


Abstract
Protein-primed replication constitutes a generalized mechanism to initiate DNA or RNA synthesis in a number of linear genomes of viruses, linear plasmids and mobile elements. By this mechanism, a so-called terminal protein (TP) primes replication and becomes covalently linked to the genome ends. Bam35 belongs to a group of temperate tectiviruses infecting Gram-positive bacteria, predicted to replicate their genomes by a protein-primed mechanism. Here, we characterize Bam35 replication as an alternative model of protein-priming DNA replication. First, we analyze the role of the protein encoded by the ORF4 as the TP and characterize the replication mechanism of the viral genome (TP-DNA). Indeed, full-length Bam35 TP-DNA can be replicated using only the viral TP and DNA polymerase. We also show that DNA replication priming entails the TP deoxythymidylation at conserved tyrosine 194 and that this reaction is directed by the third base of the template strand. We have also identified the TP tyrosine 172 as an essential residue for the interaction with the viral DNA polymerase. Furthermore, the genetic information of the first nucleotides of the genome can be recovered by a novel single-nucleotide jumping-back mechanism. Given the similarities between genome inverted terminal repeats and the genes encoding the replication proteins, we propose that related tectivirus genomes can be replicated by a similar mechanism.
INTRODUCTION
Tectiviruses infecting Bacillus cereus sensu lato species include temperate phages Bam35, GIL01, GIL16, AP50 or Wip1 (1), which are related to the B. cereus linear plasmid pBClin15 (2). The interest in these viruses has recently increased, due to the narrow host specificity of some of them for dangerous human pathogens (3,4), their complex diversity and variability patterns (5,6), as well as their possible phylogenetic relationship with some groups of eukaryotic viruses and mobile elements (7,8). Tectiviruses also include a different group of viruses, infecting Gram-negative bacteria and represented by PRD1 and closely related phages that can only replicate in lytic cycle within cells harboring certain conjugative plasmids (9). Both tectivirus groups have an almost indistinguishable morphology and a linear dsDNA of about 15 Kbp with a protein, named terminal protein (TP), covalently linked at both 5′-ends (10). These structural common features and a similar genome organization, with inverted terminal repeats (ITRs), suggest a common ancestor for all tectiviruses (10,11), prior to the diversification of the Gram-negative and Gram-positive bacteria, even though they share almost no sequence identity at the nucleotide level (10,12,13).
TPs become covalently attached to the genome by means of their primary function serving as primers for DNA synthesis (14). This mechanism has been characterized in several virus groups, including adenovirus (15), PRD1 (16,17) and especially in the podovirus Φ29 (14,18). Replication of those linear genomes starts from both ends of the terminal protein-containing DNA (TP-DNA) by the formation of a phosphoester bond of the first nucleotide to an OH group of a serine, threonine or tyrosine residue of the TP and progresses asymmetrically from both ends. Processive genome replication can be carried out in vitro often only with two proteins, the DNA polymerase (DNAP) and the TP, although additional factors, like single- and double-stranded DNA binding proteins strongly stimulate the replication and are essential in vivo (14,18–21). In some systems, these DNA binding proteins are indeed required for genome replication in vitro (22,23). In the case of Φ29, the TP and the DNA polymerase form a complex that recognizes the TP-containing genome ends. Then, replication initiation occurs at the second nucleotide of the terminal repetition of the template strand (3′TTT). To perform TP-DNA full-length synthesis, the TP-dAMP initiation product translocates backward one position to recover the template information corresponding to the first 3′T, in a so-called sliding-back mechanism (see below and Supplementary Figure S1). Thus, the terminal repetition permits, prior to DNA elongation, the asymmetric translocation of the initiation product, TP-dAMP, to be paired with the first T residue (24). Likewise, PRD1 replication initiates at the fourth nucleotide of a 3′CCCC repetition (17), and it requires three consecutive sliding-back steps to recover the DNA end information. In a further level of complexity, the adenovirus genome ends present a reiteration, 3′GTAGTA, and the formation of a TP-CAT initiation product is directed by the positions fourth to six in the template strand. Thus, recovery of the information comprising the three first nucleotides at the 3′ end is performed by a single jump, after which TP-CAT is paired with the terminal 3′GTA (jumping-back) (25). After the recovery of the 3′ ends information, the DNA polymerase continues to elongate the template still in complex with the TP but subsequent dissociation of the TP/DNA polymerase complex is required to allow the DNA polymerase to resume the replication which, as has been shown for Φ29, can be highly processive and coupled to strand displacement (26).
DNA polymerases involved in protein-primed DNA replication belong to the B family and present two specific amino acid regions, TPR1 and TPR2, involved in the interaction with the TP and in processivity and strand displacement capacity, respectively (27,28). In addition to meeting these characteristics, Bam35 DNA polymerase (B35DNAP) is able to processively bypass abasic sites in DNA (29). These features, along with the presence of ITRs and a TP covalently linked to the viral genome (10), strongly suggest that Bam35 genome replication is performed by a protein-primed mechanism. However, until now, this mechanism has not been described and the ORF that codes for the TP has not been characterized. Contrary to DNA polymerases, TP sequences are poorly conserved, even among members of the same viral family, like PRD1 and Bam35. Thus, the putative TP that would prime the replication process of Gram-positive tectiviruses cannot be directly inferred by sequence homology. However, we and others have previously suggested the ORF4 as a putative TP (6,30,31), based in the synteny with DNA polymerase and the accumulation of positive charged residues at the N-terminal half of the amino acid sequence.
In this work, we show that the Bam35 protein 4 is indeed the viral TP that can be specifically deoxynucleotidylated by the B35DNAP either in the absence or presence of a DNA template containing the sequence of the viral genome ends. This reaction occurs at tyrosine 194 of the TP, which is conserved in all tectivirus orthologs. We have also shown that both, the Bam35 TP and DNA polymerase, are required and sufficient for replication of full-length viral TP-DNA genome in vitro, in a protein-primed mechanism that is initiated at the third base of the template strand. Moreover, a subsequent third-to-first template base single-nucleotide jumping-back process can be responsible for the recovery of the information of the 3′ end sequence. This novel jumping-back alternative was unpredicted and unexpected in bacteriophages and might be a simplified reminiscent of the Adenovirus replication mechanism. These findings are discussed in the context of other protein-primed viral genome replication mechanisms and the similarities between the sequence of the origins and the TPs sequence homologies among Bam35 and related tectiviruses.
MATERIALS AND METHODS
Nucleotides and DNAs
Nucleotides were purchased from GE Healthcare. [α-32P]dATP (3000 Ci/mmol), [α-32P]dTTP (3000 Ci/mmol), [α-32P]dCTP (3000 Ci/mmol) and [α-32P]dGTP (3000 Ci/mmol) (1 Ci = 37 GBq) were supplied by PerkinElmer Inc. DNA oligonucleotides (Supplementary Table S1) were from Sigma.
Bam35 virus stock and its host, Bacillus thuringiensis HER1410, were provided by Prof. Jaana K. Bamford (University of Jyväskylä, Finland). Gene 4 of Bam35 DNA was amplified by PCR using as a template the Bam35 genome, B35TP-fw, B35TP-rv primers (Supplementary Table S1) and Vent DNA polymerase (New England Biolabs), and cloned into NdeI-BamHI sites of pT7-7 plasmid (32) to obtain the Bam35 TP expression vector (pT7-7::B35TP). The Bam35 TP mutants were obtained by directed mutagenesis using the pT7-7::B35TP plasmid as the template, the oligonucleotides indicated in Supplementary Table S1 and the Pfu DNA polymerase (New England Biolabs). The YFP fusion to the Bam35 TP (YFPTP) was produced from a vector, obtained by cloning the TP PCR product into the EcoRI/BamHI sites of pStrep-YFP plasmid (33). The resulting vector, pStrep-YFP::B35TP, was used to express a N-terminal strep-tagged YFP fusion terminal protein (YFPTP). In all cases, the complete Bam35 TP gene was sequenced.
