were performed on RAG1 binding data in thymocytes from WT, R2ΔC and cR1 mice, focusing on the RAG1 peaks defined by Clusters 1 and 2 and not the many additional peaks that appear in the mutant backgrounds (22).
WT thymocytes exhibited slightly higher levels of RAG1 binding in Cluster 1 compared to Cluster 2 (Figure 5B; P < 1e-5) and strikingly, this Cluster 1 bias became much stronger in cR1 thymocytes (P < 1e-35). In contrast, RAG1 binding in R2ΔC thymocytes re-distributed toward Cluster 2 (H3K27Ac-driven) at the expense of Cluster 1 (H3K4me3-driven; P < 1e-8), consistent with the hypothesis that RAG1 binding to Cluster 1 is strongly dependent on RAG2–PHD interactions with H3K4me3. These results strongly suggest that the WT RAG1 binding pattern arises from a mixture of RAG2/H3K4me3-dependent and -independent modes, with the former decoupled in the absence of the RAG2 PHD finger and the latter decoupled in the absence of the RAG1 non-core regions.
To explore this idea further, we determined how the correlations between RAG1 levels and H3K4me3, H3K27Ac and DNaseI-HS levels were affected in the mutant genotypes compared to WT. Strikingly, the moderate correlation that exists between RAG1 and H3K4me3 in WT thymocytes is substantially strengthened in the cR1 genotype and virtually ablated in the R2ΔC genotype (Figure 5C top row; Supplementary Table S3). This indicates that in the absence of the RAG1 non-core regions, H3K4me3 becomes even more dominant in directing RAG1 binding, consistent with the loss of a competing mode of chromatin association. The opposite pattern was observed for H3K27Ac, in which the correlation with RAG1 levels was weakened in the cR1 genotype and substantially strengthened in the R2ΔC genotype (Figure 5C middle row; Supplementary Table S3). Thus, in the absence of the RAG2 C-terminal region, a recruitment mode that correlates with H3K27Ac levels becomes dominant. Interestingly, the correlation between RAG1 binding and DNaseI-HS strengthened in both cR1 and R2ΔC compared to WT (Figure 5C, bottom row and Supplementary Table S3), suggesting that the absence of either of the two presumed chromatin ‘anchor’ domains for RAG1 (the RAG2 PHD and RAG1 N-terminal region) allows for increased non-specific DNA binding.
Interestingly, the distribution of RAG1 in R2-/- mouse thymocytes closely resembled that seen in R2ΔC mouse thymus, reflected in both the same preferential redistribution of RAG1 binding to Cluster 2 (Supplementary Figure S3A; Wilcoxon test; P < 1e-9) and strengthening of the correlation, relative to WT, of RAG1 binding intensity with H3K27Ac and DNaseI-HS over H3K4me3 (Supplementary Figure S3B). The RAG1 binding levels, however, are significantly lower in the total absence of RAG2 compared to both WT and R2ΔC (Supplementary Figure S3C; Paired Wilcoxon test; P = 0). This suggests that while the PHD domain strongly dictates RAG1 distribution, the RAG2 core region strengthens the overall RAG1 ChIP-seq signal (by an as yet unknown mechanism) but has little influence on RAG1 distribution (Supplementary Figure S3D).
In summary, RAG1 binding patterns are driven by RAG2–H3K4me3 dependent and -independent interactions, mediated, at least partially, by the RAG2–PHD and the non-core domain of RAG1, respectively (see Figure 5A for schematic representation). These interactions drive RAG1 to different types of elements in the genome: promoters and enhancers.
RAG1 levels, measured and predicted, correlate with RAG1-mediated vulnerability
RAG1 off-target cleavage activity is not randomly distributed, but is focused to promoters and enhancers (22,24–26). Our recent study, showing that RAG1 binds predominantly to such regions, provided a mechanistic rationale for this preference (22). However, a direct link between RAG1 binding and RAG1 off-target activity has not been demonstrated to date. To explore this link, we looked for a correlation between RAG1 levels and the frequency of RAG1-mediated structural variants (SVs). We calculated the enrichment of SVs in an increasing number of H3K4me3 peaks, ordered by either H3K4me3 or RAG1 levels for the top ranked 500–3000 peaks. We examined a set of 107 translocation hotspots identified in an Abelson-transformed mouse pre-B cell line (v-abl), all of them harboring heptamer-like sequences (24) using a bootstrapping approach (resampling data with replacement). RAG1 levels correlated well with SV enrichment (Figure 6A, filled blue boxes; Spearman correlation coefficient ρ = 1). Ranking the peaks by H3K4me3 (Figure 6A, orange boxes) yielded a much lower level of enrichment (paired t-test P < 1e-10 for the top ranked 500 peaks), and did not show the same level of correlation as RAG1-based ranking (Spearman correlation coefficient ρ = 0.25).
