


Abstract
Copy Number Variants (CNVs) are structural rearrangements contributing to phenotypic variation that have been proved to be associated with many disease states. Over the last years, the identification of CNVs from whole-exome sequencing (WES) data has become a common practice for research and clinical purpose and, consequently, the demand for more and more efficient and accurate methods has increased. In this paper, we demonstrate that more than 30% of WES data map outside the targeted regions and that these reads, usually discarded, can be exploited to enhance the identification of CNVs from WES experiments. Here, we present EXCAVATOR2, the first read count based tool that exploits all the reads produced by WES experiments to detect CNVs with a genome-wide resolution. To evaluate the performance of our novel tool we use it for analysing two WES data sets, a population data set sequenced by the 1000 Genomes Project and a tumor data set made of bladder cancer samples. The results obtained from these analyses demonstrate that EXCAVATOR2 outperforms other four state-of-the-art methods and that our combined approach enlarge the spectrum of detectable CNVs from WES data with an unprecedented resolution. EXCAVATOR2 is freely available at http://sourceforge.net/projects/excavator2tool/.
INTRODUCTION
Copy Number Variants (CNVs) are structural rearrangements involving DNA segments of at least 50 bp (1,2) that can be present with an altered copy number compared to the reference genome. CNVs are unbalanced events, i.e. they alter the total number of base pairs in a genome since they involve loss (deletion) or gain (duplication) of segments. During the last 10 years the advances in genomic technologies have enhanced the ability to detect these alterations, revealing that they are spread along the human genome (3–7). Latest comprehensive study estimated that from 4.8% to 9.5% of the genome contributes to CNVs of size 50 bp–3 Mb (7). Moreover, the total amount of bases associated with CNVs exceeds the number of bases involved in single nucleotides polymorphisms (SNPs) by an order of magnitude (8). CNVs may participate to phenotypic variation, underlying both adaptive traits and different classes of diseases (9). Pathogenic CNVs have been found so far associated with autism (10), neurological disorders (11), Crohn's disease (12) and cancer (13,14) and they are also suspected to be associated with complex diseases such as type I diabetes (15) and cardiovascular disease (16,17).
Over the past decade, the introduction of next generation sequencing (NGS) technologies has greatly improved CNVs detection, allowing base-pair resolution of breakpoints (18). The advent of these platforms favoured the accomplishment of large-scale re-sequencing projects, such as the 1000 Genomes Project and The Cancer Genome Atlas, but the cost and the computational complexity of downstream analysis still limit the routine use of whole-genome sequencing (WGS) to large scale projects. Whole-exome sequencing (WES), on the other hand, represents a cost-effective alternative to WGS for the study of disease-associated variants affecting the coding regions of the genome (10,19–22) and is extensively used for diagnostic purposes.
Most of the computational methods developed for the discovery of CNVs from WES data are based on read count (RC) approach (2,20,23–25). All the methods implement different strategies to normalise the depth of coverage (DoC), which is known to be heavily biased by uneven capture and enrichment efficiency across exons. In nearly every case, the available algorithms exploit solely the DoC of captured regions, limiting the possibility to investigate variations along the full-length genome. Therefore alterations in intergenic regions are completely excluded from the investigation loosing the opportunity to study the impact of CNVs on non coding genome from WES so far. Moreover, since the exons only encompass a sparse 1% of the genome, most breakpoints fall out of the targeted exons and therefore they are usually misplaced by current algorithms.
Almost all commercial enrichment kits lack specificity for the target regions, resulting in up to 60% of produced reads which map to sequences outside the target design (26). These off-target reads are generally ignored by classical computational methods for the analysis of WES data that focus on the on-target reads only. Recently, Kuilman et al. introduced CopywriteR (27), an R based method that makes use of off-target reads to build uniformly distributed DNA copy number profiles. Here, we show, for the first time, that the distribution of In- and Off-Target RCs have similar statistical properties. We demonstrate that bias reduction methods previously developed for targeted regions also work for Off-target data and that properly normalized Off-Target RCs allow for the prediction of the absolute number of DNA copies with an accuracy comparable to In-Target RCs.