To obtain Bam35 TP-DNA, a Bacillus thuringiensis HER1410 culture was grown in Luria Broth medium (LB) at 30°C up to a cell density of 3.6 × 107 bacteria per ml. Then, 10 mM MgCl2 was added to the culture and it was infected with Bam35 bacteriophage at a multiplicity of infection of 1 pfu/cell. When the cells lysed, the debris was removed by centrifugation at 12 000 × g for 10 min and the viral particles were precipitated by addition of 6% polyethylene glycol 6000 (Serva) for 2.5 h and subsequent centrifugation (25 000 × g for 20 min). The precipitate was dissolved in 6 ml of 10 mM potassium phosphate buffer (pH 7.2) and 10 mM MgCl2, at 4°C overnight. Viral suspension was further clarified by centrifugation at 25 000 × g for 20 min, and the viral particles from the supernatant were partially purified by consecutive phosphocellulose (P11, Whatman) and Q-Sepharose (GE Healthcare) chromatographic steps. Then, the eluted Bam35 bacteriophage particles were concentrated by precipitation, as described above. To disrupt the Bam35 viral particles, the pellet was resuspended in a buffer containing 4 M guanidine chloride, 25% (v/v) glycerol, 200 mM NaCl, 50 mM Tris-HCl, pH 7.5, 10 mM dithiothreitol (DTT), 1 mM EDTA and 0.05% (v/v) Tween® 20 (Sigma) for 90 min on ice. Finally, the solution was dialyzed against the same buffer without guanidine chloride and clarified by centrifugation. Final preparations containing Bam35 TP-DNA were analyzed by 0.7% alkaline agarose gel electrophoresis (34) followed by ethidium bromide staining.
Proteins
Restriction enzymes and T4 DNA ligase were from New England Biolabs. Proteinase K was from Affymetrix. Wild type Φ29DNAP, wild type Φ29TP and wild type Bam35 DNA polymerase (B35DNAP) were from the laboratory stocks and purified as described (29,35,36).
To produce the Bam35 TP as a fusion protein with the YFP (YFPTP), a culture of Escherichia coli Bl21(DE3) carrying the pStrep-YFP::B35TP vector was grown in LB medium with 150 μg/ml ampicillin at 30°C. When the OD600 was 0.3, 0.25 mM IPTG was added and the culture was grown at 24°C for 30 min. The cells were collected and the YFPTP protein was purified using Strep-Tactin resin (IBA GmbH), according to the manufacturer's instructions. Genes of wild type TP and mutants were also expressed in E. coli Bl21(DE3) harboring the corresponding expression vector in ZYM-5052 auto-induction medium (37) with 100 μg/ml ampicillin. The cultures were grown for 18 h at 27°C and the cells were collected and disrupted by grinding with alumina and suspended in buffer A (50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 7 mM β-mercaptoethanol, 5% (v/v) glycerol) containing 0.5 M NaCl. Alumina and cell debris were removed by centrifugation and the absorbance at 260 nm was adjusted to 120 units/ml prior to DNA precipitation with 0.3% polyethyleneimine and 1 M NaCl. After centrifugation at 14 000 × g for 20 min, ammonium sulfate was added to the supernatant to 65% saturation and centrifuged at 25 000 × g for 45 min. The pellet, containing either the wild type or mutant TP, was resuspended in buffer A at an ionic strength of about 0.4 M and applied to a Q-sepharose column (GE Healthcare). The flow-through containing the TP was then applied to a Whatman® P11 phosphocellulose column (Sigma). After an extensive washing with 0.5, 0.6 and 0.7 M NaCl in buffer A, proteins were eluted with 0.8 M NaCl and dyalized against a buffer containing 50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 7 mM β-mercaptoethanol, 50% (v/v) glycerol and 1 M NaCl. Final purity of the proteins was estimated to be >95% by SDS–polyacrylamide gel electrophoresis followed by Coomassie blue staining. The proteins were kept in the presence of 50% (v/v) glycerol at −20°C, or at −70°C for long-term storage.
Protein-primed initiation assays
The standard reactions contained 12.5 nM (25 ng) of B35DNAP and 34 nM (25 ng) of TP (or YFPTP) and were pre-incubated for 10 min at room temperature to allow them to interact. Then, the TP/B35DNAP complexes were added to a mixture that contained, in a final volume of 25 μl, 50 mM Tris-HCl (pH 7.5 or 8.5 as indicated), 1 mM DTT, 4% (v/v) glycerol, 0.1 mg/ml of bovine serum albumin (BSA), 0.05% (v/v) Tween® 20, 0.2 μM of the corresponding dNTP supplemented with 1 or 2 μCi of the same [α-32P]dNTP for template-independent or template-dependent reactions, respectively. When initiation assays were carried out in the presence of template, 315 nM of the indicated 29-mer single stranded oligonucleotide (Supplementary Table S1) or 75 ng of the B35 TP-DNA were also added. Reactions were triggered by the addition of either 1 mM MnCl2 or 10 mM MgCl2, as indicated. Unless otherwise stated, standard assays were incubated for 30 min at 37°C and stopped by adding 10 mM EDTA and 0.1% SDS (final concentrations). Finally, reaction products were subjected to SDS-12% polyacrylamide gel electrophoresis in MiniProtean III Gels (BioRad), using SeeBlue® Plus2 Pre-Stained Protein Standards (Invitrogen) as molecular weight markers. Reaction products were detected by autoradiography.
When used as a control, the Φ29 initiation reactions were carried out as described before (38), with 4 μCi [α-32P]dATP, 1 mM MnCl2 and 29-mer double stranded oligonucleotide with the Φ29 genome right end sequence as the template (Supplementary Table S1). The reactions were incubated for 30 min at 30°C and the samples were processed and analyzed as described above.
TP-DNA replication assay
A total of 11 nM of B35DNAP and 133 nM of Bam35 TP were incubated for 10 min at room temperature before addition to an incubation mixture that contained, in 25 μl, 50 mM Tris-HCl, pH 7.5, 1 mM DTT, 4% (v/v) glycerol, 0.1 mg/ml of BSA, 0.05% (v/v) Tween® 20, 150 mM NaCl, 20 mM ammonium sulfate, 20 μM each of the four dNTPs, 1 μCi [α-32P]dATP and 100 ng of the Bam35 TP-DNA. The reactions were triggered by addition of 1 mM MnCl2 or 10 mM MgCl2, incubated for the indicated times at 37°C and stopped as indicated above prior to gel filtration through Sephadex G-50 spin columns containing 0.1% SDS. The TP-DNA replication products were analyzed by 0.7% alkaline agarose gel electrophoresis and detected by autoradiography. The lambda DNA ladder used as a size marker was labeled by gap-filling in the presence of [α-32P]dATP (39).
Cell-free extracts assays and mapping of the priming residue
In cell-free extracts assays, E. coli Bl21(DE3) cells harboring either empty pT7.7 or the indicated pT7.7::B35TP for expression of wild type or mutant TPs were grown for 18 h at 27°C in ZYM-5052 auto-induction medium (37) with 100 μg/ml ampicillin. Cells from 1 ml culture were harvested by centrifugation and resuspended in 0.5 ml of buffer A containing 0.5 M NaCl. After sonication and centrifugation for 10 min at 20 000 x g (4°C), the supernatants containing the soluble proteins were diluted 1:1 with glycerol. Similar level of expression and solubility of all TP variants was verified by SDS-PAGE and Coomassie staining (Supplementary Figure S2).
The nature of the priming residue was determined by alkali treatment of the initiation product. Thus, a template-independent initiation assay was carried out as described above but with increased concentration of labeled [α-32P]dATP (4 μCi) and the samples were treated with 100 mM NaOH for 6 min at 95°C. Subsequently, 100 mM HCl was added to neutralize the NaOH.
The position of the priming residue was delimited by protein cleavage with cyanogen bromide. To do so, template-independent initiation reaction was performed with higher concentration of TP (272 nM) and longer incubation time (overnight at 37°C) to maximize the reaction yield. Then, reaction products were treated with 1.2 mM of cyanogen bromide (CBrN from Sigma) in the presence of 200 mM HCl and incubated for 20 h at room temperature (40). Finally, samples were neutralized and analyzed by SDS-18% polyacrylamide gel electrophoresis.
Analysis of the TP/DNA polymerase interaction by a functional assay
We designed a functional assay to compare the interaction of wild type and mutant TPs with the B35DNAP. Namely, we determined the capacity of Bam35 TP mutants to inhibit the deoxyadenylation of the wild type YFPTP fusion protein. The reactions were carried out essentially in the same conditions described for the template-independent initiation assay with 1 mM MnCl2, and using a fixed amount of B35DNAP (12.5 nM) and different concentrations of wild-type and Bam35 TP mutants. After 2.5 min, 34 nM of YFPTP was added and reactions were resumed for an additional 2.5 min. Total protein amount in the samples was kept constant by supplemental addition of BSA when required. Reactions were stopped by adding 10 mM EDTA and 0.1% SDS and were processed and analyzed as described above. Gel bands were detected by autoradiography and quantified with ImageJ software (41).
Single stranded oligonucleotide truncated replication assay
The reactions were carried out essentially in the same conditions described above for the oligonucleotide-mediated initiation assay, but in the presence of 5 μM of the indicated dNTP(s) and 100 μM ddNTP to obtain truncated replication products. To allow separation of the different replication intermediates, the samples were analyzed by high-resolution SDS-15% polyacrylamide gels (245 × 300 × 0.5 mm) and detected by autoradiography.