Figure 6.

RAG1 levels, but not H3K4me3 levels, correlate with RAG1-mediated genomic instability. H3K4me3 levels (orange boxes), predicted RAG1 levels (empty blue boxes) and experimental RAG1 levels (filled blue boxes) in v-abl or REH cells, were calculated for H3K4me3 peaks. The top 3000 peaks were ranked according to H3K4me3, actual RAG1 levels or predicted RAG1 levels. A subset of the breakpoints (30 for v-Abl and 100 for B-ALL) were randomly sampled, and the enrichment of these breakpoint was calculated for each ranked group. Each boxplot represents the value distribution of 20 iterations.
We next determined if our RAG1 targeting model could capture the correlation between predicted RAG1 binding sites and RAG-mediated off-target cleavage events. Two datasets of RAG-mediated breakpoints were used for this purpose: the first contained the 107 breakpoints isolated from v-abl cells (24), and the second contained 434 breakpoints from RAG-mediated deletions identified in human patients with ETV6–RUNX1 B-acute lymphoblastic leukemia (26).
We applied the RAG1 targeting model to predict RAG1 binding sites in v-abl cells and a human B-ALL cell line (REH), which expresses RAG and performs V(D)J recombination (46). For v-abl cells, we used pro-B cell H3K4me3 and H3K27Ac ChIP-seq datasets (47) and pre-B ATAC-seq data (22,40) as inputs for the model. For REH, we generated an H3K27Ac ChIP-seq dataset, and combined it with previously published data for H3K4me3 ChIP-seq and DNaseI-HS-seq (48,49), along with TSS proximity and %CA. The output of the model was H3K4me3 peaks ranked by their predicted RAG1 occupancy. We tested the correlation between predicted RAG1 level ranking and RAG-mediated breakpoint enrichment. Similar to what was observed for experimentally-determined RAG1 levels, predicted RAG1 levels (empty blue boxes; Figure 6), show higher enrichment and better correlation with SVs than do H3K4me3 levels in both v-abl cells (paired t-test P < 1.8e-4 for the highest ranked 500 peaks and Spearman Correlation coefficient ρ = 0.96 and 0.25 for RAG1 and H3K4me3, respectively) and REH (P < 1e-4 for the highest ranked 500 peaks and ρ = 0.94, −0.28 for RAG1 and H3K4me3, respectively). The predicted RAG1 levels in v-abl cells were not as strongly correlated with SVs as were actual RAG1 levels, nor were they as well correlated with SVs as in REH (Figure 6), which might be due to our use of epigenetic data from primary pro/pre-B cells rather than v-abl cells themselves.
These results suggest that while promoters and enhancers are in general strong candidates for RAG-mediated genomic instability, RAG1 binding levels provide substantial addition information regarding their vulnerability to RAG off-target cleavage. Importantly, H3K4me3 levels alone were insufficient to predict sites of off-target RAG activity. Furthermore, we demonstrate that the RAG1-targeting model described here can be used to identify candidate sites for RAG1-mediated genomic instability in normal and cancer cells.
DISCUSSION
RAG1–chromatin modes of interaction
There are at least four ways by which proteins can be targeted to chromatin: (i) direct binding to a specific DNA sequence motif, (ii) indirect binding through a chromatin-bound protein partner, (iii) binding to histones, and (iv) non-specific binding to DNA. Our data suggest that RAG1 localization to chromatin is a mixture of all the above (Figure 1A) and below we consider the evidence for each mode of interaction.
