


Abstract
Helicases couple ATP hydrolysis to nucleic acid binding and unwinding via molecular mechanisms that remain poorly defined for most enzyme subfamilies within the superfamily 2 (SF2) helicase group. A crystal structure of the PriA SF2 DNA helicase, which governs restart of prematurely terminated replication processes in bacteria, revealed the presence of an aromatic-rich loop (ARL) on the presumptive DNA-binding surface of the enzyme. The position and sequence of the ARL was similar to loops known to couple ATP hydrolysis with DNA binding in a subset of other SF2 enzymes, however, the roles of the ARL in PriA had not been investigated. Here, we show that changes within the ARL sequence uncouple PriA ATPase activity from DNA binding. In vitro protein-DNA crosslinking experiments define a residue- and nucleotide-specific interaction map for PriA, showing that the ARL binds replication fork junctions whereas other sites bind the leading or lagging strands. We propose that DNA binding to the ARL allosterically triggers ATP hydrolysis in PriA. Additional SF2 helicases with similarly positioned loops may also couple DNA binding to ATP hydrolysis using related mechanisms.
INTRODUCTION
Helicases are motor proteins that convert the chemical energy of nucleoside triphosphate (often adenosine triphosphate (ATP)) hydrolysis to the mechanical energy needed to unwind nucleic acids (1). This activity is essential for cellular pathways such as DNA replication or RNA splicing that rely on single-stranded (ss) nucleic acids templates in synthesis or strand pairing reactions. Nucleic acid binding and ATP hydrolysis activities in helicases are mediated by evolutionarily conserved ‘helicase motifs’ that reside within the helicase core domains of the enzymes.
Based on the sequences of helicase motifs, these enzymes can be organized into several superfamilies (2,3). Superfamily 1 (SF1) and superfamily 2 (SF2) are the two largest helicase groups, and they share similar bi-lobed helicase core domains that are comprised of a pair of RecA-like folds. These RecA-like folds position helicase motifs I, Ia, II, III, IV, V and VI between the subdomains and along their DNA or RNA binding surfaces (1,4–6). Between SF1 and SF2 helicases, motifs directly involved in nucleoside triphosphate binding and hydrolysis (I, II and VI) are highly conserved. However, motifs involved in nucleic acid binding and coupling binding with ATP hydrolysis are divergent, suggesting distinct coupling mechanisms. Motif III is a key example of these differences. In SF1 helicases, motif III is directly involved in both DNA binding and ATP hydrolysis that allows the motif to couple the two activities (7–11). In SF2 helicases, however, the motif III consensus sequence is much shorter and lacks the segment that participates in ssDNA-binding in SF1 enzymes (6,12). Motif III in SF2 enzymes is directly involved in ATP hydrolysis, but its involvement in nucleic acid binding and/or unwinding has been less clear. Interestingly, in a few SF2 enzymes, an element upstream of motif III and immediately C-terminal to motif II has been shown to directly bind ssRNA/DNA (13–15). This segment in RecQ DNA helicases was termed an aromatic-rich loop (ARL) or motif IIa, and it has been shown to couple DNA-binding with ATP hydrolysis (13,16,17). Similarly positioned segments have been implicated in nucleic acid binding and/or ATPase activities in a small number of other SF2 subfamily helicases (18–21), but whether ARL/motif IIa elements are generally involved in the helicase mechanisms of other SF2 enzymes is not known.
In this study, we examine the roles of a newly identified ARL/motif IIa region in the E. coli PriA DNA helicase. PriA enzymes are SF2 helicases that recognize branched DNA structures and initiate DNA replication restart at abandoned replication forks and DNA recombination intermediates in bacteria (22). A recent X-ray crystal structure of PriA revealed a prominent ARL/motif IIa element that shares sequence and structural similarity with the ARL in RecQ (Figure 1) (4,23). However, the possible functions of this region in PriA DNA binding and in coupling binding to ATP hydrolysis have not been examined. To better understand the function of the PriA ARL and of SF2 ARL/motif IIa regions in general, we measured the effects of sequence changes within the PriA ARL on its DNA binding and DNA-dependent ATP hydrolysis activities. Consistent with the PriA ARL serving a coupling role similar to that of the RecQ ARL, PriA variants with altered ARLs were deficient in DNA-dependent ATPase and DNA helicase function. An in vitro protein–DNA crosslinking approach mapped interactions between the ARL and DNA, along with additional DNA interactions at sites outside of the ARL. PriA residues within the ARL crosslinked to the junction site where parental, leading and lagging strands intersect in synthetic DNA replication fork structures, whereas residues outside of the ARL mapped specifically to the leading or lagging strands. The patterns and intensity of crosslinks varied with nucleotide binding, which is consistent with ATPase-dependent conformational changes in the PriA structure both within the ARL and, more globally, within the helicase core domain. The addition of the PriB protein, which binds to PriA/DNA complexes during the DNA replication restart process, influenced PriA ARL DNA crosslinking as well. Based on these data, a model of the functions of the PriA ARL in coupling DNA-binding and ATP hydrolysis is proposed.
Figure 1.

Crystal structure of PriA reveals an aromatic-rich loop (ARL) at a similar location within the helicase lobes as the ARL of RecQ. (A) Schematic PriA domain structure highlighting the positions of helicase motifs (black bars). (B) Klebsiella pneumonia PriA structure (PDB 4NL4 (23)) with ADP (red sticks) and zinc (grey spheres) bound. Domain coloring for 3′ binding domain (3′BD), winged helix (WH), helicase lobes, cysteine-rich region (CRR) and C-terminal domain (CTD) is the same in both images. Inset: PriA ARL residue side chains. (C) Sequence alignment of helicase motif II-III (grey) in K. pneumonia PriA, Cronobacter sakazakii RecQ and representative SF1 helicases. Pink highlighting corresponds to proposed ARL sequences, including the C-terminal portion of motif III in SF1 helicases. (D) Structural alignment of the helicase lobes for PriA (colored as in B, with ADP in grey sticks) with RecQ: ARL from ATPγS-bound apo E. coli RecQ in magenta (PDB 1OYY (34)) and ARL from partial duplex DNA-bound C. sakazakii RecQ in orange with DNA rings and base-stacking phenylalanine shown in black (PDB 4TMU (13)).
MATERIALS AND METHODS
Protein purification
E. coli PriA was purified as described previously (23). Briefly, Rosetta2 E. coli transformed with pET15-EcPriA (containing the priA open reading frame) or pET15-EcPriA single-site alanine variants was grown in Luria Broth supplemented with 100 μg/ml ampicillin and 50 μg/ml chloramphenicol to mid-log phase, and PriA expression was induced with 1 mM IPTG for 3 h. PriA was purified from cell lysate via nickel-affinity, ion exchange and size exclusion chromatography and dialyzed into PriA storage buffer (10 mM HEPES–HCl, pH 7.0, 0.5 M NaCl, 50% glycerol and 10 mM dithiothreitol). E. coli SSB and PriB were purified as described previously (24,25).
For expression of PriA incorporated with ρ-benzoyl-L-phenylalanine (Bpa), the priA open reading frame was subcloned from pET15-EcPriA into pBAD/His B vector and subjected to site-directed mutagenesis to substitute an amber stop codon (TAG) at specific EcPriA codons targeted for Bpa substitution (26). BL21 (DE3) E. coli were cotransformed with the pBAD-EcPriA variant plasmids and pSup-MjTyrRS-6tRNA (27). Liquid growth of cotransformants was limited to reduce mutation occurrences. Autoinduction media (ZYM-5052 (28)) supplemented with 75 μg/ml ampicillin, 37.5 μg/ml chloramphenicol, 0.3% arabinose and 1 mM Bpa (BACHEM F-2800: 100 mM stock in 1 M NaOH) was inoculated with cells directly from transformant plates (to 0.1–0.3 OD600) and grown overnight at 37°C. PriA variants were purified as described above, except that exposure to light was minimized and the size exclusion purification step was omitted.
DNA-binding fluorescence anisotropy assay
PriA or PriA variant (0.1–5000 nM) was dialyzed into 20 mM HEPES, pH 7.0, 5% glycerol, 75 mM potassium glutamate, 10 mM dithiothreitol and incubated for 30 min at ambient temperature with 0.1 mg/ml BSA and 1 nM fluorescein-labelled replication fork substrate (60 bp complementary ‘parental’ region and 38 nt noncomplementary ‘lagging’ and ‘leading’, regions (Supplementary Table S1 structure #1; (29,30))) before measuring fluorescence anisotropy, as described previously (23). Fluorescence intensities increased by ∼2-fold across the range of PriA concentrations tested and fluorescence anisotropy values were corrected for this change (31). KD and error (1 SD) were determined by fitting the data to a single site-specific model using GraphPad Prism.