RESULTS AND DISCUSSION
ORF4 of the Bam35 genome encodes a functional terminal protein
The initiation step of protein-primed DNA replication involves the linking of the first deoxynucleotide to the TP, a reaction directed by a complementary nucleotide in the template strand and performed by the replicative DNA polymerase. Additionally, template-independent TP-deoxynucleotidylation may be detected in vitro, preferentially with manganese as a divalent metal cofactor (16,42). Thus, to confirm if Bam35 protein encoded by ORF4 can be a functional TP we first analyzed whether the purified recombinant protein could be substrate for deoxyadenylation by the viral DNA polymerase (B35DNAP). As shown in Figure 1, this reaction gave rise to a radiolabeled product only when both proteins were present and the band obtained had the expected electrophoretic mobility of purified TP, as indicated by Coomassie staining (about 32 kDa, lanes 1 and 5). Moreover, this band migrates slower when an YFP fusion protein (YFPTP) was used as substrate (59 kDa, lanes 2 and 7). Note that, despite the presence of contaminant bands in the YFPTP that may be degradation products, only one band of TP-dAMP product is detected, which suggest that those contaminant bands would correspond to degraded fusion proteins that would contain the strep-tag used for affinity chromatograpy purification but not the TP portion. In both cases, the product band disappeared after proteinase K treatment, confirming that they correspond to 32P-dAMP labeled protein. This reaction could be detected with either magnesium or manganese, although manganese was more efficient as cofactor (Supplementary Figure S3A), as previously shown in other systems (16,43). The pH also affects the Bam35 TP deoxyadenylation, being more efficient at slightly basic pH (Supplementary Figure S3B), in agreement with the high isoelectric point of the protein (about 10.6). In the manganese-mediated reaction this effect was not very high, but when magnesium was used the efficiency of the reaction increased significantly, since at pH 7.5 the deoxynucleotidylated TP was almost undetectable. Thus, we used preferentially manganese as metal cofactor for initiation reactions, in a reaction buffer at pH 7.5 and, when indicated, the effect of magnesium was analyzed in a reaction buffer adjusted to pH 8.5 to facilitate the detection of the reaction products.
Figure 1.
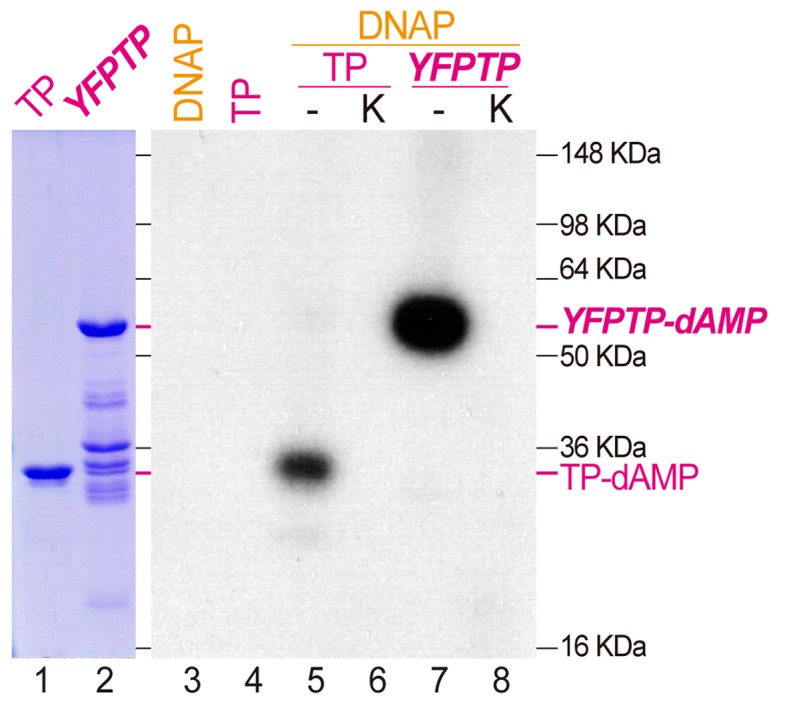
Bam35 TP functional assay. Lanes 1 and 2 show 1 μg of purified TP and YFPTP, respectively. The Bam35 TP deoxyadenylation assay (primer-independent initiation, lanes 3–8) was carried out incubating 12.5 nM B35DNAP (except in lane 4) and 34 nM of either Bam35 TP (lanes 5 and 6) or YFPTP (lanes 7 and 8). Reactions were triggered by 1 mM MnCl2 and incubated for 30 min at 37°C. After the assay, samples of lanes 6 and 8 were treated with 0.5 μg/μl of proteinase K and 0.1% of SDS for 20 min at 37°C. Finally, all reaction products were analyzed by 12% SDS-PAGE. See Materials and Methods for details.
In order to analyze whether, besides template-independent deoxyadenylation, Bam35 TP could serve as primer for viral DNA replication in vitro, we incubated the Bam35 TP-DNA with the TP and the DNA polymerase in the presence of dNTPs using magnesium as cofactor, obtaining a band of the expected size of the Bam35 genome (about 15 kB, Figure 2A, lanes 4–5). Control reactions without the DNA polymerase or the TP (lanes 1 and 2, respectively) did not result in any replication product. However, the reaction was not very efficient in these conditions and long incubation times were required to reach the genome full-length size, as compared with PRD1 or Φ29 TP-DNA genome replication (16,26). Thus, the possible role of other accessory proteins might be required for efficient replication, as has been shown for other protein-primed viral genomes (21,22). Interestingly, whereas manganese is a more efficient cofactor for the template-independent deoxyadenylation, magnesium was required to obtain full-length genome molecules (Figure 2B), since the use of manganese as cofactor, alone or together with magnesium resulted in an increase of the total product signal but with less capacity to reach the full-length TP-DNA, most likely due to an impairment of DNA polymerase processivity. These results are in agreement with other systems, like PRD1 (16) or Φ29 (24,43), in which initiation is favored with manganese but processive full-length genome replication requires magnesium. The use of each of these metal cofactors for DNA polymerases is controversial and strongly affects their performance and fidelity (44,45). Moreover, its availability in vivo, particularly of manganese, is highly regulated in bacteria (46). Nevertheless, although the biological role of manganese or magnesium mediated reactions in vivo may be uncertain, we have been able to show TP-deoxynucleotidylation and full-length genome replication in vitro with only two purified proteins, the TP and the DNAP, which allows us to conclude that the Bam35 protein encoded by ORF4 is the viral TP, essential for protein-primed genome replication.
Figure 2.
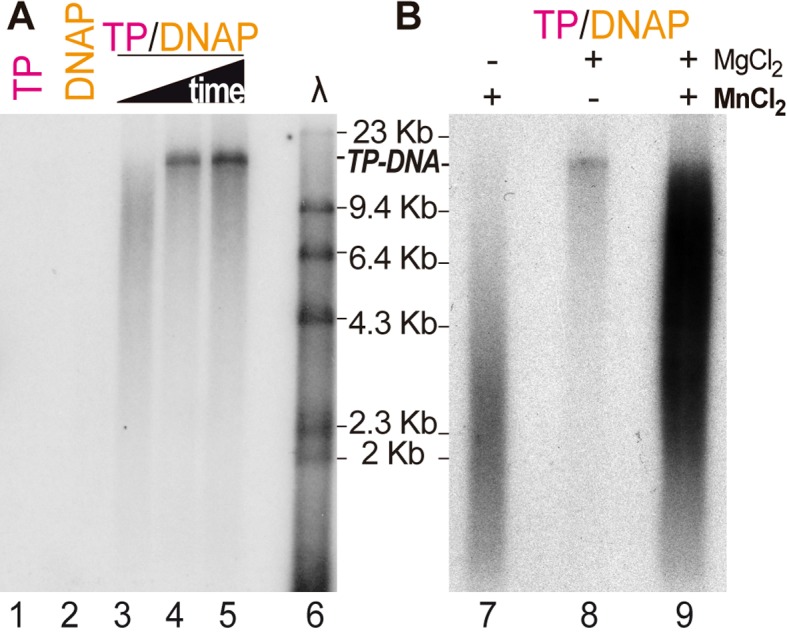
Bam35 protein-primed genome replication. Alkaline agarose gel electrophoresis of TP-DNA replication products. Samples contained 11 nM B35DNAP and 133 nM TP, as indicated, and 100 ng of Bam35 TP-DNA. See Materials and Methods for details. (A) Reactions were triggered by addition of 10 mM MgCl2 and incubated for 1, 2 and 4 h (lanes 3–5). (B) Reactions were triggered by 10 mM MgCl2 and/or 1 mM MnCl2 as indicated and incubated for 2 h. A λ-HindIII DNA ladder was loaded as a size marker, and the expected size of the Bam35 TP-DNA product is also indicated.