Direct DNA binding
Sequence-specific recruitment of RAG1 and RAG2 to bRSSs occurs both in vivo and in vitro (30,50,51). It has been proposed that preferential targeting of RAG1 to antigen receptor genes can be attributed to the combined signature of high H3K4me3 density and strong RSSs (52). We found that the cumulative RAG1 binding sites in the antigen receptor loci are indeed RSS-centric. In the recombination centers spanning Jα and Jκ segments, where RAG1 is thought to bind initially, the combination of high H3K4me3 levels and strong RSSs allows RAG2-bound RAG1 to interact with the RSSs. Strikingly, the strength of the RSS-centrism is less pronounced at gene segments within the recombination centers, which are contained within a region that is broadly marked by high H4K4me3 density and high chromatin openness, than at gene segments outside of recombination centers. This, combined with the strong resemblance between RAG and H3K4me3 profiles in recombination centers (22), strongly suggests that chromatin structure and histone modifications are important in determining the RAG binding pattern in recombination centers, with sequence-specific DNA interactions also contributing. The balance between the various modes of RAG recruitment to chromatin in recombination centers remains to be determined. After the initial binding of RAG to the recombination center, partner RSSs in the V regions (Vα and Vκ) are thought to be captured in a synaptic complex. Unlike the RSSs in recombination centers, the Vα and Vκ RSSs are less enriched in H3K4me3, making the RSSs the core elements for RAG1 binding. Indeed, although RAG1 binding levels at Vα and Vκ regions barely exceed background (Figure 2A), they show the strongest RSS-centered peak. Recent results from our laboratory show that a 12/23 RSS pair recruits RAG1 better than a single RSS or a 12/12 or 23/23 RSS pair (30). This supports the idea that the observed RSS specificity of RAG1 recruitment at H3K4me3-low V regions is achieved through synapsis.
RAG1 targeting outside of the antigen receptor genes, however, appears to differ substantially from that of classical DNA binding proteins (such as transcription factors), whose genome-wide binding patterns typically show strong correlations to preferred DNA binding motifs. The cumulative RAG1 binding patterns genome-wide (exclusive of the antigen receptor genes) showed no evidence of cRSS-centrality. This is completely consistent with our previous analyses, in which we found that cRSSs are actually depleted in the vicinity of off-target RAG1 binding sites (22). In addition, we show here that the presence of an RSS sequence is neither sufficient nor required for RAG1 binding to chromatin. Thus, the sequence-specific RAG1 binding profile appears to be a distinctive feature of the antigen receptor genes and the high-quality RSSs contained therein, and the RAG1 recruitment patterns genome-wide must arise primarily from one or more sequence-independent mechanisms.
While RAG1 binding is generally sequence-independent, some minor sequence preferences are observed. In an earlier study, we compared sequence features of RAG1(+) H3K4me3 peaks in thymocytes and pre-B cells to similarly-ranked H3K4me3 peaks from other tissues (22). We found that while RAG1(+) H3K4me3 peaks in lymphocytes are depleted in heptamer-like sequences, they are enriched in nonamer-like sequences, suggesting a limited role for nonamers in RAG1 recruitment. Interestingly, in the current RAG1-targeting model, nonamer concentration was not selected as an informative feature, as its removal did not affect the performance of prediction. This discrepancy suggests that while, on average, RAG1 binding regions have a higher nonamer concentration than expected, the difference is too subtle to be captured at the peak level. In addition, in both clusters (particularly Cluster 2), H3K4me3 levels inversely correlate with nonamer density (Supplementary Figure S4A), consistent with our previous observation that nonamer density is lower in the immediate vicinity (±1 kb) of TSSs compared to more distal regions (22), with TSS proximal regions typified by particularly high H3K4me3 levels. We do not know whether this inverse correlation reflects evolutionary selection against nonamers in regions of high H3K4me3 or other functional constraints on the sequences in these regions. Interestingly, a positive correlation is seen between H3K27Ac levels and nonamer density in Cluster 2 peaks, but not in Cluster 1 (Supplementary Figure S4A). Hence, Cluster 2 contains a subset of peaks with high H3K27Ac, a high relative density of nonamers and high levels of RAG1 binding. It is possible that nonamers contribute to RAG1 binding in in Cluster 2 but that this contribution is masked in our analysis by the much stronger predictive signal provided by H3K27Ac levels.