ATP hydrolysis assay
PriA or PriA variant (50 nM) was incubated with 0.1–5000 nM dT28 DNA for 10 min at ambient temperature, in 20 mM HEPES–HCl, pH 8.0, 50 mM sodium chloride, 1 mM β-mercaptoethanol, 5 mM magnesium chloride, 0.1 mg/ml BSA, 2 mM phosphoenolpyruvate, 0.2 mM nicotinamide adenine dinucleotide, 3 U/ml Pyruvate Kinase and 4.5 U/ml Lactate Dehydrogenase. ATP (1 mM) was added and A340 nm was monitored for 1 h at 25°C. Data were analyzed as previously described (13).
Helicase assay
PriA unwinding of a synthetic DNA replication fork structure (comprised of a 5′ 32P-labeled template lagging strand annealed in the three-armed DNA structure from the DNA-binding fluorescence anisotropy assay which was gel purified (Supplementary Table S1, structure #1 and #2 as indicated in figure legend)) was measured as previously described (23), with minor alterations. Briefly, 0–1.2 nM PriA was incubated with 1 nM synthetic replication fork substrate in 50 mM HEPES-HCl, pH 8.0, 0.04 mg/mL BSA, 2 mM dithiothreitol, 2 mM ATP and 4 mM magnesium acetate for 30 min at 37°C. The reactions were terminated by addition of STOP buffer (20 mM ethylenediaminetetracetic acid, 0.5% SDS, 0.2 mg/ml proteinase K and 2.5 ng/µl cold trap oligonucleotide (3L-98) (final concentrations)) and incubating for 30 min at 37°C. When measuring the effects of SSB and PriB on PriA helicase activity, 250 nM SSB or 10 nM PriB were preincubated with the synthetic replication fork at ambient temperature for 30 or 10 min, respectively, prior to addition of PriA (1.2 nM). Samples were resolved by 10% native polyacrylamide gel electrophoresis (PAGE), fixed, dried, exposed to a PhosphoImager screen, imaged with a Typhoon FLA 9000, and quantified using ImageQuant (GE Healthcare). Fraction DNA unwound was calculated from the intensity of the single unwound band over the total intensity of that lane.
PriABpa-DNA UV crosslinking
Crosslinking experiments with Bpa-incorporated PriA were adapted from an established method (32) to examine PriA binding to synthetic DNA replication fork structures. DNA substrates were branched structures with 60 bp long leading, lagging and parental arms (Supplementary Table S1 structure #3 or structure #4 (Supplementary Figure S5 only)) and one of the four oligonucleotides was 5′ 32P-labeled for electrophoretic mobility shift assays (EMSAs). This extended fork for crosslinking also included the corresponding ‘nascent lagging’ and ‘nascent leading’ strand oligonucleotides to yield 60 bps of double-stranded (ds) parental template, 60 bps of ds leading strand and a lagging strand arm of 60 nt (55 bps dsDNA with 5 nt ss gap at the fork junction).
Crosslinking fork (1 nM) was incubated in 50 mM Tris-HCl, pH 8.0, 0.1 mg/ml BSA, 2 mM dithiothreitol, 5 mM ethylenediaminetetracetic acid, 6% glycerol, 50 mM sodium chloride (with 5 mM magnesium acetate and 2 mM ADP or ATPγS when indicated) with 3.5 nM PriA variant for 25 min on ice. A single PriA–DNA fork shift was observed in EMSAs under these conditions. Higher order PriA/DNA complexes were observed at higher PriA concentrations and lower NaCl concentrations than those used here. PriB (100 nM) was incubated with the DNA for 3 min at ambient temperature prior to PriA addition in indicated samples. Reactions were incubated at ambient temperature for 5 min then exposed to ultraviolet light (UV; handheld 365 nm wavelength lamp). UV treatment comprised of 3 × 5 min exposures at ambient temperature with 2 min incubations on ice between UV exposures to prevent heating, followed by 15 min of UV exposure on ice. Samples were analyzed by native and denaturing EMSAs or primer extension.
Electrophoretic mobility shift assays (EMSAs)
Native EMSAs were preformed as previously described (33). Samples were resolved through 4% PAGE in 0.5x TBE at ambient temperature for 2.5 h at 80 V, before fixing, drying, exposing and imaging as indicated above.
Denaturing EMSAs were performed by adding equal volume of 2x SDS sample buffer (125 mM Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 1.43 mM β-mercaptoethanol and bromophenol blue) to crosslinking samples, with 10 nM trap oligonucleotide addition (unlabeled version of the labeled oligonucleotide in the sample) followed by heat denaturation (100°C for 10 min) of half the sample. Samples were immediately resolved through 4–5% PAGE in 1x TBE at 80–100 V. Gels were fixed, dried, exposed and imaged as above.
PriA–DNA crosslink primer extension assay
Primer extension (PE) was done as previously described (32), with minor changes. Briefly, 2.25 μl PriA–DNA crosslinked sample, 1.25 U Taq DNA Polymerase (NEB), 1X Taq buffer (NEB), 200 μM of each dNTP, 5% DMSO, 2 M betaine and 320 nM 5′ 32P-labeled primer, which anneals 60–80 nt away from the fork junction (Supplementary Table S1), were mixed. PE reactions were initially heat denatured (95°C for 30 s) and cycled 20 times (95°C for 30 s, 48°C for 30 s and 68°C for 15 s). Crosslinked samples were generated as above, except that unlabeled crosslinking fork, 2 nM DNA and 7 nM PriA were used. An equal volume of loading buffer (8 M urea, 0.5x TBE, bromophenol blue and xylene cyanol) was added, and PE products were resolved through 12% polyacrylamide, 8M urea gel electrophoresis in 1x TBE for 3 h at 35–40 W, exposed to a PhosphoImager screen, imaged with a Typhoon FLA 9000, and quantified using ImageQuant. Sequencing ladders were generated using the same primers (229 nM), 7.7 nM DNA template (the oligonucleotide to be extended on in the annealed fork), and the Thermo Sequencing Kit (Affymetrix). Sequencing ladders were cycled with the PE samples, diluted 4x in loading buffer and resolved alongside PE samples. To confirm nucleotide length determination, the run-off band from the corresponding 61 nt length of the template lagging strand was use to calibrate the ddNTP-terminated sequence ladder.
RESULTS
PriA loop sequence and location aligns with RecQ aromatic-rich loop
The recently determined X-ray crystal structure of PriA revealed a surface-exposed loop within the helicase core domain that was similar in sequence and position to the aromatic-rich loop (ARL) found in RecQ helicases (Figure 1) (16,23,34–36). This element is adjacent to motif II in both proteins. The PriA ARL sequence is highly-conserved across bacterial phyla (Supplementary Figure S1A) and it contains Trp, Tyr and Arg residues that are also found in the RecQ ARL and in the C-terminal, DNA-binding portion of motif III in SF1 helicases (Figure 1C). Additionally, the PriA and RecQ ARLs structurally align with one another within the helicases, although the loops assume different orientations in each structure relative to the helicase core (Figure 1D). While sequence conservation of this loop is high within PriA homologues and partially conserved with the RecQ ARL, any conservation is not clear across all SF2 enzymes. However, elements directly following motif II, called ‘motif IIa’ in several other SF2 helicases, have identified residues that appear to be important for their helicase functions (Supplementary Figure S1B) (13,15–21). Given the similarities between the PriA ARL and RecQ ARL, we hypothesized that the PriA ARL could provide a critical element for coupling DNA binding to ATPase in the enzyme.
Sequence substitution within the PriA ARL disrupts PriA ATPase and helicase functions
To examine the function of residues within the ARL, E. coli PriA variants carrying individual alanine substitutions within the loop (K328A, Q329A, Q330A, W333A, R334A and Y335A) were constructed, purified and tested for activity. An additional PriA alanine variant, substituting a lysine from motif I that is essential for ATPase activity (K230A), was also purified for use as an ATP hydrolysis negative control.