Following these observations we developed a novel release of our EXCAVATOR tool (20), names EXCAVATOR2. EXCAVATOR2 enhances the identification of genomic CNVs (overlapping or non-overlapping exons) from WES data by integrating the analysis of In-targets and Off-targets reads. It extends the RC approach to the whole genome sequence and exploits the Shifting Level Model (SLM) algorithm (28,29) to segment the two combined profiles. Thereafter, the FastCall algorithm (30) allows to classify each segmented region into five possible states (two-copy deletion, one-copy deletion, normal, one-copy duplication and multiple-copy amplification). In order to show the versatility of our tool in different experimental settings, we applied it to two WES data sets: a population study and a cancer genomic study. As a first step we evaluated the performance of EXCAVATOR2 to identify genomic CNVs in a data set made of HapMap samples sequenced by the 1000 Genomes Project and we compared it to other four state-of-the-art methods for WES data that include CoNIFER, XHMM and the recently published CODEX and CopywriteR tools. As a further step, EXCAVATOR2 and CopywriteR were used for the analysis of the WES data of 14 matched tumor/control of Bladder samples, and their results were compared with those obtained from high-density SNP-array assays. The new methodological advances have been included in a new version of the tool that is freely available at http://sourceforge.net/projects/excavator2tool/.
MATERIALS AND METHODS
WES data inspection, WMRC description and biases normalization
In order to study the distribution of reads generated by WES experiments we used a Survey Data set made of 30 WES samples sequenced by using three capture kits: NimbleGen SeqCap EZ Exome v2.0 44 Mb, Agilent SureSelect Human All Exon 50 Mb and Illumina TruSeq Exome Enrichment 62 Mb. This data set includes both public available data generated by two previous comparative studies (31,32) and exomes sequenced in our laboratories. More details are available in Section 2.1 and Table S1 of Supplementary Material.
Although commercial WES kits are designed to capture specific genomic regions, mainly transcribed sequences, we found that a substantial fraction of reads systematically aligned outside the designed targeted regions (see Results). In order to extend the RC approach to Off-target regions and exploit this measure to increase the potentiality of identifying regions with altered copy number in intronic/intergenic DNA segments we divided the genome into two distinct classes of genomic features: (i) all the regions targeted by WES kits (according to respective manufacturers design) and (ii) non-overlapping genomic windows that belong to intronic/intergenic regions. We defined the Window Mean Read Count (WMRC) measure that expands the Exon Mean Read Count (20). Specifically, WMRC corresponds to:
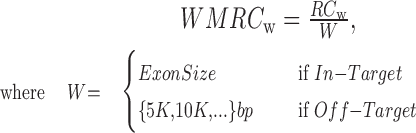 |
(1) |
and RC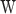 is the number of reads aligned to a genomic region of length W. W varies according to the size of each designed targeted DNA segment or, in case of Off-target, it corresponds to the selected fixed size of non-overlapping windows in which the intergenic chromosome is divided.
is the number of reads aligned to a genomic region of length W. W varies according to the size of each designed targeted DNA segment or, in case of Off-target, it corresponds to the selected fixed size of non-overlapping windows in which the intergenic chromosome is divided.
We studied the relationship between WMRC data and classical genomic systematic biases: (i) local GC-content and (ii) mappability, that have already been proved to influence read depth in targeted regions (33,34). We applied previously adopted RC median normalization approach introduced by Yoon et al. and extended in our previous work (34) for mitigating the effect of both CG-content and mappability in WMRC, separately for In- and Off-Target windows (details are in Supplementary Section 1).
Integration into EXCAVATOR2
The new WMRC was introduced into a new version of EXCAVATOR tool (20). Starting from the .bed file of the target regions, EXCAVATOR2 first creates the new pseudo-target splitting intronic/intergenic regions into non-overlapping windows of fixed length and skipping 200 bp stretches flanking the target exons. WMRC biases are corrected for In- and Off-Target regions independently exploiting the median normalisation approach (Supplementary Section 1). We used the log-transformed ratio (log2 ratio) between the WMRC values of test and control samples. WMRC ratio is median-normalised for both In- and Off-Target. Then normalised WMRC is segmented by means of the previously described Heterogeneous Shifting Level Model (HSLM) segmentation algorithm (28,29). Finally, FastCall algorithm (30) classifies each segmented regions as one of the five possible discrete states (two-copy deletion, one-copy deletion, normal, one-copy duplication and multiple-copy amplification). The FastCall calling procedure takes into account sample heterogeneity and exploits the Expectation Maximisation algorithm to estimate the parameters of a five gaussian mixture model and to provide the probability that each segment belongs to a specific copy number state. The new functionalities were included in a new version of the tool. EXCAVATOR2 is freely available at http://sourceforge.net/projects/excavator2tool/.