Indirect recruitment of RAG1 to H3K4me3-rich chromatin via RAG2
The model of RAG1 targeting presented here describes general principles that underlie RAG1 targeting to chromatin. The dominant mode of RAG1–chromatin interaction is mediated by RAG2, which essentially acts as an adaptor to direct RAG1 to H3K4me3. Several studies have demonstrated the role of the RAG2 C-terminus, specifically the PHD finger, in regulating both RAG1 binding and activity (20,53–55). Our non-linear regression model for RAG1 occupancy suggests that, for 88.5% of RAG1hi peaks (Cluster 1), H3K4me3 is by far the most important factor mediating RAG1 binding, consistent with the predictions of a prior theoretical analysis (52). Deletion of the RAG2 C-terminus decouples the indirect link between RAG1 and H3K4me3, selectively depletes RAG1 binding in the H3K4me3-driven Cluster 1 (Figure 5A) and allows for expanded RAG1 binding at thousands of new sites characterized by lower H3K4me3 density (22). Interestingly, this uncoupling allows for secondary, RAG2-independent modes of RAG1–chromatin interactions to become dominant, as discussed below.
RAG1 binding to enhancers (perhaps through direct interaction with histones)
For the majority of RAG1 binding sites, H3K27Ac density is the secondary correlate, after H3K4me3 density. However, we have also identified a subset of RAG1 binding sites (Cluster 2, representing 11.5% of the RAG1 peaks analyzed) for which H3K27Ac is the primary correlate of RAG1 binding. This subset of RAG1 binding sites reflects a RAG2-independent mode of interaction that depends, at least partially, on the non-core regions of RAG1, as manifested by depletion of RAG1 in the H3K27Ac-driven cluster of peaks in the absence of the non-core RAG1 regions.
Previous studies have implicated the RAG1 N-terminal E3 ligase domain as a module that directly interacts with and ubiquitylates histone H3 (10,28). It remains unknown whether this chromatin ‘writing’ activity of RAG1 is relevant to its function in V(D)J recombination. The preferential association of off-target RAG1 binding at H3K27Ac(+) enhancers raises the possibility that RAG1 may function as a direct ‘reader’ of the histone code. Indeed, histone acetylation has been reported to stimulate the efficiency of V(D)J recombination (56–58). However, there exists no experimental evidence that RAG1 directly binds to H3K27Ac, and it is possible that the correlation simply reflects a propensity of RAG1 to associate with some unidentified factor or feature correlated with the chromatin state found at active enhancers. We note that there are other chromatin modifications that, because of their association with promoters and enhancers, correlate with H3K4me3 and therefore with RAG1. We tested two such marks, H3K4me1 and H3K9Ac, for their correlation with RAG1. To generate estimates for H3K4me3-independent RAG1 correlations, we determined the Spearman correlation coefficient (ρ) for the histone mark with RAG1 and also with H3K4me3, and then calculated the RAG1 correlation score as the ratio ρ(RAG1): ρ(H3K4me3). While for H3K27Ac this ratio is 1.42, it drops to 0.89 for both H3K4me1 and H3K9Ac (Supplementary Figure S4B), suggesting that among these three marks, only H3K27Ac correlates with RAG1 in an H3K4me3-independent manner. Moreover, when we repeated this analysis using data from R2ΔC thymocytes, the ratio increased significantly relative to WT thymocytes for H3K27Ac but not for H3K4me1 and H3K9Ac (Supplementary Figure S4B; one-tailed Paired t-test (resampling); P = 4e-61, 1 and 1, respectively), suggesting a biologically significant correlation between RAG1 and H3K27Ac.
This secondary mode of RAG1 recruitment predominates in the absence of the RAG2 PHD, such that the RAG1 binding pattern shifts away from H3K4me3 and toward H3K27Ac. Recent studies have shown that the absence of the RAG2 C-terminal region results in elevated RAG-mediated genome instability (25,59). These findings together are consistent with the idea that the secondary mode of RAG1 recruitment to H3K27Ac-rich regions of the genome is more likely to result in off target RAG activity than the primary, H3K4me3-driven mode of recruitment. Interestingly, the H3K27Ac-driven cluster of RAG1 binding sites shows a much higher density of CA dinucleotides (Figure 4B; t-test; P < 1e-56) and heptamers (Supplementary Figure S4C; Wilcoxon test P < 1e-8) than does the H3K4me3-driven cluster, which should result in a higher density of potential RAG cleavage sites (24).