Each of the variants and wild-type (wt) PriA were first tested for activity in DNA-dependent ATPase assays. ATPase activity was undetectable for wt PriA in the absence of DNA but was stimulated by the addition of dT28 ssDNA (Figure 2A) as has been previously observed (37). The ATP hydrolysis rate at saturating ssDNA concentrations (kmax) was 130 ± 5 min−1 and the concentration of dT28 required for 50% stimulation (KDNA) was 100 ± 20 nM. As expected, ATPase activity was undetectable at all dT28 concentrations for the negative control K230A PriA variant. For the PriA ARL variants, kmax values were reduced relative to wt PriA values. The kmax values for W333A and Y335A were 11 ± 1 min−1 and 9 ± 1 min−1 (12- and 14-fold reduced), respectively, which were the lowest levels measured among the ARL variants. The K328A, Q330A and R334A variants had modest 3–4 fold reductions in kmax, whereas PriA Q329A had the highest level of activity for the ARL variants, with kmax ∼2-fold reduced relative to wt PriA kmax. The KDNA values for all variants were within error of that for wt PriA, except for the KDNA value for the W333A variant, which was reduced ∼3-fold. These results were consistent with roles for the ARL in stimulating DNA-dependent ATPase activity but not in stabilizing PriA/DNA complex formation.
Figure 2.

Alanine substitutions within the PriA ARL reduce PriA ATPase activities. (A) Coupled spectrophotometric ATPase assay monitoring DNA-dependent ATP hydrolysis. kmax and KDNA are shown for each PriA variant. K230A is a Walker A mutant used as a negative control for ATPase and helicase activity in this study. (‘ND’ indicates none detected). (B) Equilibrium binding of PriA variants to a 5′-fluorescein labeled DNA (depicted to right: the DNA is comprised of two DNA oligonucleotides with 60 nt of complementary DNA and 38 nt of non-complementarity (structure #1 in Supplementary Table S1)). Inset shows apparent KD derived from fits to a single-site binding model. (C) DNA helicase assay measuring unwinding of 1 nM 5′ 32P-labeled DNA fork (as in part B: Supplementary Table S1 structure #1) by PriA variants. Inset shows example PAGE (lanes: Boiled DNA, 0, 0.15, 0.3, 0.6 and 1.2 nM PriA) and graph shows quantification of unwound DNA band. All data are mean of a minimum of three replicates ± SD.
A fluorescent anisotropy assay was next used to directly test whether alanine substitutions in the ARL altered the equilibrium DNA-binding properties of PriA. Based on the KDNA values from the ATPase experiment, it was predicted that the sequence changes would have little effect on overall DNA-binding affinity. PriA concentration-dependent changes in fluorescence anisotropy were measured with a fluorescently-labeled synthetic DNA fork structure (Figure 2B and Supplementary Figure S2). All variants bound to this DNA substrate with apparent dissociation constants (KDs) that were within 2.2-fold of the KD observed for wt PriA. Thus, DNA-binding was not significantly affected in the variants and the ATPase defects in the ARL variants observed above were not a result of decreased overall DNA-binding affinity. These results parallel the previously observed results of alanine substitutions with the RecQ ARL (13,16).
The impact of ARL sequence changes on the DNA unwinding functions of the variants was measured next. The first assay measured PriA unwinding of a radiolabeled two-stranded synthetic DNA fork structure (Figure 2C). Wt PriA unwound the DNA fork in an enzyme concentration-dependent manner. A maximum of ∼60% of the DNA substrate was unwound over the enzyme concentration range tested, and addition of higher concentrations of PriA resulted in apparent helicase inhibition or DNA reannealing (previously suggested (38)). No unwinding was observed with the negative control, K230A PriA variant. In comparison to wt PriA, the PriA ARL variants each had reduced DNA unwinding activities. Q329A, Q330A and R334A variants had 1.5- to 6-fold reductions in the fraction DNA unwound at the highest enzyme concentrations tested, whereas W333A, Y335A and K328A had no detectable helicase activity. These observed reductions in helicase activity correlated with the ATPase results: variants with reduced ATPase activity had similarly reduced helicase function.
The unwinding activities of the variant panel were next tested using a four-stranded replication fork mimic substrate and the effects of adding SSB or PriB were examined. This substrate adds nascent leading and lagging strands to the simple fork structure used above. The two-stranded DNA fork used above is a simple DNA fork that PriA binds and unwinds robustly. Adding a synthetic nascent leading strand increases PriA affinity for the DNA fork and orients PriA to preferentially unwind the nascent lagging strand (39,40). The nascent lagging strand is 5 nt shorter than the template non-complementary region, leaving a 5 nt gap on the lagging strand adjacent to the fork junction (29) that enhances PriA helicase activity ((39,41) and data not shown). PriA preferential unwinding of the nascent lagging strand in this substrate can be stimulated by the addition of either SSB or PriB (29,33,38,39). At a 1.2 nM PriA concentration (the concentration for maximum DNA unwinding from the previous assay), wt PriA unwound ∼50% of the substrate (Figure 3, top). Addition of SSB or PriB stimulated wt PriA helicase activity, resulting in unwinding of the nascent lagging strand from ∼80% of substrate molecules (Figure 3, middle and bottom). Consistent with the results obtained using the simple two-stranded substrate (Figure 2), all of the PriA variants had reduced helicase activity compared to wt PriA in the absence of SSB and PriB. SSB significantly stimulated DNA unwinding by K328A, Q329A, Q330A and R334A PriA variants, but DNA unwinding by the K230A, W333A or Y335A PriA variants remained at background levels. PriB stimulated unwinding in several variants to a greater extent than SSB, with the fraction DNA unwound by K328A, Q330A and R334A rising to wt PriA levels, and the fraction of DNA unwound by Q329A and W333A at ∼2- and ∼4-fold lower than wt PriA. In contrast, the addition of PriB failed to stimulate helicase activity in the Y335A PriA variant that resembled the K230A negative control variant. In aggregate, these results point to the importance of the PriA ARL element in coupling ATPase and DNA unwinding functions and to an essential role for Y335 in PriA helicase activities.
Figure 3.
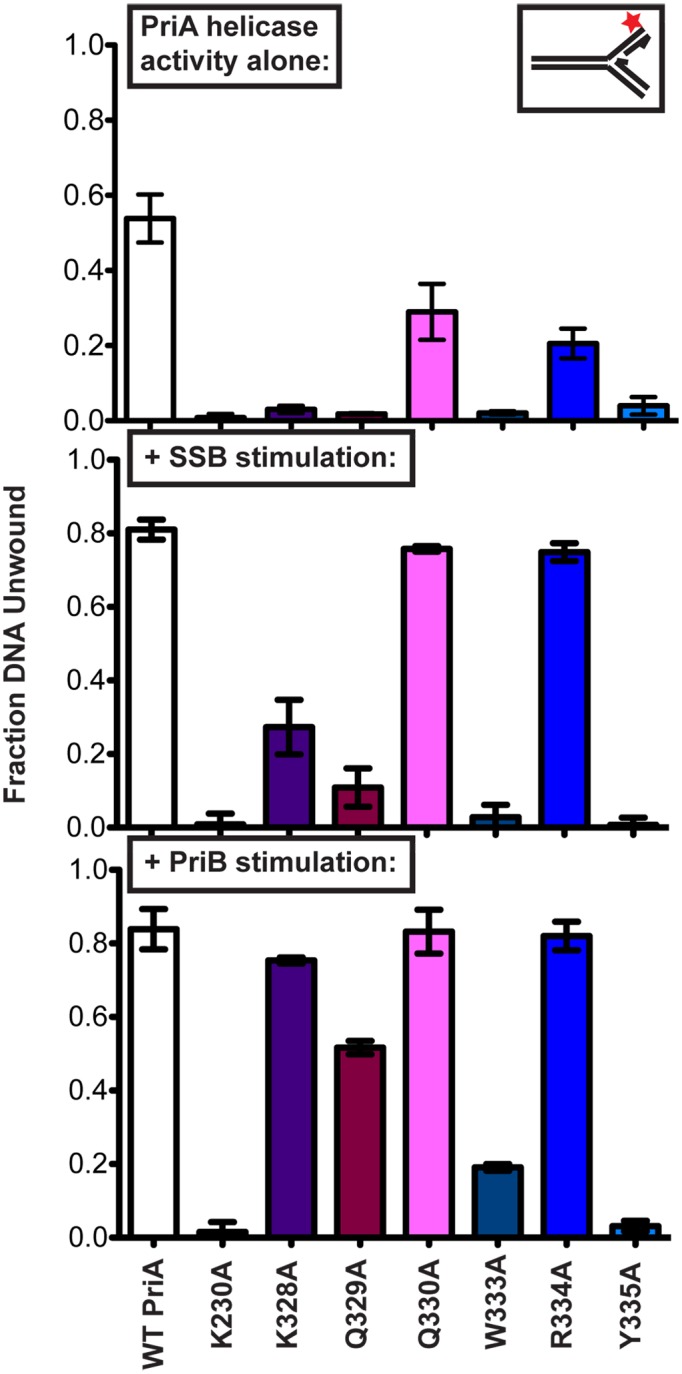
Impact of individual ARL sequence changes on PriA DNA unwinding in isolation and in the presence of SSB or PriB. Fraction of DNA unwound (synthetic nascent lagging strand unwound) measured after incubating 1.2 nM PriA variants with 1 nM 5′ 32P-labeled synthetic DNA fork (depicted in upper inset and Supplementary Table S1 (structure #2, which is comprised of four oligonucleotides, with a 5 nt ssDNA gap between parental duplex and nascent lagging strand 5′ end). Top: PriA in isolation. Middle: PriA unwinding with DNA prebound by SSB (250 nM). Bottom: PriA unwinding with DNA prebound by PriB (10 nM). Data are mean of three replicates ± SD.