Validation data sets
To evaluate the accuracy of our novel computational approach for predicting the absolute number of DNA copies of genomic regions from WES data in a population study, the exomes of 8 healthy individuals sequenced by the 1000 Genomes Project (Supplementary Table S2) were selected. They were previously genotyped by both McCarroll (35) and Conrad (36) using array-based technologies. Specifically, McCarroll estimated regions with altered copy number in 270 individuals by using SNP 6.0 arrays, performing all the experiments in duplicates, using two different computational approaches for analysing array data and confirming the presence of CNV at 27 loci in 30 individuals using q-PCR. In Conrad's study on the other hand, a customised high-density tiling CGH array was exploited to accurately detect the genotype of common CNV loci in 450 HapMap samples (inferring integer copy numbers only in the range of 0 to 5). With this technology the power of detection and resolution improved significantly.
We also built a gold standard data set made of 100 healthy individuals randomly picked out from the catalogue of the 1000 Genomes Project and belonging to 3 different populations (37 CEU-Utah residents with northern and western European ancestry, 17 JPT-Japanese people from Tokyo, 32 YRI-Yoruba people from Ibadan and 14 CHB-Han Chinese individuals from Beijing). The exomes were sequenced during phase3 of the Project in 2 different Centres using various sequencing libraries and WES enrichment kits (see Supplementary Table S3 for details). All the individuals were genotyped by the International HapMap Consortium (37) and also by the 1000 Genomes Consortium (6). The HapMap Consortium (HapMap) made use of two distinct SNP arrays (Affimetrix 6.0 and Illumina 1M) and regional PCR-Sanger sequencing to outline alterations of copy number at genomic level for both common and rare alleles. In addition, the sequencing-based genotypes of CNVs identified by the Pilot 1 and 2 of the 1000 Genomes Project (1KG Pilot) for those individuals were considered to assess the ability of our method to detect CNVs. We compared our results to recently published CopywriteR (27) and to a selection of three existing RC-based computational methods, largely used for CNV identification from WES data (i.e. CoNIFER, XHMM and CODEX). A complete description of the data set and running parameters of each tool are available in Supplementary Section 2.2 and 3.
Finally, to test the performance of our method in detecting somatic CNVs we analysed 28 WES from Urothelial bladder cancer samples (Supplementary Table S4). The somatic data set comprises 14 pairs of tumor and matched normal samples (38) enriched using the Agilent SureSelect Human All Exon plus v3 50 Mb or v4 51 Mb. Same samples were also genotyped by F.X. Real's laboratory using Illumina HumanOmniExpress-12 v1.0 SNP-array.
RESULTS
WES read count in Off-Target regions
To study the distribution of the reads aligned along the genome we used a WES Survey Data set (see Materials and Methods) made of 30 sequencing experiments and we split the genome into regions of three different categories: In-Target, Off-Target or Flanking. Following Asan (39) we considered as Flanking stretches of 200 bp adjacent to each of the boundaries of the targeted regions. The 30 WES experiments were chosen to balance the three enrichment kits (10 SureSelect, 10 SeqCap and 10 TruSeq) and with diversified sequencing throughput in order to have a general overview of reads distribution. All reads were aligned to the human reference genome (hg19). Reads overlapping the targeted regions of each exome kit (In-Target), reads surrounding enriched regions (Flanking) and reads mapping outside those regions (Off-Target) were counted separately (see Supplementary Section 2.1 for more details). Despite the sequencing throughput variability, the overall mean percentage of reads that unambiguously maps to Off-Target regions is nearly 30% for all three different enrichment kits (see Figure 1). Specifically, 23–43% of reads for TruSeq, 20–50% for SeqCap and 21–35% for SureSelect (see Supplementary Figure S1 for per sample details).
Figure 1.

Mapping statistics. The distribution of mapped reads into In-Target, Flanking and Off-Target regions are shown for the 30 WES samples belonging to the Survey Data set. They are split per enrichment kit. TS62M states for Illumina TruSeq Exome Enrichment 62 Mb, SS50M is Agilent SureSelect Human All Exon 50 Mb and SCV2 is the NimbleGen SeqCap EZ Exome v2.0 44 Mb. The absolute and relative means of mapped reads are respectively showed on the left and right.