Nonspecific RAG1 binding to DNA
Our findings also suggest that binding of RAG1 to accessible, non-specific DNA sequences also contributes to its genome-wide localization. We have shown previously that mutation of key DNA-interacting residues in the RAG1 NBD disrupts both sequence-specific and non-specific RAG1 binding, indicating that the key DNA-binding module of RAG1 has an inherent, non-specific interaction with DNA (22). Such non-specific binding is further enhanced in the absence of RAG2 (31). Consistent with the idea that non-specific interactions with accessible DNA drive RAG1 localization, we find that chromatin openness (measured by DNaseI-HS) constitutes an informative feature for predicting RAG1 binding and that the degree of openness correlates with RAG1 levels. The predictive importance of DNAseI-HS increases in the absence of either the non-core domains of RAG1 or the RAG2-PHD finger (Figure 5C).
From binding to activity
Candidate cRSSs are found in large numbers throughout the genome, ranging from thousands to millions, depending on the criteria by which they are defined (24–27). Most of them, however, show no evidence for RAG activity. The cRSSs that are subject to off-target RAG activity map to promoters and enhancers (24–26), where RAG binding is enriched (22). Our findings here directly link RAG1 binding and RAG activity. Using data from v-abl cells (22,24), we show that RAG1 levels, but not H3K4me3 levels, correlate with enrichment in RAG1-mediated SVs. The strong agreement between sites of experimentally-identified RAG-dependent SVs and the sites of RAG1 binding predicted by our RAG1-targeting model (in both mouse v-abl pre-B cells and a human B-ALL cell line) demonstrate that the model captures this binding-activity correlation. Hence, common epigenetic and genetic features can be used to predict candidate targets of aberrant RAG activity. While this model narrows down the list of candidate cRSSs more than other existing definitions, its rate of false-positives (falsely identified cRSSs) and false-negatives (real cRSSs that were overlooked) are high when it comes to predicting specific cleavage sites of RAG in vivo. Features that capture the cross-talk between pairs of RSSs might provide some of the missing criteria for predicting RAG1 activity. In this regard, Hu et al. suggest that the initiation of RAG activity genome-wide requires paired RSSs that occupy the same topological activation domain and lie in convergent orientation with respect to one other (24).
Supplementary Material
Acknowledgments
The authors wish to thank Yoram Louzoun and Gilad Liberman for valuable insight and advice regarding computational approaches used in this study, and to Lindsay Cowell and Min Kim for genome-wide RSS scanning.
ACCESSION NUMBERS
The accession numbers for ChIP-seq datasets for RAG1 in WT mouse thymocytes and for H3K27Ac in REH cells are in Gene Expression Omnibus (GEO): GSE84052.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R37 AI32524 to D.G.S.]; NRSA Institutional Postdoctoral Training Grant [T32 AI007019]; Cancer Research Institute Irvington Postdoctoral Fellowship [to G.T]. Funding for open access charge: National Institutes of Health [R37 AI32524 to D.G.S.]. D.G.S. is an investigator of the Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1.Ru H., Chambers M.G., Fu T.M., Tong A.B., Liao M., Wu H. Molecular mechanism of V(D)J recombination from synaptic RAG1-RAG2 complex structures. Cell. 2015;163:1138–1152. doi: 10.1016/j.cell.2015.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swanson P.C. The bounty of RAGs: recombination signal complexes and reaction outcomes. Immunol. Rev. 2004;200:90–114. doi: 10.1111/j.0105-2896.2004.00159.x. [DOI] [PubMed] [Google Scholar]
- 3.Schatz D.G., Swanson P.C. V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 4.Fugmann S.D., Schatz D.G. Identification of basic residues in RAG2 critical for DNA binding by the RAG1-RAG2 complex. Mol. Cell. 2001;8:899–910. doi: 10.1016/s1097-2765(01)00352-5. [DOI] [PubMed] [Google Scholar]