Creation of a panel of benzophenone-substituted PriA variants
The experiments described above suggest a model in which the PriA DNA-binding status is communicated to the ATPase site via the ARL. A simple mechanism to support this coupling would be that the ARL forms a DNA binding site in PriA. However, individual sequence changes to residues in the ARL had no apparent effects on DNA binding affinity (Figure 2). In RecQ, the ARL plays a similar coupling role and, although RecQ ARL variants also have wt DNA binding affinities (16), a structure of the RecQ/DNA complex has recently shown that the RecQ ARL directly interacts with ssDNA (13). Thus, it is possible that the PriA ARL could directly bind DNA and that individual residue changes are not sufficient to impact DNA binding affinity. Since no structure of the PriA/DNA complex is currently available, we developed an in vitro method to test the possibility that the PriA ARL could contact DNA.
Our approach for testing an ARL/DNA interaction examined whether crosslinks could be formed between specific residues in PriA and a replication fork DNA substrate. A panel of PriA variants in which the UV-activatable crosslinking group Bpa (26,27) was substituted for individual residues was created and purified as described in Materials and Methods. Bpa can form crosslinks with macromolecules that are within ∼10 Å of the substituted side chains (42,43) and has been used for identifying region-specific interactions between labeled proteins and their macromolecular binding partners. PriA sites within the ARL and in other domains were chosen to test the ARL interactions and to begin to map global PriA–DNA interactions. These included substitutions of K328, Q329, Q330, E331, W333, R334 and Y335 from the PriA ARL; D17, which is important in binding to the 3′ end of the nascent leading strand of replication forks (44); and E492 from the second helicase lobe, which is predicted to be near the lagging strand based on comparisons of PriA with the structures of DNA-bound forms of other SF2 helicases (Figure 4A) (13,45). Variants are referred to as ‘residueBpa’ (e.g. K328Bpa) throughout.
Figure 4.

PriA–DNA fork crosslinking maps strand- and residue-specific interactions. (A) Model of PriA domains predicted to bind to specific DNA regions of an abandoned DNA fork (23). Stars note the location of Bpa incorporation: D17 within the 3′BD, Q329-E331 within the ARL of helicase lobe 1, and E492 within the helicase lobe 2. (B) Synthetic DNA fork substrate used in crosslinking experiments. The substrate is lengthened relative to that used in Figure 3 but it retains the 5 nt nt ssDNA gap between parental duplex and nascent lagging strand 5′ end (Supplementary Table S1 structure #3). (C) Denaturing PAGE separation of DNA-bound, Bpa-incorporated PriA variants after exposure to ultraviolet light. DNA forked substrate in B was individually 5′-radiolabeled on each of the 4 strands, yielding the set of 4 adjacent samples. Half of each sample was additionally heat-denatured, yielding the second set of 4 adjacent samples marked by the red delta. Arrows point to covalently-crosslinked PriA-shifted bands. Data are representative gels from among three replicates.
Prior to carrying out crosslinking assays, the impact of Bpa substitutions on PriA biochemical activities was examined to ensure that the changes did not block protein folding and function. DNA binding by the PriABpa variants was tested using the 4-stranded replication fork DNA substrate for crosslinking studies (Figure 4B), which was similar to that used for DNA binding and helicase studies with the alanine-substitution panel except that the non-complementary arms were extended. This extended substrate was used to allow for later analysis of crosslinked PriA/DNA complexes. All of the PriABpa variants bound the replication fork structure with similar apparent affinities to that of wt PriA, indicating that Bpa incorporation did not block DNA binding (Supplementary Figure S3).
DNA-dependent ATP hydrolysis activity was also measured for all PriABpa variants. As was the case with the alanine-substituted PriA variants, several of the Bpa variants had reduced ATPase rates. However, the specific sites that had the lowest ATPase activities when substituted with alanine (W333 and Y335) had only modest or no reduction in ATPase activity when substituted with Bpa (Supplementary Table S2). Moreover, the Q329Bpa variant had the most strongly reduced ATPase levels across the ARL panel (16-fold lower than wt PriA) whereas alanine substitution at this residue resulted in only a 2-fold loss in activity (Supplementary Table S2). It could be that the aromatic nature of the Bpa allowed for substitutions for Tyr and Trp side chains in the ARL, whereas Bpa substitution had a more dramatic impact at non-aromatic positions within the ARL. Nonetheless, ATPase activity was measurable for each of the PriABpa variants, which provided confidence in the PriABpa variants utility in crosslinking studies.
Protein–DNA crosslinking defines strand-specific interactions for the PriA ARL, 3′BD and helicase core with a DNA replication fork substrate
An EMSA was first used to test whether PriABpa variants could crosslink to DNA and to map the strand(s) of a four-stranded replication fork structure to which specific Bpa side chains crosslink. Individual PriABpa variants were incubated with substrate and exposed to UV light conditions that induce crosslinking. The complexes were then treated with SDS and analyzed by PAGE to detect covalent complexes, which are resolved as shifted DNA bands. The crosslinking experiments were performed using four 32P radiolabeled versions of the four-stranded replication fork structure, in which one of the four strands in the substrate was labeled – this allowed us to determine the strand(s) to which a given Bpa site was covalently attached (Figure 4B). Half of each SDS-denatured crosslinking sample was heat-denatured to resolve individual DNA oligonucleotides crosslinked to PriA. Thermal denaturation of the substrate was modestly reversible, even with addition of unlabelled trap oligonucleotide, so shifted bands in PriA–DNA gels were compared with control experiments in which PriA was omitted.
Among the PriABpa variants tested, reproducible crosslinks to the four-stranded DNA substrate were observed with D17Bpa, Q329Bpa (low level), Q330Bpa, E331Bpa and E492Bpa (Figure 4C). Crosslinks were not observed in the absence of UV exposure, with wt PriA, or with the remaining UV-exposed Bpa variants (K328Bpa, W333Bpa, R334Bpa and Y335Bpa). Crosslinks were detected for D17Bpa in both the non-heat denatured samples (Figure 4C, black arrow) and in the lane resolving the nascent leading strand after heat denaturation (red arrow). The D17Bpa data are consistent with previous observations suggesting that D17 is important for recognition of the 3′ end of the nascent leading strand (40,44). Among the PriA ARL Bpa variants, Q329Bpa-DNA crosslinked bands were detected in both the non-heat denatured samples and in the lane resolving the heat denatured, 32P labeled template lagging strand at low levels. Q330Bpa-DNA crosslinked bands were strongly detected in both the non-heat denatured samples and in the lane resolving the heat denatured, 32P labelled template lagging strand. E331Bpa-DNA crosslinked bands were also present in the non-heat denatured samples but, unlike Q330Bpa, E331Bpa crosslinked with both the template lagging and leading strands, with a somewhat more intense band in the heat denatured, leading strand sample. Finally, DNA crosslinked complexes of E492Bpa were observed in both the non-heat denatured samples and in the lane resolving the heat denatured template lagging strand. The E492Bpa data are consistent with a model that suggested the lagging strand would bind across the RecA-like folds of PriA (23).