In order to exploit this large amount of extra-target reads, we studied the properties of RC distribution across Off-Target regions and how it is influenced by classical sources of bias, like GC content and mappability. To this end, we calculated the WMRC (see Materials and Methods) for different window sizes and we found that they are similarly influenced by local nucleotide content and sequence uniqueness in Off-Target regions (see Figure 2, panel A and C for Off-Target and Supplementary Figure S2 panel C and E for In-Target) as it has been already proved for targeted regions in our previous study (20). The median normalisation approach allowed to mitigate both GC-content and uniqueness biases in Off-Target regions (Figure 2, panel B and D, respectively) as well as in In-Target regions (Supplementary Figure S2 panel D and F).
Figure 2.

Pre- and post-normalisation WMRC distribution in Off-Target regions. The influence of mappability and GC content percentage on WMRC are shown in (A) and (C) and compared with normalised signals (B) and (D).
Copy number resolution
To measure the capability of our method to correctly predict the absolute number of DNA copies of each genomic region, we calculated the normalised WMRC for In-Target and Off-Target regions from eight WES data from the 1000 Genomes Project. The ratio of normalised WMRC between each test and the control (NA10847) was compared to the copy number ratio of regions inferred by McCarroll and Conrad studies (35,36). McCarroll identified an average of 112 amplifications (with size of around 50 Kb on average) and 123 deletions (with size of around 33 Kb on average) per sample (Supplementary Table S2), while Conrad recognised in those samples 356 duplications and 441 deletions (with size of around 10 Kb on average for both variants).
The WMRC-based copy number profiles are highly correlated with McCarroll prediction for both In-Target and Off-target regions (Pearson R = 0.9 and R = 0.86, respectively; see Figure 3 panel B and C) and the correlation of combined WMRC reaches R = 0.89 (Figure 3, panel A). Interestingly, the analyses performed on Conrad data set demonstrate that the use of Off-Target reads can improve the prediction capability of RC data: the correlation between real CN and WMRC for Off-Target is larger than for In-Target regions (Supplementary Figure S3). In conclusion, these results clearly demonstrate the capability of WMRC to accurately predict copy numbers from WES data in Off-Target as well as in In-Target regions.
Figure 3.

Copy number correlation with McCarroll calls. Boxplots summarize the capability of WMRC data to predict the exact number of DNA copies of a CNV region. Normalised WMRC ratio were calculated for eight samples using NA10847 as control and compared with copy number ratios from McCarroll characterisation. R is the Pearson correlation coefficient. (A) all genomic regions, (B) In-Target regions and (C) Off-Target regions.
1KG data analysis
In order to evaluate the capability of our method to identify CNVs in a population study we analysed the WES data of a cohort of 100 healthy individuals from the catalogue of the 1000 Genomes Project. We analysed the same data set also with four other different tools that include XHMM (25), CoNIFER (23), the recently published CODEX (21) and CopywriteR (27) using the collections of all genomic CNVs genotyped by HapMap or by 1KG Pilot projects for the inspected 100 individuals as a gold standard (see Supplementary Section 2.2 and 3 for details).
To compare the performance of the five methods in identifying (i) all genomic CNVs and (ii) CNVs in targeted regions, we calculated precision (P) and recall (R) rates. For each tool P corresponds to the fraction of calls overlapping the reference set (TP) and the total number of calls (TP + FP), while R is the fraction of TP calls with respect to the whole reference set of CNVs (TP + FN). For the comparison at genomic level, the gold standard consisted of the whole set of CNVs identified by HapMap/1KG Pilot projects for the selected 100 individuals while, to measure the performance in targeted exons, the gold standard was restricted to the HapMap/1KG Pilot CNVs overlapping regions covered by each enrichment kit. To classify the calls made by each tool, we used the approach previously described by Yoon et al. (40) and Magi et al. (29) for calls classification: a detected segment is considered a true positive (TP) if there is at least 10% overlap with the gold standard CNVs for the same sample identified by HapMap/1KG Pilot, and is considered a false positive (FP) if there is no overlap or is smaller than 10%. Since the capability of detecting regions with altered copy number is influenced by the length of the segment, we distinguished three classes of events: Small (length ≤ 20Kb), Medium (length > 20 Kb and ≤ 100 Kb) and Large (length > 100 Kb). Figure 4 summarises the results using the F-measure (the harmonic mean of precision