Conserved Tyrosine 194 of Bam35 TP is the priming residue for DNA replication
As mentioned above, the initiation reaction in protein-primed DNA replication consists in the addition, catalyzed by the DNA polymerase, of one deoxynucleotide to the hydroxyl group provided by a serine, threonine or tyrosine of the TP. We performed a hot alkali treatment of the initiation product that is known to disrupt the phosphoester bond of a serine or threonine residue to the dAMP, whereas a phosphoester bond to a tyrosine would be resistant to this treatment (14,49). As shown in Figure 3B, Bam35 TP-dAMP complex is resistant to alkaline conditions, whereas Φ29 TP-dAMP product (formed through the TP serine 232) is sensitive (50), strongly suggesting that the priming residue is a tyrosine. Bam35 TP amino acid sequence contains 15 tyrosine residues (Figure 3A), although only two, in positions 172 and 194 are conserved among all related sequences, as expected for a priming residue. To delimit the location of the priming tyrosine residue, we performed a cyanogen bromide (CNBr) cleavage that breaks polypeptide chains after each methionine. Bam35 TP only has two methionines, at positions 1 and 190; thus only two fragments of 22.4 and 6.2 KDa would be generated by CNBr cleavage. As shown in Figure 3C, only a labeled fragment of about 6 kDa was detected, indicating that the priming tyrosine residue is in the C-terminal portion of the TP comprising residues 191–245. This result pointed to Y194 as the putative priming residue. In order to confirm that, we performed mutagenesis to phenylalanine of all the tyrosine residues in this fragment (highlighted in pink in Figure 3A).
Figure 3.

Mapping Bam35 TP priming residue. (A) Multiple sequence alignment of Bam35 TP and related TPs. Sequences used were from putative TPs (proteins encoded by ORF4) of representative related Gram-positive tectiviruses Bam35 (NCBI ID NP_943750.1, 10), GIL16 (YP_224102.1, 47), AP50 (YP_002302516.1, 30), as well as other BLAST-retrieved orthologous sequences from NR protein database and tentatively annotated as bacterial proteins from Bacillus cereus (WP_001085581.1), Streptococcus pneumoniae (WP_050224775.1), Exiguobacerium antarticum (WP_026829749.1), Bacillus flexus (WP_025907183.1) and Brevibacillus sp. CF112 (WP_007784052.1). These bacterial proteins may correspond to TPs from uncharacterized tectivirus-like prophages or linear plasmids from Gram-positive hosts. Sequences were aligned with MUSCLE algorithm implemented in Geneious R8 software (48). The C-terminal fragment of all proteins that corresponds with the bromide cyanogen cleavage product is shadowed in blue and the tyrosine residues present in the Bam35 portion are highlighted in pink. Conserved Y172 and Y194 residues are marked with an asterisk above the sequences. (B) Determination of the nature of the Bam35 TP priming residue by alkali treatment. Control initiation reactions with Φ29 DNA polymerase and TP were performed in parallel. After the initiation reaction, samples were incubated for 6 min at 95°C in the absence or presence of 100 mM NaOH, and subsequently neutralized and analyzed by SDS-PAGE and autoradiography. (C) Mapping the Bam35 TP priming residue. The TP-AMP complexes were performed as described and afterward treated with 1.2 mM of cyanogen bromide (CNBr) and 200 mM HCl for 20 h at room temperature. Finally, the samples were neutralized and analyzed by SDS-18% polyacrylamide electrophoresis. (D) Identification of Y194 as the priming residue by TP-deoxyadenylation assays with 0.5 or 2 μl of cell-free extracts of bacterial cultures expressing the TP variants. Extracts prepared from bacteria harboring the empty plasmid (lanes 1, 2) and the wild type TP expression vector (lanes 3, 4) were also used as negative or positive controls, respectively. See Materials and Methods for details.
Given that the initiation reaction requires the specific interaction of TP and DNA polymerase, template-independent initiation assays could be performed using cell-free extracts of cells overexpressing each of the TP mutants (Figure 3D). All TP mutants except Y194F were deoxyadenylated, suggesting again that this residue may be the priming residue. This was confirmed using purified Y194F and Y194A TPs as substrate (Figure 4A). In order to verify that the mutation of Y194 was only affecting the priming residue and the protein functionality was maintained, we examined whether those mutants were able to interact with the DNA polymerase in a functional assay, as compared with the wild type YFPTP. To do so, we analyzed the decrease in the YFPTP deoxyadenylation product by previous incubation of the DNA polymerase with limiting amounts of either wild type or Y194F or Y194A TP mutants (Figure 4B and C). Wild type and Y194F TPs interact similarly with the DNA polymerase since increasing amounts of these proteins gave rise to a very similar inhibition pattern in YFPTP template-independent priming (Figure 4B lanes 2–5 and 7–10 and Figure 4C, black and red lines). On the other hand, Y194A TP required slightly higher concentration to reduce YFPTP deoxyadenylation (Figure 4B lanes 11–14 and Figure 4C green line). Therefore, these results showed that mutations in tyrosine 194 only affect the DNA priming capacity of the TP, thus indicating the role of this residue as an anchor for the first genome nucleotide.
Figure 4.
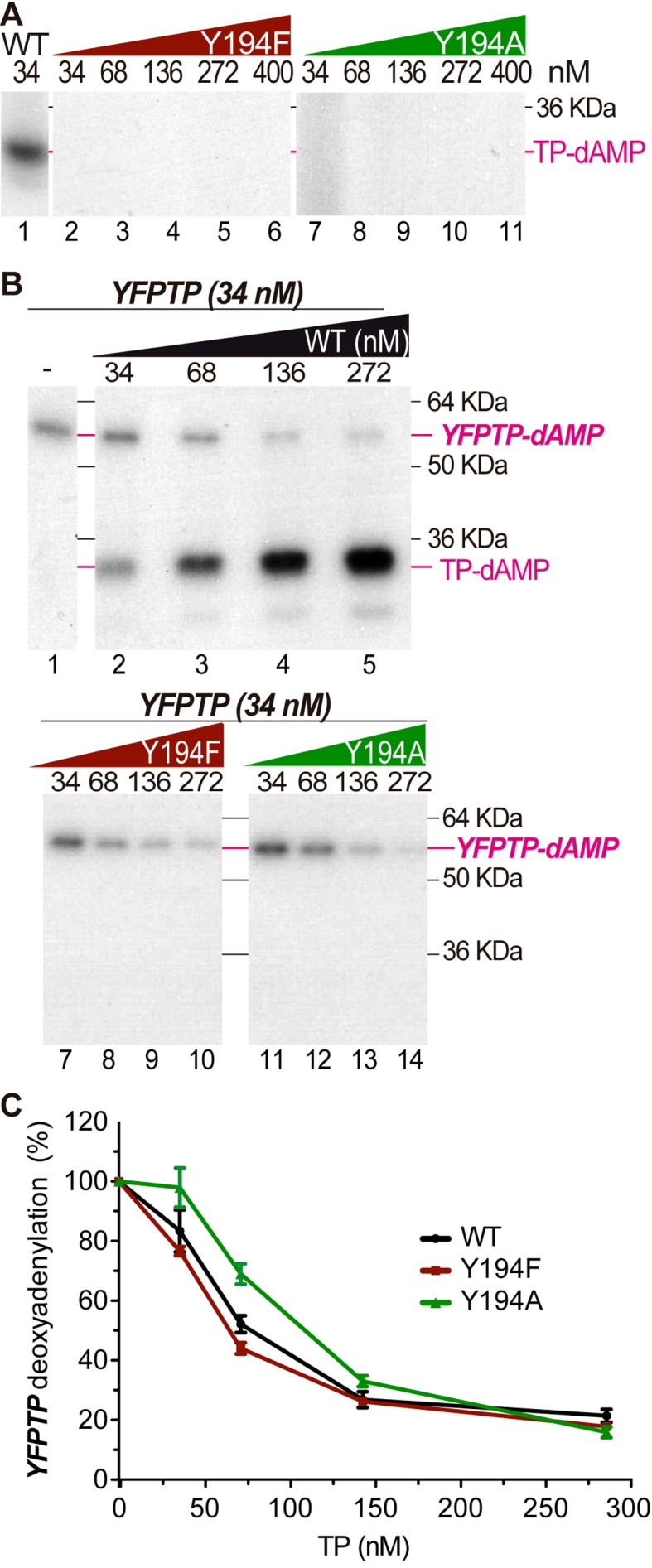
Functional characterization of mutants in the Y194 priming residue. (A) Template-independent TP-deoxyadenylation products of wild type (lane 1) and increasing concentrations of Y194F and Y194A TP mutants. Reactions were carried out triggered with 1 mM MnCl2 and incubated for 30 min. (B) Comparative analysis of wild type and Y194A and Y194F mutant TPs interaction with the DNA polymerase. The reactions were triggered with 1 mM MnCl2 in the presence of the indicated TP variant and, after 2.5 min, the competitor YFPTP fusion protein was added and the samples were incubated again for 2.5 min. See Materials and Methods for details. The effect of the TP variants concentration on the relative YFPTP deoxyadenylation, from three independent experiments (mean and standard error), is shown in panel C.