Primer extension analysis maps the sites bound by PriABpa variants within a DNA replication fork substrate and can detect nucleotide- and protein cofactor-induced changes in crosslinking positions
Primer extension (PE) assays were next performed on the PriABpa–DNA crosslinked samples to map the nucleotide location of the crosslinks. Nucleotide-resolution Bpa-DNA crosslink mapping has previously been performed in RNA polymerase, where mapping showed that PE is halted at the expected crosslinked base (32). PriABpa variants crosslinked to the four-stranded DNA substrate (unlabeled) were analyzed by PE using radiolabeled primers that anneal to the parental strands within the fork (Figure 5A, Supplementary Figure S4, Table S1). In the absence of UV treatment, the PE reaction produced full-length products (Figure 5B, left). However, UV crosslinking of Q330Bpa, E331Bpa and E492Bpa to the DNA led to the formation of sample-specific PE products that were shorter than full length (Figure 5B, right). Significant non-full length bands, enhanced over background ‘No PriA’ or ‘No UV’ controls, were detected only in samples that were previously observed to crosslink strongly in the gel shift assay (Figure 4). Moreover, the observed bands were highly reproducible and specific to the template lagging or leading strand for which crosslink formation was observed with in the gel shift assay (Figure 5B and Supplementary Figure S4). PE along the nascent leading strand, which would allow D17Bpa crosslink mapping, was not conducted, since the primer for that strand would anneal across the first ∼20 nts of the nascent leading strand at the fork junction where D17 would interact with the 3′ nascent leading strand end (40,44).
Figure 5.

Primer extension maps PriABpa–DNA fork crosslink sites at the nucleotide-level. (A) Diagram of primer extension assay on the crosslinking DNA fork (Figure 4B and Supplementary Table S1 structure #3). Red arrows represent 5′ 32P-labelled primers that allow extension along the template lagging (primer 1) or leading strands (primer 2; Supplementary Table S1). ‘X’ indicates possible location of block to primer extension by a covalent PriA–DNA crosslink. (B) Lagging strand PE products resolved by urea-denaturing PAGE with DNA sequencing ladders in middle and right. T,C,A or G at bottom of sequencing ladder lane denote halted base identity in primer extension product sequence, Thymidine nt # 52–66 are labeled on gel, to right of T lane, corresponding to numbering in panel D. Labeling to left of gel indicates full-length (FL) product, fork junction location and primers (run off). Data are representative gel of greater than three replicates. (C) Overlay of helicase domains from: PriA (multi-coloring as in Figure 1, (23)) and partial duplex DNA-bound RecQ (PDB 4TMU (23)) and Hel308 (PDB 2P6R (45)) structures, showing just the DNA from RecQ structure (black) and Hel308 structure (grey). (D) Sequence of the crosslinking DNA fork at the fork junction with primer extension truncated products for each variant mapped onto DNA fork substrate. Dotted lines connect the structural location of the Bpa-substituted residue in panel C with the mapped nucleotide crosslink location in panel D. As indicated earlier, the DNA includes a 5 nt ssDNA gap present between the parental duplex and nascent lagging strand 5′ end.
Comparisons of the positions of prematurely terminated PE products to a sequencing ladder generated using the PE primers facilitated residue-specific mapping of the PriABpa–DNA crosslink sites. Using a primer that annealed to the lagging strand template, prematurely terminated PE products were observed with three of the PriABpa variants. Crosslinked E492Bpa (E492X) strongly halted PE at nucleotides (nts) 65 and 66 as measured from the template lagging strand 3′ end, which corresponded to a crosslink within the lagging strand arm of the replication fork structure 5 or 6 nts away from the fork junction (Figure 5B–D). Weaker crosslinks were observed for E492X to nucleotides 62–64 as well. Crosslinks formed by ARL residues E331X and Q330X halted PE at nts 60–61 and 60–63 on the lagging strand template, respectively. These positions map directly at the replication fork junction in the substrate or 1–3 nts into the lagging strand arm of the structure (Figure 5D). Using a primer that annealed to the leading strand template revealed prematurely terminated PE products only for PriA E331Bpa variant (Supplementary Figure S4, left). E331X halted PE on the leading strand template 61 nts downstream from the leading strand primer 5′ end (leading strand 3′ end). This position maps to the branch junction of the replication fork, indicating that Bpa-incorporated at E331 can crosslink to either nucleotide in the final parental duplex base pair prior to the leading and lagging strand branch point.
To test whether the crosslinking observed with the four-stranded DNA substrate described above was due to the overall replication fork structure and not to the specific sequence of the substrate, a second version of the 4-stranded DNA substrate was tested in the PE assay (Supplementary Figure S5A). In particular, the thymine nucleotides in the fork junction region were changed to cytosine since thymine is known to have enhanced UV reactivity in Bpa-nucleotide crosslinking experiments (46). Crosslinks with the second fork structure were observed for Q330X and E492X in very similar positions to those mapped with the first substrate (Supplementary Figure S5B). Q330X crosslinked most strongly to C61 on the lagging strand template of the second fork, whereas it crosslinked strongly to both A60 and T61 in the first substrate. E492X crosslinked most strongly to C63 and C64 in the lagging strand template of the second substrate. E492X crosslinks to both of these positions in the first substrate but these were more minor compared to C65 and T66 crosslinked bands (Supplementary Figure S5B). E331X crosslinks, which produced weaker PE bands than E492X or Q330X crosslinks with the first substrate, were too weak to detect in the second substrate. The modest differences observed in crosslinked products between the two replication fork substrates tested indicated that the results using the first substrate were enhanced by the abundance of thymine. However, crosslinks for Q330X and E492X that were found in both substrates correlated well, indicating that the overall crosslinking pattern was preserved, in a structure-specific manner, with the more subtle differences being sequence-specific.
We next examined whether the positions of PriABpa crosslinks were sensitive to the addition of ADP or ATPγS that could alter the positions of the ARL and/or the RecA folds in the helicase core domain as has been observed in other DNA helicases (47–50). Introduction of ADP or ATPγS in the E492Bpa crosslinking reaction resulted in a bimodal pattern of PE bands with a new significant PE band at 62 nt of the lagging strand, along with the previously mapped crosslink at 65–66 nts (Figure 6 and Supplementary Figure S6). The appearance of new ADP/ATPγS-dependent crosslinks between a residue in the C-terminal-most helicase lobe and positions closer to the branch junction of the replication fork is consistent with the predicted rotation of this domain in inchworm models of SF1/SF2 helicase lobe dynamics (1). This model relies on ATP hydrolysis cycle-dependent movements between the RecA-like folds of the helicase core domain to form the ATP binding pocket and to couple these motions to directional translocation and DNA unwinding. The ADP/ATPγS-dependent crosslinking patterns revealed alteration in the crosslinking efficiency but not an accumulation of new crosslinking sites in the PriA ARL Bpa variants (Figure 6). For E331X a greater bias toward crosslinking with lagging strand nts 60 and 61 was observed in the presence of ADP or ATPγS relative to reactions where the cofactors were omitted. However, on the template leading strand, E331Bpa crosslinks were not altered by the presence of ADP but ATPγS addition reduced E331Bpa crosslinking intensity (Supplementary Figure S4, right). For Q330X inclusion of ADP led to a PE banding pattern similar to that without nucleotide but with decreased intensity (Figure 6). Addition of ATPγS in the Q330BPA crosslinking reaction further decreased the crosslinking intensity to background levels. Crosslinks remained undetected in the remaining Bpa variants (K328Bpa, W333Bpa, R334Bpa and Y335Bpa) upon incubation with the additional factors in this study.
Figure 6.