and recall). We run our tool with 3 different windows size for Off-Target regions (5 Kb, 10 Kb, 20 Kb) and we obtained the best results with the 20 Kb windows. Overall, EXCAVATOR2 outperforms all the other tools with the highest F-measure with respect to HapMap and 1KG Pilot calls sets (Figure 4, panel (A,C) and (B,D), respectively). Specifically, EXCAVATOR2 has the highest precision rates in identifying genomic CNVs (panel A for HapMap, panel B for 1KG Pilot) as well as in identifying CNVs that overlap only targeted exonic regions (panel C for HapMap, panel D for 1KG Pilot). CopywriteR shows higher recall than EXCAVATOR2 only for Large and Medium genomic events in the HapMap data set (panel A), while CODEX's recall rates are only higher with respect to CNVs overlapping targeted exons for all except Large events (panel C and D). The precision levels of XHMM and CONIFER are similar to those of CopywriteR and CODEX but with significant lower recall rates. As expected, the best absolute performance in terms of precision and recall involves Large CNVs. Remarkably, Figure 4 also shows that the two gold standard data sets used in this comparison give different results in terms of algorithm performance. In particular, almost all tools obtain lower sensitivity in recognising Medium and Small CNVs from HapMap than from 1KG Pilot. These differences can be mainly ascribed to the different nature of CNV events comprised in the two sets. HapMap data set was generated by using Affimetrix 6.0 and Illumina 1M array platforms, while the 1KG Pilot data set includes structural variants identified with three different computational approaches (split read, paired-end and depth of coverage) from low-coverage whole genome sequencing data.
Figure 4.

CNV calls assessment for the population data set. CNV events identified in 100 samples from 1000 Genomes Project catalogue by using Codex, CONIFER, CopywriteR, EXCAVATOR2 and XHMM were validated respect to the (A and C) HapMap Consortium and (B and D) 1KG Pilot genotyping calls. Results are reported from the comparison with all (A and B) genomic CNVs and restricted to only the targeted regions (C and D). Precision-recall plots are shown with light grey curves representing F-measure levels. CNV events are distinguished based on their size (Small: ≤ 20 Kb, Medium > 20 Kb and ≤ 100 Kb and Large > 100 Kb).
Somatic calls
To evaluate whether our method can give insight into cancer genomic studies we used EXCAVATOR2 and CopywriteR to analyse WES data from 28 Urothelial bladder cancer samples. To measure the accuracy and resolution of the two methods in discovering CNVs, same samples were also genotyped by F.X. Real and his group using high-resolution Illumina HumanOmniExpress SNP-array. First, we observed that CopywriteR produced a signal characterized by higher variance than EXCAVATOR2 (see Figure 5, panel B for EXCAVATOR2 and C for CopywriteR) compared to the SNP-array (Figure 5, panel A). To measure the noise level generated by the two approaches we calculated the Median Absolute Deviation (MAD) values of their segmented signals. As shown in Figure 5D, CopywriteR MAD (mean = 0.43, range = 0.02–0.98) was higher than that of EXCAVATOR2 (mean = 0.26, range = 0.13–0.65) while the mean MAD for SNP-array was smaller at 0.09 (see Figure 5D). This can be mainly ascribed to the normalization approaches adopted by the two tools and to the capability of segmentation algorithms to discriminate between the real biological signals and the experimental noise. Moreover tumor samples are characterised by clonal heterogeneity and somatic CNVs belonging to subclones with different percentages produce signals that can be confounded with the experimental noise of normal copy state. Thus, P and R are not the most appropriate statistics to compare somatic CNVs with a SNP-array gold standard. Hence, we studied the correlation between the SNP-array segmented profiles and those inferred by EXCAVATOR2 and CopywriteR along the genome. To this end, for each paired tumor/control samples we juxtaposed the median value of each region segmented by our tool and by CopywriteR (see Supplementary Sec. 2.3 and 3) with the log2 ratio median values of the SNP-array signal and calculated the global and per CNV-size correlations. The table in Figure 5E indicates that segmented profiles inferred by EXCAVATOR2 well correlates with those from SNP-array, and outperforms CopywriteR irrespective of the CNV size. In addition, the use of combined signals from In- and Off-Target allows our method to better detect CNVs in exon-rich regions and to be more accurate in breakpoints detection with respect to CopywriteR. As an example, we reported a 20 Kb deletion involving an exon-rich region detected by SNP-array (see panel F of Figure 5) that was correctly identified by EXCAVATOR2 with similar breakpoints (panel G) but completely missed by CopywriteR (panel H). Additional examples are shown in Supplementary Figure S4.