Conserved tyrosine 172 of Bam35 TP is not required for priming but it is involved in the interaction with B35DNAP
As mentioned above, besides Y194, Y172 was the only conserved tyrosine in all Bam35 related TP sequences (Figure 3A). Thus, we wondered if this residue might serve as a secondary priming residue or be involved in a different function. We analyzed the capacity of Y172F and Y172A TP mutants to be deoxyadenylated by the B35DNAP. Figure 5A shows that Y172A was almost inactive and the labeled protein band was only detected at the highest concentration used (lane 8). On the other hand, the deoxyadenylation of the Y172F conservative mutant was somewhat enhanced as compared with the wild type TP (lanes 9–12 versus 1–4). Thus, we can conclude that residue Y172 is not directly involved in the TP deoxynucleotidylation. To further examine the role of this conserved residue, we analyzed the functional interaction of the TP mutants with DNA polymerase (Figure 5B), and we found that at the concentrations analyzed, the Y172A mutant was not able to reduce the YFPTP deoxyadenylation below 70–80%, suggesting that this mutant may have an impaired interaction with B35DNAP. Strikingly, preincubation with the Y172F mutant decreased the YFPTP product much faster than the wild type TP, with a reduction up to 70% with equimolar concentrations of both proteins, suggesting an enhanced interaction of the Y172F variant with the B35DNAP, as compared with the wild type TP. Therefore, Bam35 TP tyrosine 172 may be involved in a hydrophobic interaction with the B35DNAP, that may be impaired by the elimination of the aromatic moiety (Y172A mutant) and somewhat strengthened by the elimination of the hydroxyl group (Y172F). Furthermore, we investigated whether this apparently tighter interaction of Y172F mutant and the B35DNAP may impair the dissociation that, as has been shown in the case of Φ29 (51), would be required to resume replication after the first replication steps. Thus, we analyzed template-dependent initiation and subsequent DNA replication with a 29-mer single stranded oligonucleotide containing the origin sequence. Figure 5C shows that with both proteins, wild type and Y172F mutant, the B35DNAP was able to replicate the oligonucleotide at a similar rate, suggesting that the improvement in the interaction of Y172F TP mutant with B35DNAP does not significantly affect subsequent steps of replication. Altogether, these results indicate that conserved Y172 in Bam35 TP could be involved in the interaction with the B35DNAP and underline the role of conserved Y194 as the only priming residue for the first nucleotide insertion in replication of Bam35, and likely in related tectiviral genomes.
Figure 5.
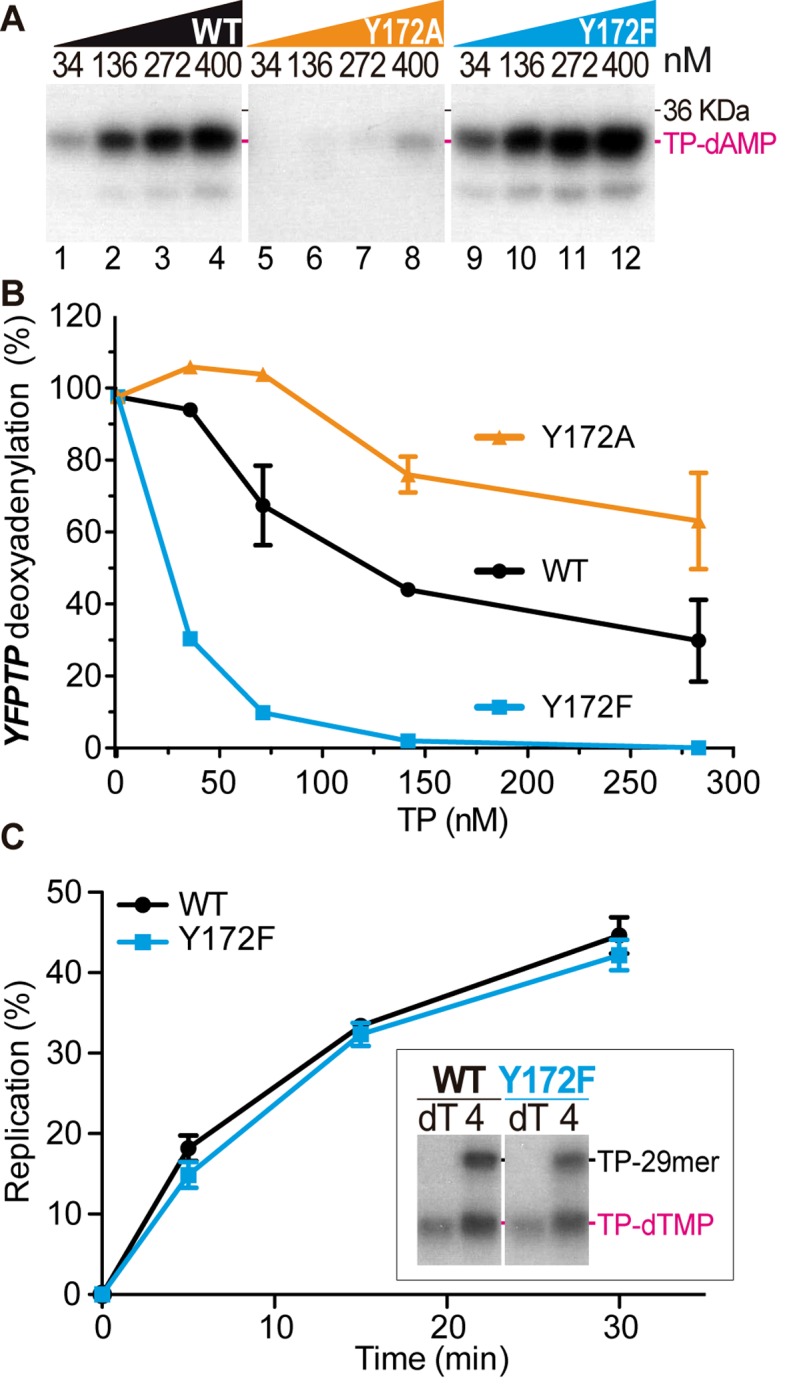
Role of conserved residue Y172 in the interaction with the DNAP. (A) Template-independent initiation products of increasing concentrations of wild type and Y172F and Y172A TP mutants. Reactions were carried out with the indicated concentrations of TP and triggered with 1 mM MnCl2. (B) Comparative analysis of wild type and Y172A and Y172F mutant TPs interaction with the DNA polymerase. Shown are mean and standard error of three independent experiments. See Materials and Methods for details. (C) TP-primed replication of 29-mer single stranded oligonucleotide template containing the Bam35 genome origin sequence, using either wild type or Y172F TPs as primer. Time-course of full-length replication, relative to the initial events. Shown are mean and standard error of three independent experiments. The inset panel shows a representative SDS-PAGE of initiation (with dTTP) and replication (with all 4 dNTPs) products primed by the wild type or Y172F TPs after 30 min of reaction.
Initiation of Bam35 DNA replication is mainly directed by the third nucleotide at the 3′ end of the template strand
To further characterize the early steps of the Bam35 genome replication we analyzed the deoxynucleotide preference for TP deoxynucleotidylation in the absence or presence of TP-DNA template. Template-independent TP deoxynucleotidylation was mainly dependent on dATP, either with manganese (Figure 6A) or with magnesium (Supplementary Figure S4A), and only a slight incorporation of dCMP could be detected using manganese as cofactor (Figure 6A, lane 4). A strong preference for dATP in template-independent TP-deoxynucleotidylation assays has been also reported for PRD1 (16), whereas adenovirus has a strong dCTP preference (25) and Φ29 shows a low base specificity for this reaction (42). Thus, the dAMP preference may be a common feature for tectivirus, although the possible biological meaning of this reaction is unclear, since the base preference seems to be different to the first base in genome replication initiation: dGMP for PRD1 TP-DNA replication (16) or dTMP in the case of Bam35 (see below).
Figure 6.