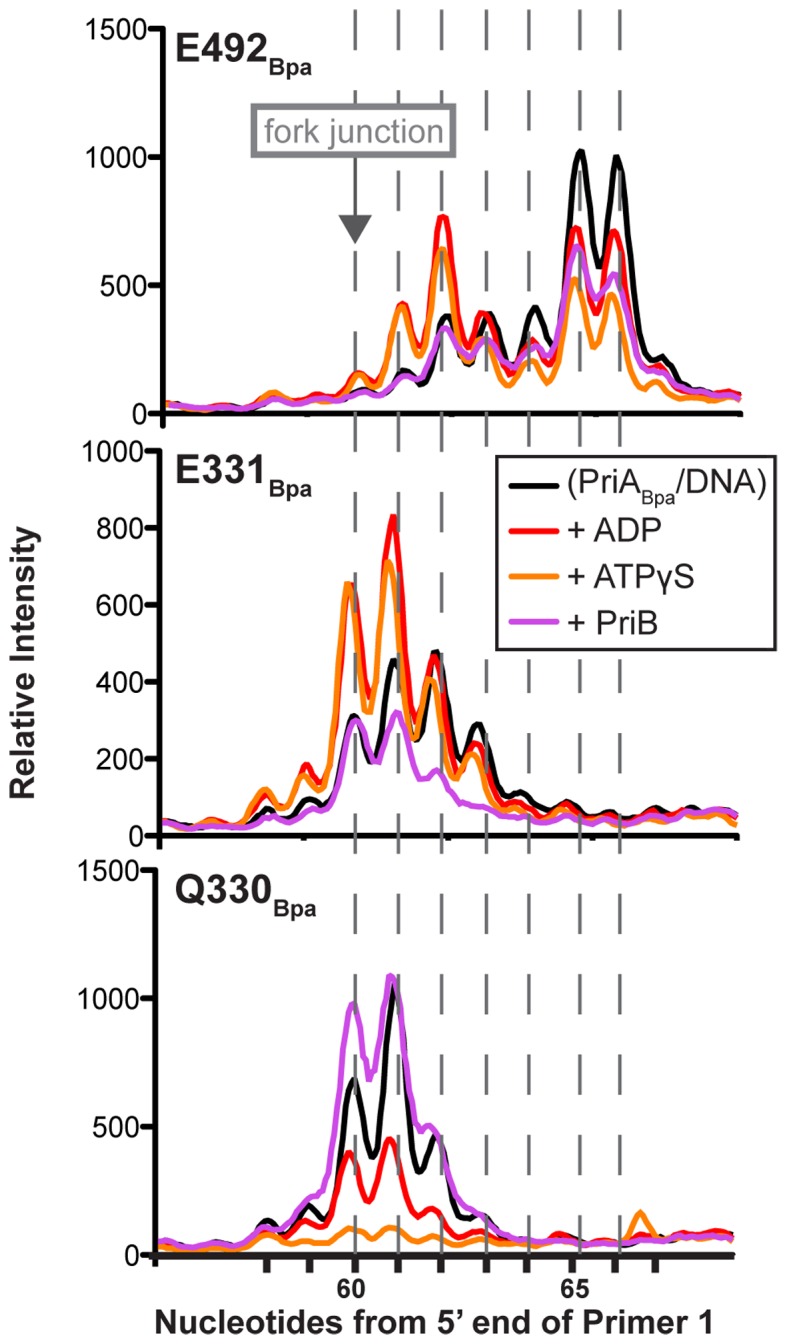
Nucleotide and PriB alter PriABpa–DNA fork crosslinks. Densitometry profiles of nucleotide crosslink sites on the template lagging strand, from primer extension gels as in Figure 5B, for PriA variants with Bpa incorporated at E491 (top), E330 (middle) and Q329 (bottom), when crosslinks are formed in the presence of no additional factors, ADP, ATPγS or PriB. Data are quantified from a representative gel of greater than three replicates.
Finally, we also tested whether the PriA/DNA crosslink patterns were altered by the addition of the PriB protein that binds to PriA/DNA complexes in formation of the replication restart primosome (51–53). In contrast to ADP or ATPγS addition, PriB addition resulted in a similar E492X banding pattern but with lower intensity (Figure 6). Addition of PriB in E331BPA crosslinking reaction shifted the lagging strand banding pattern similarly to that with addition of nucleotide, but without the increased crosslinking efficiency (Figure 6), and did not alter the leading strand banding pattern (Supplementary Figure S4, right). In contrast, addition of PriB in the Q330BPA crosslinking reaction increased the intensity of the 60 nt band (0 nt into the lagging strand) within the 60–63 nt crosslinked banding pattern (Figure 6). Taken together, these results are consistent with nucleotide- and PriB-binding status influencing the architecture of the PriA/DNA complex.
DISCUSSION
We have examined the structural and functional roles of the ARL in the helicase mechanism of PriA. Similarities between the PriA ARL and a related loop found in RecQ helicases led to the hypothesis that the PriA ARL binds DNA and couples this binding to enhanced ATP hydrolysis. In agreement with this model, PriA variants with alanine substitutions within the ARL had significantly reduced DNA-dependent ATPase rates and helicase activities. However the variants displayed no reduction in DNA binding affinity. Similarly contradictory results had been previously observed with RecQ ARL variants, but a later crystal structure of the RecQ/DNA complex showed that the ARL forms a direct interaction with ssDNA (13,16). A crosslinking approach was therefore developed to test directly whether the ARL is in close proximity to DNA in PriA/DNA complexes. Consistent with a RecQ-like DNA-binding strategy for PriA, PriA–DNA crosslinks were formed by multiple residues in the ARL, indicating that the loop engages DNA. The crosslinking approach identified residue- and nucleotide-specific interfaces between DNA and the ARL and other sites within PriA. These crosslinks revealed a DNA binding arrangement for PriA in which different regions of the protein interact with the fork junction as well as the leading and lagging strands of replication fork structures.
How might DNA binding to the ARL influence PriA ATPase functions? Our crosslinking studies have mapped interactions between the PriA–ARL and DNA replication fork junctions. The positions and apparent robustness of these crosslink are sensitive to the presence of nucleotides (ADP versus ATPγS, Figure 6), which is consistent with a linkage between the ARL and the ATP binding/hydrolysis site. These sites are ∼20 Å apart in the apo PriA structure, making it likely that such interdependence arises from allosteric effects. A similar relationship between the ARL and ATPase sites has been noted for RecQ helicases in which structural studies have shown that DNA binding at the ARL is coordinated with structural rearrangement of the enzyme's ATPase active site that appears to poise RecQ for ATP hydrolysis (13). This communication in RecQ appears to be facilitated in part by a helix that directly connects motif II in the ATPase site to the ARL. PriA has a similar helix between its ARL and ATPase sites (Figure 1), so it is possible that a related ARL/DNA-dependent conformational change could alter the position of motif II in a manner that promotes ATP hydrolysis. This allosteric relationship conversely implies that the nucleotide state in the ATP binding/hydrolysis site might also affect the structure of the ARL, as we have observed in crosslinking experiments. Such an effect could have evolved to aid in coupling the chemical energy derived from ATP hydrolysis to translocation of PriA along DNA and/or DNA unwinding. Consistent with this model, single-site alanine substitutions in the ARL are sufficient to reduce or abolish DNA unwinding (Figures 2 and 3).
The apparent shared functions of the PriA and RecQ ARLs lead to the question of whether similarly positioned loops mediate analogous roles in other SF2 helicases. Examination of the 16 available SF2 DNA or RNA helicases for which crystal structures have revealed nucleic acid binding to the helicase domain shows that all have a loop or helix immediately C-terminal to motif II that is near DNA or RNA (Supplementary Figure S7) (18,45,47,54–65). In these loops, the side chain of at least one residue extends toward the nucleic acid, although the distances between the residues and the bound DNA or RNA are too great to infer direct interactions in many instances. In the 16 structures, base-stacking (3.3–3.6 Å) is observed with phenylalanines in 3 structures, hydrogen bonding (2.6–2.9 Å) from at least one residue is observed in 9 other structures, other van der waal interactions (3.0–3.9 Å) are observed with these and 3 more structures, and the remaining structure motif IIa region does not interact with DNA (6.3 Å separation; this helicase, Cas3, forms its own, recently added, SF2 subfamily (66)). This survey suggests that many SF2 helicases could contain nucleic acid-interacting motif IIa-like segments that may be important for linking binding to ATP hydrolysis. Additionally, ARL-like segments are highly conserved within PriA homologues and in a small number of other SF2 subfamilies but similar sequence elements are not common across the superfamily (6). Thus, this position within the helicase core domains could be conserved for DNA or RNA interaction but the aromatic nature of the loop that is observed in PriA and RecQ is not universal.
Investigations into the roles of ARL/motif IIa elements in SF2 enzymes have substantiated the broad importance of this element in nucleic acid binding. In the RecQ subfamily of SF2 enzymes, the ARL interacts with ssDNA to structurally couple DNA-binding with ATP hydrolysis in bacteria enzymes; the same loop has been implicated in helicase function in the S. cerevisiae RecQ protein, Sgs1 and human RecQ proteins, RECQ1 and BLM (13,16,17,35,36). In the DEAD-box subfamily of RNA helicases, motif IIa (also referred to as ‘post-II’) forms a highly conserved RNA binding surface that appears to clash with dsRNA and may force strand separation (15,21,54). Interestingly, our crosslinking results suggest a similar position of the ARL/motif IIa in PriA as motifs IIa in DEAD-box RNA helicases at ss/ds nucleic acid junctions. In the Swi2/Snf2 helicase Rad54, this highly conserved element (switch region/motif IIa) is implicated in DNA binding and ATP hydrolysis (18). This region is also conserved in the RIG-I-like RNA helicase subfamily, and sequence changes within this region abolished RNA-dependent ATPase activity (19). Finally, in the DEAH/RHA subfamily member, Prp28p, mutations in this region can suppress mutations in motif III and V (20). Thus the ARL/motif IIa region appears to be broadly important for helicase function in a range of SF2 enzymes.