Figure 5.

Analysis on somatic data set. A total of 14 WES tumour/control samples pairs of Urothelial Bladder cancer were analysed using EXCAVATOR2 and CopywriteR, and results were compared to the SNP-array genotyping. Copy number profiles for the chromosome 5 of patient 251 with segmentation values depicted in red are shown for (A) SNP-array, (B) EXCAVATOR2 and (C) CopywriteR. MAD values for all 14 samples pairs are represented in (D). Table (E) shows the Spearman correlation coefficient values for different CNV sizes between the array-based profiles and those resulting from EXCAVATOR2 and CopywriteR. Finally, the ‘zoomed’ in region containing a potential deletion in the chromosome 5 of the patient 64 is showed with CN profiles and calls from (F) SNP-array, (G) EXCAVATOR2 and (H) CopywriteR.
DISCUSSION
Recent studies have remarked the increasingly relevance of CNVs identification for elucidating the molecular mechanisms underlying several diseases. Germline CNVs contribute to phenotypic variation and recurrent somatic CN alterations (gains and losses) in tumor genomes often involve genes with key-roles as oncogenes or oncosuppressors. Thus, improving the accuracy of breakpoint mapping and copy number prediction is fundamental for straightforward genotype–phenotype correlations and diagnostic classification of CNVs. The last years have seen the emergence of several tools for the detection of CNVs from the analysis of WES data. The first generation of these tools, as CoNIFER, XHMM, ExomeCNV, EXCAVATOR and CODEX, exploits only coverage information from targeted regions. All these tools use a similar procedure consisting of two stages: a normalization step to mitigate systematic biases due to GC content, mappability and capture efficiency, and a segmentation step for the identification of the boundaries of the altered region and the estimation of the absolute or relative number of DNA copies. ExomeCNV is the first tool implemented for the detection of CNVs from WES data. It estimates copy number values by using uncalibrated read-depth. CoNIFER and XHMM employ singular value decomposition (SVD) and principal-component analysis (PCA) techniques to identify and remove the principal sources of variation underlying the non-uniform read-depth among captured regions. SVD and PCA normalization procedures require the analysis of many samples at once, thus limiting their application to large-scale sequencing projects. CODEX, as CoNIFER and XHMM, uses latent factor models to remove systematic biases (GC content, mappability, capture and amplification efficiency) assuming a Poisson log-linear model that is more suitable for discrete count data rather than the continuous Gaussian model employed by the other two methods. In this way CODEX estimates a ‘control coverage’ for no CNV that it compares with the observed coverage for each exon and each samples.
The first release of EXCAVATOR combines a three-step normalization procedure with a novel heterogeneous hidden Markov model algorithm and a calling method that classifies genomic regions into five copy number states. CopywriteR is instead the first published method that makes direct use of off-target reads to build uniformly distributed genomic copy number profiles. It uses MACS-based peak calling to identify all regions that are well covered by reads (peaks) and keeps only the background reads which are used to build a compensated binned Depth of Coverage profile. The compensated DoC is normalized using loess-based corrections for mappability and GC content and finally segmented by means of circular binary segmentation method. In this way the authors claim to reduce the noise and they show that the segmented profile built from reads after peaks removal is ‘close’ to the one from low-coverage WGS. This choice, together with a filtering step aiming at removing extreme values for coverage, length, mappability and GC content, allowed the method to (globally) outperform both CoNIFER and XHMM and also our previous version of EXCAVATOR when compared with calls from the International HapMap Consortium, McCarroll and Conrad genotyping studies (27). In this work, we present the first computational approach that combines together reads aligned to In- and Off-targeted regions of WES experiments to identify CNVs. This approach significantly improves the detection of CNVs in targeted regions with respect to all other state-of-the-art computational methods and (globally) outperforms CopywriteR in identifying CNVs at genomic-level. We proved that around 30% of the reads produced by WES experiments align outside the targeted regions. These reads are often regarded as the ‘junk’ of WES experiments, because they consume a substantial amount of sequencing throughput but are of no value for the common scope of WES, that is, identifying variants in targeted exons. In order to exploit this amount of extra-exonic data, we measured the RC in these regions and we found that they is affected by the same sources of biases and is able