- 5.Jones J.M., Simkus C. The roles of the RAG1 and RAG2 ‘non-core’ regions in V(D)J recombination and lymphocyte development. Arch. Immunol. Ther. Exp. (Warsz) 2009;57:105–116. doi: 10.1007/s00005-009-0011-3. [DOI] [PubMed] [Google Scholar]
- 6.Dudley D.D., Sekiguchi J., Zhu C.M., Sadofsky M.J., Whitlow S., DeVido J., Monroe R.J., Bassing C.H., Alt F.W. Impaired V(D)J recombination and lymphocyte development in core RAG1-expressing mice. J. Exp. Med. 2003;198:1439–1450. doi: 10.1084/jem.20030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horowitz J.E., Bassing C.H. Noncore RAG1 regions promote Vbeta rearrangements and alphabeta T cell development by overcoming inherent inefficiency of Vbeta recombination signal sequences. J. Immunol. 2014;192:1609–1619. doi: 10.4049/jimmunol.1301599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones J.M., Gellert M. Autoubiquitylation of the V(D)J recombinase protein RAG1. Proc. Natl. Acad. Sci. U.S.A. 2003;100:15446–15451. doi: 10.1073/pnas.2637012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yurchenko V., Xue Z., Sadofsky M. The RAG1 N-terminal domain is an E3 ubiquitin ligase. Gene Dev. 2003;17:581–585. doi: 10.1101/gad.1058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grazini U., Zanardi F., Citterio E., Casola S., Goding C.R., McBlane F. The RING domain of RAG1 ubiquitylates histone H3: a novel activity in chromatin-mediated regulation of V(D)J joining. Mol. Cell. 2010;37:282–293. doi: 10.1016/j.molcel.2009.12.035. [DOI] [PubMed] [Google Scholar]
- 11.Jones J.M., Bhattacharyya A., Simkus C., Vallieres B., Veenstra T.D., Zhou M. The RAG1 V(D)J recombinase/ubiquitin ligase promotes ubiquitylation of acetylated, phosphorylated histone 3.3. Immunol. Lett. 2011;136:156–162. doi: 10.1016/j.imlet.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Sutherland H.G., Mumford G.K., Newton K., Ford L.V., Farrall R., Dellaire G., Caceres J.F., Bickmore W.A. Large-scale identification of mammalian proteins localized to nuclear sub-compartments. Hum. Mol. Genet. 2001;10:1995–2011. doi: 10.1093/hmg/10.18.1995. [DOI] [PubMed] [Google Scholar]
- 13.Musselman C.A., Kutateladze T.G. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi X.B., Kachirskaia I., Walter K.L., Kuo J.H.A., Lake A., Davrazou F., Chan S.M., Martin D.G.E., Fingerman I.M., Briggs S.D., et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 2007;282:2450–2455. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taverna S.D., Ilin S., Rogers R.S., Tanny J.C., Lavender H., Li H.T., Baker L., Boyle J., Blair L.P., Chait B.T., et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pena P.V., Davrazou F., Shi X., Walter K.L., Verkhusha V.V., Gozani O., Zhao R., Kutateladze T.G. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Ilin S., Wang W., Duncan E.M., Wysocka J., Allis C.D., Patel D.J. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimazaki N., Tsai A.G., Lieber M.R. H3K4me3 stimulates the V(D)J RAG complex for both Nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol. Cell. 2009;34:535–544. doi: 10.1016/j.molcel.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grundy G.J., Yang W., Gellert M. Autoinhibition of DNA cleavage mediated by RAG1 and RAG2 is overcome by an epigenetic signal in V(D)J recombination. Proc. Natl. Acad. Sci. U.S.A. 2010;107:22487–22492. doi: 10.1073/pnas.1014958107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu C., Ward A., Bettridge J., Liu Y., Desiderio S. An autoregulatory mechanism imposes allosteric control on the V(D)J recombinase by histone H3 methylation. Cell Rep. 2015;10:29–38. doi: 10.1016/j.celrep.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji Y., Resch W., Corbett E., Yamane A., Casellas R., Schatz D.G. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng G., Maman Y., Resch W., Kim M., Yamane A., Qian J., Kieffer-Kwon K.R., Mandal M., Ji Y., Meffre E., et al. RAG represents a widespread threat to the lymphocyte genome. Cell. 2015;162:751–765. doi: 10.1016/j.cell.