Deoxynucleotide specificity for Bam35 TP initiation reaction. B35DNAP and TP were incubated in template-independent (A) or TP-DNA directed (B) initiation assays. Template sequence determination of Bam35 TP initiation reaction is shown in (C). Initiation assays either in the absence of template (panel C, lanes 1, 8, 15 and 22) or with single stranded 29-mer oligonucleotide template containing the sequence of the genome left end or variants of this sequence (Supplementary Table S1). The deoxynucleotide used as well as the first six nucleotides of the template oligonucleotide sequence (in the 3′-5′ direction) are indicated above the gels. Reactions were triggered with MnCl2 (see Materials and Methods). Longer autoradiography exposition times, related to the dATP assays, are indicated for each provided deoxynucleotide.
When TP-DNA was used as template for the initiation reaction (Figure 6B), incorporation of dAMP was again more efficient than that of the other dNMPs (compare lanes 1–2 with the overexposed lanes 3–8). However, the presence of TP-DNA gave rise to a reduction of the dAMP and dCMP linking to the TP (lanes 2 and 4, as compared with 1 and 3), whereas dTMP incorporation was detected only when the TP-DNA was present in the reaction (lane 8). These results suggest that dTMP incorporation is directed by the TP-DNA template, whereas dAMP and dCMP linking to the TP might be—at least partially—a template-independent reaction, which would be reduced because the TP or the DNA polymerase may bind to the TP-DNA in a non-productive manner. However, only dAMP incorporation was detected when magnesium was the cofactor (Supplementary Figure S4B), indicating that, with or without template, the initiation step is more efficient with manganese.
As mentioned in the Introduction, protein-primed initiation is often directed by the second, third or fourth nucleotide of the template strand, and the full-length genome replication involves a backward mechanism to recover the information of the template. The first nucleotides of the template strand of both Bam35 genome ends are 3′ATAATACCATGGGG… Thus, dAMP may be incorporated opposite the second or fifth nucleotides of the template strand and dTMP if the first, third or fourth template position directs the initiation. We analyzed in detail which nucleotide from the template strand is directing the incorporation of the first nucleotide to the TP. To do so, we used a single stranded oligonucleotide containing the first 29 nucleotides of the template strand from the genome left end. Moreover, we designed oligonucleotides in which a C –absent from the first six positions – was placed in either first, second, third, fourth or fifth position. We performed an initiation assay without template or with each of these templates, using either dATP, dCTP, dGTP or dTTP as the initiator nucleotide and manganese (Figure 6C) or magnesium (Supplementary Figure S4C) as cofactor. Again, dATP was the most efficiently incorporated nucleotide with all templates (lanes 2–7), at a similar extent than in the template-independent reaction (lane 1). A slight reduction of intensity was detected when C was in the second position of the template, which was not detected with magnesium (Supplementary Figure S4C). Thus, Bam35 TP deoxyadenylation may be mainly a template-independent reaction. On the other hand, as shown above (Figure 6C, lane 8), dCMP only gave rise to a minor TP labeling in the absence of template, but no product could be detected with the template oligonucleotides (Figure 6C, lanes 9–14), indicating that dCMP cannot be used for replication initiation with any of these substrates. On the contrary, dGMP, which could not be incorporated in the absence of template (lane 15), could be more efficiently linked to the TP when a C was placed in the third position of the template (lane 19). A minor initiation product could be detected when a C was in the first, second and fourth positions (lanes 17, 18 and 20). These results indicate that the third position of the template strand is the major determinant for the deoxynucleotide specificity of the initiation step. This could be further confirmed when dTTP was provided to the reaction. In this case, the reaction product could not be detected without template (lane 22), but a product band was obtained with all the oligonucleotide templates, except with the oligonucleotide that had a C in the third position (lanes 23–25 and 27–28 versus lane 26) that prevents the incorporation of dTMP opposite to this position. Importantly, and despite the different efficiency, this specificity is not modified by the use of manganese or magnesium as cofactor (compare Figure 6C and Supplementary Figure S4C). Therefore, by gain (with dGTP) and loss (with dTTP) of function, we have been able to show that the third position of a template containing derived sequences of the Bam35 genome end is directing the replication initiation step, which suggests that TP-DNA replication is initiated at the third position of the template strand.
Jumping-back after protein-priming to recover the genetic information of the genome end
Protein-primed genome replication usually entails a terminal repetition that allows the recovery of the 3′-end information in the template strand after initiation in an internal position, by means of a backward translocation of the primed TP/DNAP complex (24). Based on the Bam35 ends sequence (3′ATAATA…), it can be readily hypothesized that the initiation opposite to the third terminal base that we have found should be resumed by a single-nucleotide jumping-back mechanism of the TP-dTMP/DNAP complex to the first base, and thus avoiding the loss of information. To disclose whether the information of the genome ends may be recovered or not by a backward translocation mechanism, we analyzed the replication of oligonucleotide templates using truncated replication assays with dideoxynucleotides (Figure 7, see Materials and Methods for details). To facilitate the step-by-step dissection of the replication mechanism we used a cytosine modified origin 3′CTCATA… (CTC in Figure 7) and monitored its initiation and early replication steps. For comparison, a template sequence in which the first and third bases are different to downplay the putative jumping-back and favor forward replication, 3′ATCATA… (ATC in Figure 7) was also used. A scheme of the alternative replication pathways of CTC and ATC templates is shown in panel B.
Figure 7.
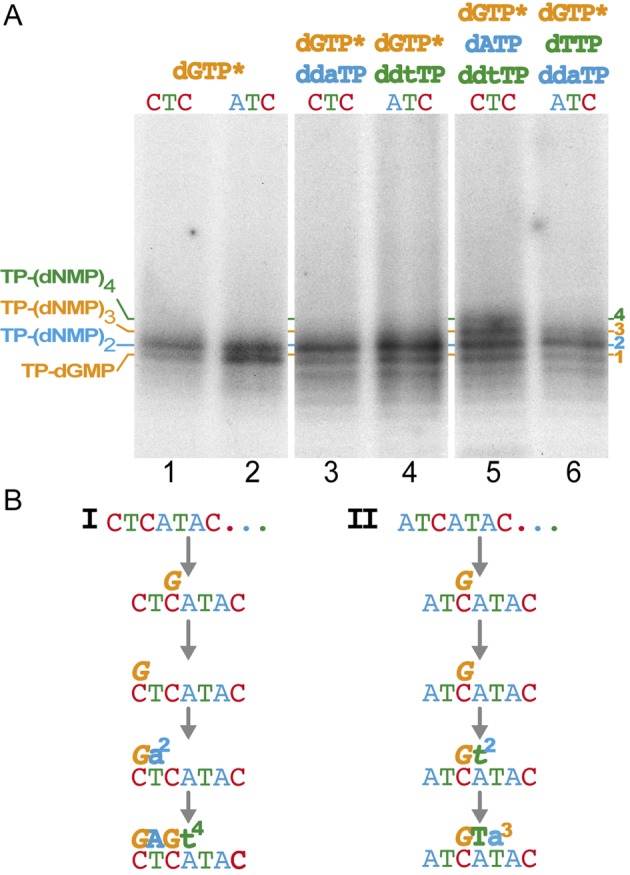
Detailed mechanism of early steps of protein-primed replication of single-stranded oligonucleotides containing Bam35 origins sequence variants. (A) High resolution electrophoresis of TP-primed replication products of 29-mer oligonucleotides. The two alternative sequences used 3′CTCATAC… and 3′ATCATAC…, respectively, are indicated as CTC and ATC above each lane. The combination of nucleotides included in each reaction is also detailed. The length of the different partially elongated products is indicated. Shorter bands that would correspond to TP degradation products (see Figure 1, lane 1) were not considered. Reactions were carried out as detailed in Materials and Methods, and triggered with manganese. Panel B show a schematic representation of alternative replication paths for CTC and ATC templates, respectively (see text for details). For clarity, initiator dGMP is highlighted in italic font and dideoxynucleotide bases are indicated in lower case.
The initiation reactions were more efficient with the ATC than with the CTC template (Figure 7A, lane 2 versus 1) and, due to the higher resolution of these gels, we detected two bands after the initiation in the presence of manganese. This could be the result of a dGMP misinsertion after the TP-dGMP complex formation, possible as consequence of a relative lower fidelity of the DNA polymerase when using manganese as cofactor. Then, when we provided a dideoxynucleotide to allow only one step of elongation, there was an increase of TP-(dNMP)2 band intensity, relative to the TP-dGMP initiation product (lanes 3 and 4), that would correspond to the TP-dGddA or TP-dGddT products for the CTC and ATC templates, respectively.