Our results also provide insight into the structure of the PriA/DNA replication fork complex. Several studies have suggested that PriA recognition of replication fork structures is a critical first step that triggers assembly of the full replication restart primosome (53,67). A model derived from the crystal structure of a bacterial PriA in the absence of DNA, earlier mutagenesis and DNA footprinting studies suggested that PriA DNA binding domains are arranged in a manner that allows binding to each arm of a branched replication fork (23,68,69). The lack of a high-resolution structure of the PriA/DNA complex has left the veracity of this model unclear. The DNA crosslinking results presented herein confirm PriA binding to at least two of the three arms of a model replication fork: the PriA 3′BD crosslinks to the nascent leading strand and several residues within the helicase domain crosslink to the lagging strand. These observations are consistent with previous reports demonstrating that the 3′BD interacts with the 3′ end of the nascent leading strand, with the PriA preference for unwinding the lagging strand, and with the position of DNA in other helicase structures (23,39,40,44). The protein–DNA crosslinking in each helicase lobe and the 3′BD in this study provides insights at residue and nucleotide resolution that support the overall DNA binding model for PriA proposed earlier (23). The results and model are also in agreement with a recent study in which bacterial primosome proteins, including PriA, were crosslinked to a synthetic DNA fork decorated with photo-reactive groups (70). In this study, PriA crosslinks with base analogs 3 nts into the lagging strand, 6–12 nts into the leading strand (which included the leading strand primer terminus) and 3 nts into the parental duplex. In sum, our study validates proposed PriA–DNA fork interaction models and provides insight into the PriA helicase mechanism in a way that may help to clarify the coupling mechanism of several SF2 helicases. Further investigation is needed to determine if all SF2 enzymes contain a motif IIa that couples DNA-binding with ATP hydrolysis.
Supplementary Material
Acknowledgments
The authors thank Basudeb Bhattacharyya, Jason Chin and Peter Schultz for plasmids, Jared Winkelman and David Brow for technical assistance, and Nick George for purifying SSB protein. The authors thank the Keck lab for critical evaluation of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation Graduate Research Fellowship [DGE-1256259 to T.A.W.]; National Institutes of Health (NIH) [GM098885]. Funding for open access charge: National Institutes of Health [GM098885].
Conflict of interest statement. None declared.
REFERENCES
- 1.Singleton M.R., Dillingham M.S., Wigley D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 2.Gorbalenya A.E., Koonin E.V. Helicases: amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 1993;3:419–429. [Google Scholar]
- 3.Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989;17:4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya B., Keck J.L. Grip it and rip it: structural mechanisms of DNA helicase substrate binding and unwinding. Protein Sci. 2014;23:1498–1507. doi: 10.1002/pro.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caruthers J.M., McKay D.B. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 6.Fairman-Williams M.E., Guenther U.P., Jankowsky E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subramanya H.S., Bird L.E., Brannigan J.A., Wigley D.B. Crystal structure of a DExx box DNA helicase. Nature. 1996;384:379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- 8.Dillingham M.S., Soultanas P., Wigley D.B. Site-directed mutagenesis of motif III in PcrA helicase reveals a role in coupling ATP hydrolysis to strand separation. Nucleic Acids Res. 1999;27:3310–3317. doi: 10.1093/nar/27.16.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soultanas P., Dillingham M.S., Wiley P., Webb M.R., Wigley D.B. Uncoupling DNA translocation and helicase activity in PcrA: direct evidence for an active mechanism. EMBO J. 2000;19:3799–3810. doi: 10.1093/emboj/19.14.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korolev S., Hsieh J., Gauss G.H., Lohman T.M., Waksman G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 11.Brosh R.M., Jr, Matson S.W. A partially functional DNA helicase II mutant defective in forming stable binary complexes with ATP and DNA. A role for helicase motif III. J. Biol. Chem. 1996;271:25360–25368. doi: 10.1074/jbc.271.41.25360. [DOI] [PubMed] [Google Scholar]
- 12.Hall M.C., Matson S.W. Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 1999;34:867–877. doi: 10.1046/j.1365-2958.1999.01659.x. [DOI] [PubMed] [Google Scholar]
- 13.Manthei K.A., Hill M.C., Burke J.E., Butcher S.E., Keck J.L. Structural mechanisms of DNA binding and unwinding in bacterial RecQ helicases. Proc. Natl. Acad. Sci. U.S.A. 2015;112:4292–4297. doi: 10.1073/pnas.1416746112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Del Campo M., Lambowitz A.M. Structure of the Yeast DEAD box protein Mss116p reveals two wedges that crimp RNA. Mol. Cell. 2009;35:598–609. doi: 10.1016/j.molcel.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen C.B., Ballut L., Johansen J.S., Chamieh H., Nielsen K.H., Oliveira C.L., Pedersen J.S., Seraphin B., Le Hir H., Andersen G.R. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 16.Zittel M.C., Keck J.L. Coupling DNA-binding and ATP hydrolysis in Escherichia coli RecQ: role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 2005;33:6982–6991. doi: 10.1093/nar/gki999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ui A., Satoh Y., Onoda F., Miyajima A., Seki M., Enomoto T. The N-terminal region of Sgs1, which interacts with Top3, is required for complementation of MMS sensitivity and suppression of hyper-recombination in sgs1 disruptants. Mol. Genet. Genomics. 2001;265:837–850. doi: 10.1007/s004380100479. [DOI] [PubMed] [Google Scholar]
- 18.Durr H., Korner C., Muller M., Hickmann V., Hopfner K.P. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell. 2005;121:363–373. doi: 10.1016/j.cell.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 19.Civril F., Bennett M., Moldt M., Deimling T., Witte G., Schiesser S., Carell T., Hopfner K.P. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011;12:1127–1134. doi: 10.1038/embor.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang T.H., Latus L.J., Liu Z., Abbott J.M. Genetic interactions of conserved regions in the DEAD-box protein Prp28p. Nucleic Acids Res. 1997;25:5033–5040. doi: 10.1093/nar/25.24.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohr G., Del Campo M., Turner K.G., Gilman B., Wolf R.Z., Lambowitz A.M. High-throughput genetic identification of functionally important regions of the yeast DEAD-box protein Mss116p. J. Mol. Biol. 2011;413:952–972. doi: 10.1016/j.jmb.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabbai C.B., Marians K.J. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharyya B., George N.P., Thurmes T.M., Zhou R., Jani N., Wessel S.R., Sandler S.J., Ha T., Keck J.L. Structural mechanisms of PriA-mediated DNA replication restart. Proc. Natl. Acad. Sci. U.S.A. 2014;111:1373–1378. doi: 10.1073/pnas.1318001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.George N., Ngo K., Chitteni-Pattu S., Norais C., Battista J., Cox M., Keck J. Structure and cellular dynamics of Deinococcus radiodurans Single-stranded DNA (ssDNA)-binding Protein (SSB)-DNA complexes. J. Biol. Chem. 2012;287:22123–22132. doi: 10.1074/jbc.M112.367573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopper M., Holton J.M., Keck J.L. Crystal structure of PriB, a component of the Escherichia coli replication restart primosome. Structure. 2004;12:1967–1975. doi: 10.1016/j.str.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Chin J.W., Martin A.B., King D.S., Wang L., Schultz P.G. Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryu Y., Schultz P.G. Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods. 2006;3:263–265. doi: 10.1038/nmeth864. [DOI] [PubMed] [Google Scholar]
- 28.Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Heller R.C., Marians K.J. Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J. Biol. Chem. 2005;280:34143–34151. doi: 10.1074/jbc.M507224200. [DOI] [PubMed] [Google Scholar]
- 30.Heller R.C., Marians K.J. The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell. 2005;17:733–743. doi: 10.1016/j.molcel.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 31.Dandliker W.B., Hsu M.L., Levin J., Rao B.R. Equilibrium and kinetic inhibition assays based upon fluorescence polarization. Methods Enzymol. 1981;74 Pt C:3–28. doi: 10.1016/0076-6879(81)74003-5. [DOI] [PubMed] [Google Scholar]