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis S.M., Agard E., Suh S., Czyzyk L. Cryptic signals and the fidelity of V(D)J joining. Mol. Cell. Biol. 1997;17:3125–3136. doi: 10.1128/mcb.17.6.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu J., Zhang Y., Zhao L., Frock R.L., Du Z., Meyers R.M., Meng F.L., Schatz D.G., Alt F.W. Chromosomal loop domains direct the recombination of antigen receptor genes. Cell. 2015;163:947–959. doi: 10.1016/j.cell.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mijuskovic M., Chou Y.F., Gigi V., Lindsay C.R., Shestova O., Lewis S.M., Roth D.B. Off-target V(D)J recombination drives lymphomagenesis and is escalated by loss of the Rag2 C terminus. Cell Rep. 2015;12:1842–1852. doi: 10.1016/j.celrep.2015.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papaemmanuil E., Rapado I., Li Y.L., Potter N.E., Wedge D.C., Tubio J., Alexandrov L.B., Van Loo P., Cooke S.L., Marshall J., et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cowell L.G., Davila M., Kepler T.B., Kelsoe G. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0072. RESEARCH0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng Z., Liu H., Liu X. RAG1-mediated ubiquitylation of histone H3 is required for chromosomal V(D)J recombination. Cell Res. 2015;25:181–192. doi: 10.1038/cr.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin F.F., Bailey S., Innis C.A., Ciubotaru M., Kamtekar S., Steitz T.A., Schatz D.G. Structure of the RAG1 nonamer binding domain with DNA reveals a dimer that mediates DNA synapsis. Nat. Struct. Mol. Biol. 2009;16:499–508. doi: 10.1038/nsmb.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shetty K., Schatz D.G. Recruitment of RAG1 and RAG2 to chromatinized DNA during V(D)J recombination. Mol. Cell. Biol. 2015;35:3701–3713. doi: 10.1128/MCB.00219-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao S.Y., Gwyn L.M., De P., Rodgers K.K. A non-sequence-specific DNA binding mode of RAG1 is inhibited by RAG2. J. Mol. Biol. 2009;387:744–758. doi: 10.1016/j.jmb.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raghavan S.C., Swanson P.C., Wu X., Hsieh C.L., Lieber M.R. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- 33.Piper J., Elze M.C., Cauchy P., Cockerill P.N., Bonifer C., Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 2013;41:e201. doi: 10.1093/nar/gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cortes C., Vapnik V. Support-Vector Networks. Mach. Learn. 1995;20:273–297. [Google Scholar]
- 36.Mittas N., Angelis L. A permutation test based on regression error characteristic curves for software cost estimation models. Empir. Softw. Eng. 2012;17:34–61. [Google Scholar]
- 37.Liang H.E., Hsu L.Y., Cado D., Cowell L.G., Kelsoe G., Schlissel M.S. The ‘dispensable’ portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 2002;17:639–651. doi: 10.1016/s1074-7613(02)00448-x. [DOI] [PubMed] [Google Scholar]
- 38.Bailey T.L., Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40:e128. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schatz D.G., Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011;11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- 40.Mandal M., Hamel K.M., Maienschein-Cline M., Tanaka A., Teng G., Tuteja J.H., Bunker J.J., Bahroos N., Eppig J.J., Schatz D.G., et al. Histone reader BRWD1 targets and restricts recombination to the Igk locus. Nat. Immunol. 2015;16:1094–1103. doi: 10.1038/ni.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwickert T.A., Tagoh H., Gultekin S., Dakic A., Axelsson E., Minnich M., Ebert A., Werner B., Roth M., Cimmino L., et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat. Immunol. 2014;15:283–293. doi: 10.1038/ni.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey T.L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pio F., Assa-Munt N., Yguerabide J., Maki R.A. Mutants of ETS domain PU.1 and GGAA/T recognition: free energies and kinetics. Protein Sci. 1999;8:2098–2109. doi: 10.1110/ps.8.10.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGrath P.T., Lee H., Zhang L., Iniesta A.A., Hottes A.K., Tan M.H., Hillson N.J., Hu P., Shapiro L., McAdams H.H. High-throughput identification of transcription start sites, conserved promoter motifs and predicted regulons. Nat. Biotechnol. 2007;25:584–592. doi: 10.1038/nbt1294. [DOI] [PubMed] [Google Scholar]