In the next step, we added 2 dNTPs and 1 ddNTP and we could detect clearly up to four bands with the CTC template (Figure 7A, lane 5), which only could be the product of the elongation of the translocated TP-dGMP complex, taking into account the very low level of initiation obtained when C is in the first position of the template (Figure 6C, lane 17), thus supporting a jumping-back (from the third to the first position, path I in panel B). On the other hand, only three bands could be detected with the ATC sequence that does not allow the backward translocation and only might be replicated straightforward (without a jumping-back, path II). This different replication pattern strongly suggests that, after initiation opposite to the third template base (Figure 6), a backward translocation to the first position can occur prior to forward replication of the CTC template. Overall, truncated replication of single stranded oligonucleotides showed that TP is substrate for protein-primed DNA replication that can undergo a third-to-first positions single-nucleotide jumping back. Given that the actual sequence of both Bam35 genome origins has the 3′ATAATA… repetition, it could be suggested that this mechanism is the favored alternative in vivo, as has been shown for adenovirus replication (25).
CONCLUSION AND PERSPECTIVES
Despite the lack of sequence conservation between different viral groups, bacteriophage genome-attached TPs have important common functional and structural hallmarks (52), like a similar modular arrangement, with the priming residue in the C-terminal half of the protein and an accumulation of positively charged residues at the N-terminal portion. We have confirmed here that the protein encoded by the ORF4 of Bam35 is the bacteriophage TP that can be used as a primer for the full-length genome replication by the viral DNA polymerase, through the formation a phosphoester bond between dTMP and the conserved tyrosine 194. This priming reaction is directed by the third base of the template and the genetic information of the genome end can be recovered by a single-nucleotide jumping-back mechanism. In contrast, previously described backward translocation mechanisms in bacteriophages (Φ29, Cp-1 or PRD1, see Introduction) consist in a single nucleotide or one-by-one nucleotide sliding back over a short template repetition. On the other hand, recent phylogenetic analysis suggested that tectiviruses would have gave rise to a number of major groups of dsDNA viruses, including Adenovirus (7,8). Therefore, since a jumping-back is required for Adenovirus replication whereas Gram-negative tectiviruses replication initiation is followed by a stepwise sliding back process (Supplementary Figure S1), it is tempting to speculate that the Bam35 replication mechanism may be at the origin of adenovirus replication mechanism.
Interestingly, the genome ends of closely related phages Gil01 and Gil16 have very similar sequence to that of Bam35, with only minor differences in the first base of one or both ends (Supplementary Figure S1). We do believe that these specific variations in close genomes may be due to the technical difficulties in sequencing the DNA ends of a TP-containing genome. Thus, it is tempting to speculate that Bam35, Gil01 and Gil16 genomes replicate by the same mechanism shown here. However, AP50 genome ends do not contain a clear ITR that may be replicated by a backward translocation. The increase of available sequences from Gram-positive tectivirus genome ends and analysis of replication mechanisms will be required to know whether the single-nucleotide jumping-back is a common feature of these viruses and eventually to gain insights into the evolutionary pathway of this mechanism, in relation with the podovirus and PRD1 sliding-back and the adenovirus triple-nucleotide jumping-back.
Finally, it should be mentioned that Φ29 or Adenovirus replication systems have been successfully engineered for the generation ad hoc of protein-capped DNAs for diverse biotechnological purposes (36,53,54). Interestingly, since Bam35 and related tectiviruses can replicate as episomal linear plasmids in bacteria, they might be manipulated or evolved both in vitro or in vivo, as per convenience.
Supplementary Material
Acknowledgments
The authors thank Dr J. K. Bamford for Bam35 phage and B thuringiensis stocks, José María Lázaro for technical assistance and Dr Miguel de Vega for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Economy and Competitiveness [BFU2014-52656P to M.S.]; ComFuturo Grant from Fundación General CSIC [NewPols4Biotech to M.R-R.]; M.B-O. was holder of a PhD fellowship [FPI, BES-2012-052228] from the Spanish Ministry of Economy and Competitiveness; Institutional grant from Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa. Funding for open access charge: Spanish Ministry of Economy and Competitiveness [BFU2014-52656P].
Conflict of interest statement. None declared.
REFERENCES
- 1.Gillis A., Mahillon J. Phages preying on Bacillus anthracis. Bacillus cereus, and Bacillus thuringiensis: past, present and future. Viruses. 2014;6:2623–2672. doi: 10.3390/v6072623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stromsten N.J., Benson S.D., Burnett R.M., Bamford D.H., Bamford J.K. The Bacillus thuringiensis linear double-stranded DNA phage Bam35, which is highly similar to the Bacillus cereus linear plasmid pBClin15, has a prophage state. J. Bacteriol. 2003;185:6985–6989. doi: 10.1128/JB.185.23.6985-6989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischetti V.A. Exploiting what phage have evolved to control gram-positive pathogens. Bacteriophage. 2011;1:188–194. doi: 10.4161/bact.1.4.17747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kan S., Fornelos N., Schuch R., Fischetti V.A. Identification of a ligand on the Wip1 bacteriophage highly specific for a receptor on Bacillus anthracis. J. Bacteriol. 2013;195:4355–4364. doi: 10.1128/JB.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillis A., Mahillon J. Prevalence, genetic diversity, and host range of tectiviruses among members of the Bacillus cereus Group. Appl. Environ. Microbiol. 2014;80:4138–4152. doi: 10.1128/AEM.00912-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jalasvuori M., Palmu S., Gillis A., Kokko H., Mahillon J., Bamford J.K., Fornelos N. Identification of five novel tectiviruses in Bacillus strains: analysis of a highly variable region generating genetic diversity. Res. Microbiol. 2013;164:118–126. doi: 10.1016/j.resmic.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Krupovic M., Koonin E.V. Polintons: a hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol. 2014;13:105–115. doi: 10.1038/nrmicro3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koonin E.V., Krupovic M., Yutin N. Evolution of double-stranded DNA viruses of eukaryotes: from bacteriophages to transposons to giant viruses. Ann. N. Y. Acad. Sci. 2015;1341:10–24. doi: 10.1111/nyas.12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olsen R.H., Siak J.S., Gray R.H. Characteristics of PRD1, a plasmid-dependent broad host range DNA bacteriophage. J. Virol. 1974;14:689–699. doi: 10.1128/jvi.14.3.689-699.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravantti J.J., Gaidelyte A., Bamford D.H., Bamford J.K. Comparative analysis of bacterial viruses Bam35, infecting a gram-positive host, and PRD1, infecting gram-negative hosts, demonstrates a viral lineage. Virology. 2003;313:401–414. doi: 10.1016/s0042-6822(03)00295-2. [DOI] [PubMed] [Google Scholar]
- 11.Krupovic M., Prangishvili D., Hendrix R.W., Bamford D.H. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. 2011;75:610–635. doi: 10.1128/MMBR.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saren A.M., Ravantti J.J., Benson S.D., Burnett R.M., Paulin L., Bamford D.H., Bamford J.K. A snapshot of viral evolution from genome analysis of the tectiviridae family. J. Mol. Biol. 2005;350:427–440. doi: 10.1016/j.jmb.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 13.Oksanen H.M., Bamford D.H. In: Virus Taxonomy: classification and nomeclature of viruses. Ninth report of the International Commitee on Taxonomy of Viruses. King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. San Diego: Academic Press; 2012. pp. 317–321. [Google Scholar]
- 14.Salas M. Protein-priming of DNA replication. Annu. Rev. Biochem. 1991;60:39–71. doi: 10.1146/annurev.bi.60.070191.000351. [DOI] [PubMed] [Google Scholar]
- 15.Hoeben R.C., Uil T.G. Adenovirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013;5:a013003. doi: 10.1101/cshperspect.a013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caldentey J., Blanco L., Savilahti H., Bamford D.H., Salas M. In vitro replication of bacteriophage PRD1 DNA. Metal activation of protein-primed initiation and DNA elongation. Nucleic Acids Res. 1992;20:3971–3976. doi: 10.1093/nar/20.15.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caldentey J., Blanco L., Bamford D.H., Salas M. In vitro replication of bacteriophage PRD1 DNA. Characterization of the protein-primed initiation site. Nucleic Acids Res. 1993;21:3725–3730. doi: 10.1093/nar/21.16.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Vega M., Salas M. Protein-Primed Replication of Bacteriophage Φ29 DNA. In: Kusic-Tisma J, editor. DNA Replication and Related Cellular Processes. Rijeka: InTech; 2011. [Google Scholar]