- 32.Winkelman J.T., Winkelman B.T., Boyce J., Maloney M.F., Chen A.Y., Ross W., Gourse R.L. Crosslink mapping at amino acid-base resolution reveals the path of scrunched DNA in initial transcribing complexes. Mol. Cell. 2015;59:768–780. doi: 10.1016/j.molcel.2015.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cadman C.J., Lopper M., Moon P.B., Keck J.L., McGlynn P. PriB stimulates PriA helicase via an interaction with single-stranded DNA. J. Biol. Chem. 2005;280:39693–39700. doi: 10.1074/jbc.M508521200. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein D.A., Zittel M.C., Keck J.L. High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 2003;22:4910–4921. doi: 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirzaei H., Schmidt K.H. Non-Bloom syndrome-associated partial and total loss-of-function variants of BLM helicase. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19357–19362. doi: 10.1073/pnas.1210304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banerjee T., Sommers J.A., Huang J., Seidman M.M., Brosh R.M., Jr Catalytic strand separation by RECQ1 is required for RPA-mediated response to replication stress. Curr. Biol. 2015;25:2830–2838. doi: 10.1016/j.cub.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shlomai J., Kornberg A. A prepriming DNA replication enzyme of Escherichia coli. I. Purification of protein n': a sequence-specific, DNA-dependent ATPase. J. Biol. Chem. 1980;255:6789–6793. [PubMed] [Google Scholar]
- 38.Cadman C.J., McGlynn P. PriA helicase and SSB interact physically and functionally. Nucleic Acids Res. 2004;32:6378–6387. doi: 10.1093/nar/gkh980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones J.M., Nakai H. Escherichia coli PriA helicase: fork binding orients the helicase to unwind the lagging strand side of arrested replication forks. J. Mol. Biol. 2001;312:935–947. doi: 10.1006/jmbi.2001.4930. [DOI] [PubMed] [Google Scholar]
- 40.Mizukoshi T., Tanaka T., Arai K., Kohda D., Masai H. A critical role of the 3′ terminus of nascent DNA chains in recognition of stalled replication forks. J. Biol. Chem. 2003;278:42234–42239. doi: 10.1074/jbc.C300285200. [DOI] [PubMed] [Google Scholar]
- 41.Szymanski M.R., Jezewska M.J., Bujalowski W. The Escherichia coli PriA helicase specifically recognizes gapped DNA substrates: effect of the two nucleotide-binding sites of the enzyme on the recognition process. J. Biol. Chem. 2010;285:9683–9696. doi: 10.1074/jbc.M109.094789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dorman G., Prestwich G.D. Benzophenone photophores in biochemistry. Biochemistry. 1994;33:5661–5673. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 43.Wittelsberger A., Mierke D.F., Rosenblatt M. Mapping ligand-receptor interfaces: approaching the resolution limit of benzophenone-based photoaffinity scanning. Chem. Biol. Drug Des. 2008;71:380–383. doi: 10.1111/j.1747-0285.2008.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasaki K., Ose T., Okamoto N., Maenaka K., Tanaka T., Masai H., Saito M., Shirai T., Kohda D. Structural basis of the 3′-end recognition of a leading strand in stalled replication forks by PriA. EMBO J. 2007;26:2584–2593. doi: 10.1038/sj.emboj.7601697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buttner K., Nehring S., Hopfner K.P. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 46.Cuquerella M.C., Lhiaubet-Vallet V., Cadet J., Miranda M.A. Benzophenone photosensitized DNA damage. Acc. Chem. Res. 2012;45:1558–1570. doi: 10.1021/ar300054e. [DOI] [PubMed] [Google Scholar]
- 47.Gu M., Rice C.M. Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. U.S.A. 2010;107:521–528. doi: 10.1073/pnas.0913380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velankar S.S., Soultanas P., Dillingham M.S., Subramanya H.S., Wigley D.B. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 49.Forne I., Ludwigsen J., Imhof A., Becker P.B., Mueller-Planitz F. Probing the conformation of the ISWI ATPase domain with genetically encoded photoreactive crosslinkers and mass spectrometry. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.012088. M111.012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee J.Y., Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J., Nurse P., Marians K.J. The ordered assembly of the phiX174-type primosome. III. PriB facilitates complex formation between PriA and DnaT. J. Biol. Chem. 1996;271:15656–15661. doi: 10.1074/jbc.271.26.15656. [DOI] [PubMed] [Google Scholar]
- 52.Ng J.Y., Marians K.J. The ordered assembly of the phiX174-type primosome. II. Preservation of primosome composition from assembly through replication. J. Biol. Chem. 1996;271:15649–15655. doi: 10.1074/jbc.271.26.15649. [DOI] [PubMed] [Google Scholar]
- 53.Lopper M., Boonsombat R., Sandler S.J., Keck J.L. A hand-off mechanism for primosome assembly in replication restart. Mol. Cell. 2007;26:781–793. doi: 10.1016/j.molcel.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mallam A.L., Sidote D.J., Lambowitz A.M. Molecular insights into RNA and DNA helicase evolution from the determinants of specificity for a DEAD-box RNA helicase. Elife. 2014;3:e04630. doi: 10.7554/eLife.04630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sengoku T., Nureki O., Nakamura A., Kobayashi S., Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 56.Collins R., Karlberg T., Lehtio L., Schutz P., van den Berg S., Dahlgren L.G., Hammarstrom M., Weigelt J., Schuler H. The DEXD/H-box RNA helicase DDX19 is regulated by an {alpha}-helical switch. J. Biol. Chem. 2009;284:10296–10300. doi: 10.1074/jbc.C900018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong E.V., Cao W., Voros J., Merchant M., Modis Y., Hackney D.D., Montpetit B., De La Cruz E.M. Pi release limits the intrinsic and RNA-stimulated ATPase cycles of DEAD-Box protein 5 (Dbp5) J. Mol. Biol. 2016;428:492–508. doi: 10.1016/j.jmb.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buchwald G., Ebert J., Basquin C., Sauliere J., Jayachandran U., Bono F., Le Hir H., Conti E. Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10050–10055. doi: 10.1073/pnas.1000993107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prabu J.R., Muller M., Thomae A.W., Schussler S., Bonneau F., Becker P.B., Conti E. Structure of the RNA helicase MLE reveals the molecular mechanisms for uridine specificity and RNA-ATP coupling. Mol. Cell. 2015;60:487–499. doi: 10.1016/j.molcel.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 60.Webster M.P., Jukes R., Zamfir V.S., Kay C.W., Bagneris C., Barrett T. Crystal structure of the UvrB dimer: insights into the nature and functioning of the UvrAB damage engagement and UvrB-DNA complexes. Nucleic Acids Res. 2012;40:8743–8758. doi: 10.1093/nar/gks633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weir J.R., Bonneau F., Hentschel J., Conti E. Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc. Natl. Acad. Sci. U.S.A. 2010;107:12139–12144. doi: 10.1073/pnas.1004953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devarkar S.C., Wang C., Miller M.T., Ramanathan A., Jiang F., Khan A.G., Patel S.S., Marcotrigiano J. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. U.S.A. 2016;113:596–601. doi: 10.1073/pnas.1515152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 64.Jedrzejczak R., Wang J., Dauter M., Szczesny R.J., Stepien P.P., Dauter Z. Human Suv3 protein reveals unique features among SF2 helicases. Acta Crystallogr. D Biol. Crystallogr. 2011;67:988–996. doi: 10.1107/S0907444911040248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huo Y., Nam K.H., Ding F., Lee H., Wu L., Xiao Y., Farchione M.D., Jr, Zhou S., Rajashankar K., Kurinov I., et al. Structures of CRISPR Cas3 offer mechanistic insights into Cascade-activated DNA unwinding and degradation. Nat. Struct. Mol. Biol. 2014;21:771–777. doi: 10.1038/nsmb.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jackson R.N., Lavin M., Carter J., Wiedenheft B. Fitting CRISPR-associated Cas3 into the helicase family tree. Curr. Opin. Struct. Biol. 2014;24:106–114. doi: 10.1016/j.sbi.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng J.Y., Marians K.J. The ordered assembly of the phiX174-type primosome. I. Isolation and identification of intermediate protein-DNA complexes. J. Biol. Chem. 1996;271:15642–15648. doi: 10.1074/jbc.271.26.15642. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka T., Mizukoshi T., Sasaki K., Kohda D., Masai H. Escherichia coli PriA protein, two modes of DNA binding and activation of ATP hydrolysis. J. Biol. Chem. 2007;282:19917–19927. doi: 10.1074/jbc.M701848200. [DOI] [PubMed] [Google Scholar]
- 69.Liu J., Marians K.J. PriA-directed assembly of a primosome on D loop DNA. J. Biol. Chem. 1999;274:25033–25041. doi: 10.1074/jbc.274.35.25033. [DOI] [PubMed] [Google Scholar]
- 70.Manhart C.M., McHenry C.S. Identification of subunit binding positions on a model fork and displacements that occur during sequential assembly of the Escherichia coli primosome. J. Biol. Chem. 2015;290:10828–10839. doi: 10.1074/jbc.M115.642066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.