- 45.Plessy C., Dickmeis T., Chalmel F., Strahle U. Enhancer sequence conservation between vertebrates is favoured in developmental regulator genes. Trends Genet. 2005;21:207–210. doi: 10.1016/j.tig.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 46.Hansenhagge T.E., Yokota S., Reuter H.J., Schwarz K., Bartram C.R. Human common acute lymphoblastic leukemia-derived cell-lines are competent to recombine their T-cell receptor delta/alpha regions along a hierarchically ordered pathway. Blood. 1992;80:2353–2362. [PubMed] [Google Scholar]
- 47.Lane A.A., Chapuy B., Lin C.Y., Tivey T., Li H., Townsend E.C., van Bodegom D., Day T.A., Wu S.C., Liu H., et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. 2014;46:618–623. doi: 10.1038/ng.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guenther M.G., Lawton L.N., Rozovskaia T., Frampton G.M., Levine S.S., Volkert T.L., Croce C.M., Nakamura T., Canaani E., Young R.A. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Gene Dev. 2008;22:3403–3408. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kreher S., Bouhlel M.A., Cauchy P., Lamprecht B., Li S., Grau M., Hummel F., Kochert K., Anagnostopoulos I., Johrens K., et al. Mapping of transcription factor motifs in active chromatin identifies IRF5 as key regulator in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E4513–E4522. doi: 10.1073/pnas.1406985111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.vanGent D.C., Ramsden D.A., Gellert M. The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell. 1996;85:107–113. doi: 10.1016/s0092-8674(00)81086-7. [DOI] [PubMed] [Google Scholar]
- 51.Hiom K., Gellert M. A stable RAG1-RAG2-DNA complex that is active in V(D)J Cleavage. Cell. 1997;88:65–72. doi: 10.1016/s0092-8674(00)81859-0. [DOI] [PubMed] [Google Scholar]
- 52.Askary A., Shimazaki N., Bayat N., Lieber M.R. Modeling of the RAG reaction mechanism. Cell Rep. 2014;7:307–315. doi: 10.1016/j.celrep.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gigi V., Lewis S., Shestova O., Mijuskovic M., Deriano L., Meng W., Luning Prak E.T., Roth D.B. RAG2 mutants alter DSB repair pathway choice in vivo and illuminate the nature of 'alternative NHEJ'. Nucleic Acids Res. 2014;42:6352–6364. doi: 10.1093/nar/gku295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y., Subrahmanyam R., Chakraborty T., Sen R., Desiderio S. A plant homeodomain in Rag-2 that binds hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity. 2007;27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthews A.G., Kuo A.J., Ramon-Maiques S., Han S., Champagne K.S., Ivanov D., Gallardo M., Carney D., Cheung P., Ciccone D.N., et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kwon J., Morshead K.B., Guyon J.R., Kingston R.E., Oettinger M.A. Histone acetylation and hSWI/SNF remodeling act in concert to stimulate V(D)J cleavage of nucleosomal DNA. Mol. Cell. 2000;6:1037–1048. doi: 10.1016/s1097-2765(00)00102-7. [DOI] [PubMed] [Google Scholar]
- 57.McBlane F., Boyes J. Stimulation of V(D)J recombination by histone acetylation. Curr. Biol. 2000;10:483–486. doi: 10.1016/s0960-9822(00)00449-8. [DOI] [PubMed] [Google Scholar]
- 58.McMurry M.T., Krangel M.S. A role for histone acetylation in the developmental regulation of VDJ recombination. Science. 2000;287:495–498. doi: 10.1126/science.287.5452.495. [DOI] [PubMed] [Google Scholar]
- 59.Deriano L., Chaumeil J., Coussens M., Multani A., Chou Y., Alekseyenko A.V., Chang S., Skok J.A., Roth D.B. The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature. 2011;471:119–123. doi: 10.1038/nature09755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.