- 19.Pakula T.M., Caldentey J., Serrano M., Gutiérrez C., Hermoso J.M., Salas M., Bamford D.H. Characterization of a DNA binding protein of bacteriophage PRD1 involved in DNA replication. Nucleic Acids Res. 1990;18:6553–6557. doi: 10.1093/nar/18.22.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pakula T.M., Caldentey J., Gutiérrez C., Olkkonen V.M., Salas M., Bamford D.H. Overproduction, purification, and characterization of DNA-binding protein P19 of bacteriophage PRD1. Gene. 1993;126:99–104. doi: 10.1016/0378-1119(93)90595-t. [DOI] [PubMed] [Google Scholar]
- 21.de Jong R.N., van der Vliet P.C., Brenkman A.B. Adenovirus DNA replication: protein priming, jumping back and the role of the DNA binding protein DBP. Curr. Top Microbiol. Immunol. 2003;272:187–211. doi: 10.1007/978-3-662-05597-7_7. [DOI] [PubMed] [Google Scholar]
- 22.Longás E., de Vega M., Lázaro J.M., Salas M. Functional characterization of highly processive protein-primed DNA polymerases from phages Nf and GA-1, endowed with a potent strand displacement capacity. Nucleic Acids Res. 2006;34:6051–6063. doi: 10.1093/nar/gkl769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dekker J., Kanellopoulos P.N., Loonstra A.K., van Oosterhout J.A., Leonard K., Tucker P.A., van der Vliet P.C. Multimerization of the adenovirus DNA-binding protein is the driving force for ATP-independent DNA unwinding during strand displacement synthesis. EMBO J. 1997;16:1455–1463. doi: 10.1093/emboj/16.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Méndez J., Blanco L., Esteban J.A., Bernad A., Salas M. Initiation of Φ29 DNA replication occurs at the second 3′ nucleotide of the linear template: a sliding-back mechanism for protein-primed DNA replication. Proc. Natl. Acad. Sci. U.S.A. 1992;89:9579–9583. doi: 10.1073/pnas.89.20.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King A.J., van der Vliet P.C. A precursor terminal protein-trinucleotide intermediate during initiation of adenovirus DNA replication: regeneration of molecular ends in vitro by a jumping back mechanism. EMBO J. 1994;13:5786–5792. doi: 10.1002/j.1460-2075.1994.tb06917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanco L., Bernad A., Lázaro J.M., Martín G., Garmendia C., Salas M. Highly efficient DNA synthesis by the phage Φ29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem. 1989;264:8935–8940. [PubMed] [Google Scholar]
- 27.Rodríguez I., Lázaro J.M., Blanco L., Kamtekar S., Berman A.J., Wang J., Steitz T.A., Salas M., de Vega M. A specific subdomain in Φ29 DNA polymerase confers both processivity and strand-displacement capacity. Proc. Natl. Acad. Sci. U.S.A. 2005;102:6407–6412. doi: 10.1073/pnas.0500597102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dufour E., Méndez J., Lázaro J.M., de Vega M., Blanco L., Salas M. An aspartic acid residue in TPR-1, a specific region of protein-priming DNA polymerases, is required for the functional interaction with primer terminal protein. J. Mol. Biol. 2000;304:289–300. doi: 10.1006/jmbi.2000.4216. [DOI] [PubMed] [Google Scholar]
- 29.Berjón-Otero M., Villar L., de Vega M., Salas M., Redrejo-Rodríguez M. DNA polymerase from temperate phage Bam35 is endowed with processive polymerization and abasic sites translesion synthesis capacity. Proc. Natl. Acad. Sci. U.S.A. 2015;112:E3476–E3484. doi: 10.1073/pnas.1510280112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sozhamannan S., McKinstry M., Lentz S.M., Jalasvuori M., McAfee F., Smith A., Dabbs J., Ackermann H.W., Bamford J.K., Mateczun A., et al. Molecular characterization of a variant of Bacillus anthracis-specific phage AP50 with improved bacteriolytic activity. Appl. Environ. Microbiol. 2008;74:6792–6796. doi: 10.1128/AEM.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redrejo-Rodríguez M., Muñoz-Espín D., Holguera I., Mencía M., Salas M. Functional eukaryotic nuclear localization signals are widespread in terminal proteins of bacteriophages. Proc. Natl. Acad. Sci. U.S.A. 2012;109:18482–18487. doi: 10.1073/pnas.1216635109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabor S., Richardson C.C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U.S.A. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Redrejo-Rodríguez M., Muñoz-Espín D., Holguera I., Mencía M., Salas M. Nuclear and nucleoid localization are independently conserved functions in bacteriophage terminal proteins. Mol. Microbiol. 2013;90:858–868. doi: 10.1111/mmi.12404. [DOI] [PubMed] [Google Scholar]
- 34.McDonell M.W., Simon M.N., Studier F.W. Analysis of restriction fragments of T7 DNA and determination of molecular weights by electrophoresis in neutral and alkaline gels. J. Mol. Biol. 1977;110:119–146. doi: 10.1016/s0022-2836(77)80102-2. [DOI] [PubMed] [Google Scholar]
- 35.Lázaro J.M., Blanco L., Salas M. Purification of bacteriophage Φ29 DNA polymerase. Methods Enzymol. 1995;262:42–49. doi: 10.1016/0076-6879(95)62007-9. [DOI] [PubMed] [Google Scholar]
- 36.Mencía M., Gella P., Camacho A., de Vega M., Salas M. Terminal protein-primed amplification of heterologous DNA with a minimal replication system based on phage Φ29. Proc. Natl. Acad. Sci. U.S.A. 2011;108:18655–18660. doi: 10.1073/pnas.1114397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Gella P., Salas M., Mencía M. Improved artificial origins for phage Φ29 terminal protein-primed replication. Insights into early replication events. Nucleic Acids Res. 2014;42:9792–9806. doi: 10.1093/nar/gku660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sambrook J., Russell D. Molecular cloning: a laboratory manual. 4th edn. NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 40.Andreev Y.A., Kozlov S.A., Vassilevski A.A., Grishin E.V. Cyanogen bromide cleavage of proteins in salt and buffer solutions. Anal. Biochem. 2010;407:144–146. doi: 10.1016/j.ab.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 41.Rasband W.S. Maryland: National Institutes of Health, Bethesda; 1997. http://imagej.nih.gov/ij/ –2016. [Google Scholar]
- 42.Blanco L., Bernad A., Esteban J.A., Salas M. DNA-independent deoxynucleotidylation of the Φ29 terminal protein by the Φ29 DNA polymerase. J. Biol. Chem. 1992;267:1225–1230. [PubMed] [Google Scholar]
- 43.Esteban J.A., Bernad A., Salas M., Blanco L. Metal activation of synthetic and degradative activities of Φ29 DNA polymerase, a model enzyme for protein-primed DNA replication. Biochemistry. 1992;31:350–359. doi: 10.1021/bi00117a006. [DOI] [PubMed] [Google Scholar]
- 44.Beckman R.A., Mildvan A.S., Loeb L.A. On the fidelity of DNA replication: manganese mutagenesis in vitro. Biochemistry. 1985;24:5810–5817. doi: 10.1021/bi00342a019. [DOI] [PubMed] [Google Scholar]
- 45.Esteban J.A., Salas M., Blanco L. Fidelity of Φ29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J. Biol. Chem. 1993;268:2719–2726. [PubMed] [Google Scholar]
- 46.Moore C.M., Helmann J.D. Metal ion homeostasis in Bacillus subtilis. Curr. Opin. Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Verheust C., Fornelos N., Mahillon J. GIL16, a new gram-positive tectiviral phage related to the Bacillus thuringiensis GIL01 and the Bacillus cereus pBClin15 elements. J. Bacteriol. 2005;187:1966–1973. doi: 10.1128/JB.187.6.1966-1973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothberg P.G., Harris T.J., Nomoto A., Wimmer E. O4-(5′-uridylyl)tyrosine is the bond between the genome-linked protein and the RNA of poliovirus. Proc. Natl. Acad. Sci. U.S.A. 1978;75:4868–4872. doi: 10.1073/pnas.75.10.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hermoso J.M., Méndez E., Soriano F., Salas M. Location of the serine residue involved in the linkage between the terminal protein and the DNA of phage Φ29. Nucleic Acids Res. 1985;13:7715–7728. doi: 10.1093/nar/13.21.7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Méndez J., Blanco L., Salas M. Protein-primed DNA replication: a transition between two modes of priming by a unique DNA polymerase. EMBO J. 1997;16:2519–2527. doi: 10.1093/emboj/16.9.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redrejo Rodríguez M., Salas M. Multiple roles of genome-attached bacteriophage terminal proteins. Virology. 2014;468–470:322–329. doi: 10.1016/j.virol.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Redrejo-Rodríguez M., Muñoz-Espín D., Holguera I., Mencía M., Salas M. Nuclear localization signals in phage terminal proteins provide a novel gene delivery tool in mammalian cells. Commun. Integr. Biol. 2013;6:e22829. doi: 10.4161/cib.22829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holkers M., Maggio I., Henriques S.F., Janssen J.M., Cathomen T., Goncalves M.A. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods. 2014;11:1051–1057. doi: 10.1038/nmeth.3075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.