to predict the exact number of DNA copies with nearly the same accuracy. For this reason we extended the EMRC that we introduced in (20) by defining the WMRC which accounts for both In-Target exons and Off-Target windows of fixed size. We integrated the WMRC into a new tool, EXCAVATOR2 that we used to analyse two different data sets and we showed that it outperforms the other state-of-the-art tools in calling genomic CNVs. In particular, EXCAVATOR2 is able to identify CNVs involving large intergenic regions with few exons that are usually missed by first generation methods, included the previous version of our tool. Indeed, only EXCAVATOR2 detected and correctly genotyped a heterozygous deletion implicated in epilepsy (11). The deletion spans around 270 kb of intergenic regions across exons 2–3 of CNTNAP2 (Contactin-associated protein-like 2; OMIM 604569) which was predicted to produce an out-of-frame transcript p.Gln33Argfs*7 (NM 014141:c.98402_del). Furthermore, as shown, EXCAVATOR2 is able to detect also CNVs belonging to exon-rich regions that CopywriteR fails to catch. This is due to the fact that it removes regions that are well covered by reads and only keeps the background to build CN profiles. All together the results obtained for the two data sets, a population-based and a cancer study, clearly prove that our combined approach improves the precision of calling CNVs overlapping targeted exons from WES data and enlarges the spectrum of detectable CNVs to off-target events. Therefore, EXCAVATOR2 can be effectively employed for the identification of CNVs in small as well as large-scale re-sequencing studies with best performance and so maximizing the utility of exome sequencing data in genetic and cancer studies. Lastly, it's of particular interest that all WES experiments can be re-analysed using our method with the beneficial effect to identify novel CNVs in extra-exonic regions by having the full-genome CN profile.
Supplementary Material
Acknowledgments
The authors wish to acknowledge E. Carrillo-de-Santa-Pau, N. Malats, and F.X. Real for providing the tumor data used in the study.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Flagship project InterOmics (PB. P05, CUP B91J12000270001), Italian Ministry for Instruction University and Research (MIUR) and CNR organisations; Italian Ministry of Health, Young Investigators Award for the Project GR-2011-02352026 ‘Detecting copy number variants from whole-exome sequencing data applied to acute myeloid leukemias’ (to A.M.). Funding for open access charge: Flagship project InterOmics (PB. P05, CUP B91J12000270001), Italian Ministry for Instruction University and Research (MIUR) and CNR organisations.
Conflict of interest statement. None declared.
REFERENCES
- 1.Alkan C., Coe B.P., Eichler E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tattini L., D'Aurizio R., Magi A. Detection of genomic structural variants from next-generation sequencing data. Front. Bioeng. Biotechnol. 2015;3:92. doi: 10.3389/fbioe.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mills R.E., Walter K., Stewart C., Handsaker R.E., Chen K., Alkan C., Abyzov A., Yoon S.C., Ye K., Cheetham R.K., et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conrad D.F., Hurles M.E. The population genetics of structural variation. Nat. Genet. 2007;39(Suppl. 7):S30–S36. doi: 10.1038/ng2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pang A.W., MacDonald J.R., Pinto D., Wei J., Rafiq M.A., Conrad D.F., Park H., Hurles M.E., Lee C., Venter J.C., et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 2010;11:R52. doi: 10.1186/gb-2010-11-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.1000 Genomes Project Consortium. Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarrei M., MacDonald J.R., Merico D., Scherer S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015;16:172–183. doi: 10.1038/nrg3871. [DOI] [PubMed] [Google Scholar]
- 8.MacDonald J.R., Ziman R., Yuen R.K.C., Feuk L., Scherer S.W. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee C., Scherer S.W. The clinical context of copy number variation in the human genome. Expert Rev. Mol. Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 10.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pippucci T., Licchetta L., Baldassari S., Palombo F., Menghi V., D'Aurizio R., Leta C., Stipa C., Boero G., d'Orsi G. Epilepsy with auditory features: A heterogeneous clinico-molecular disease. Neurol. Genet. 2015;1:e5. doi: 10.1212/NXG.0000000000000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarroll S.A., Huett A., Kuballa P., Chilewski S.D., Landry A., Goyette P., Zody M.C., Hall J.L., Brant S.R., Cho J.H., et al. Deletion polymorphism upstream of irgm associated with altered irgm expression and crohn's disease. Nat. Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell P.J., Stephens P.J., Pleasance E.D., O'Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C., et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. A coding-independent function of gene and pseudogene mrnas regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wellcome Trust Case Control Consortium. Genome-wide association study of cnvs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahed A.C., Gelb B.D., Seidman J.G., Seidman C.E. Genetics of congenital heart disease: the glass half empty. Circ. Res. 2013;112:707–720. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fakhro K.A., Choi K., Ware S.M., Belmont J.W., Towbin J.A., Lifton R.P., Khokha M.K., Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. U.S.A. 2011;7:2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiang D.Y., Getz G., Jaffe D.B., O'Kelly M.J.T., Zhao X., Carter S.L., Russ C., Nusbaum C., Meyerson M., Lander E.S. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A., et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magi A., Tattini L., Cifola I., D'Aurizio R., Benelli M., Mangano E., Battaglia C., Bonora E., Kurg A., Seri M., et al. Excavator: detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013;14:R120. doi: 10.1186/gb-2013-14-10-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Y., Oldridge D.A., Diskin S.J., Zhang N.R. Codex: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015;43:e39. doi: 10.1093/nar/gku1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magi A., D'Aurizio R., Palombo F., Cifola I., Tattini L., Semeraro R., Pippucci T., Giusti B., Romeo G., Abbate R., et al. Characterization and identification of hidden rare variants in the human genome. BMC Genomics. 2015;16:340. doi: 10.1186/s12864-015-1481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krumm N., Sudmant P.H., Ko A., O'Roak B.J., Malig M., Coe B.P., Quinlan A.R., Nickerson D.A., Eichler E.E., NHLBI Exome Sequencing Project Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sathirapongsasuti J.F., Lee H., Horst B.A.J., Brunner G., Cochran A.J., Binder S., Quackenbush J., Nelson S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: Exomecnv. Bioinformatics. 2011;27:2648–2654. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fromer M., Moran J.L., Chambert K., Banks E., Bergen S.E., Ruderfer D.M., Handsaker R.E., McCarroll S.A., O'Donovan M.C., Owen M.J., et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012;91:597–607. doi: 10.1016/j.ajhg.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samuels D.C., Han L., Li J., Quanghu S., Clark T.A., Shyr Y., Guo Y. Finding the lost treasures in exome sequencing data. Trends Genet. 2013;29:593–599. doi: 10.1016/j.tig.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuilman T., Velds A., Kemper K., Ranzani M., Bombardelli L., Hoogstraat M., Nevedomskaya E., Xu G., de Ruiter J., Lolkema M.P., et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49. doi: 10.1186/s13059-015-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magi A., Benelli M., Marseglia G., Nannetti G., Scordo M.R., Torricelli F. A shifting level model algorithm that identifies aberrations in array-cgh data. Biostatistics. 2010;11:265–280. doi: 10.1093/biostatistics/kxp051. [DOI] [PubMed] [Google Scholar]
- 29.Magi A., Benelli M., Yoon S., Roviello F., Torricelli F. Detecting common copy number variants in high-throughput sequencing data by using jointslm algorithm. Nucleic Acids Res. 2011;39:e65. doi: 10.1093/nar/gkr068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benelli M., Marseglia G., Nannetti G., Paravidino R., Zara F., Bricarelli F.D., Torricelli F., Magi A. A very fast and accurate method for calling aberrations in array-cgh data. Biostatistics. 2010;11:515–518. doi: 10.1093/biostatistics/kxq008. [DOI] [PubMed] [Google Scholar]
- 31.Clark M.J., Chen R., Lam H.Y.K., Karczewski K.J., Chen R., Euskirchen G., Butte A.J., Snyder M. Performance comparison of exome dna sequencing technologies. Nat. Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sulonen A-M., Ellonen P., Almusa H., Lepistö M., Eldfors S., Hannula S., Miettinen T., Tyynismaa H., Salo P., Heckman C., et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12:R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller C.A., Hampton O., Coarfa C., Milosavljevic A. Readdepth: a parallel r package for detecting copy number alterations from short sequencing reads. PLoS One. 2011;6:e16327. doi: 10.1371/journal.pone.0016327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magi A., Tattini L., Pippucci T., Torricelli F., Benelli M. Read count approach for dna copy number variants detection. Bioinformatics. 2012;28:470–478. doi: 10.1093/bioinformatics/btr707. [DOI] [PubMed] [Google Scholar]
- 35.McCarroll S.A., Kuruvilla F.G., Korn J.M., Cawley S., Nemesh J., Wysoker A., Shapero M.H., de Bakker P.I.W., Maller J.B., Kirby A., et al. Integrated detection and population-genetic analysis of snps and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 36.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–12. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Consortium T.I.H. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balbas-Martinez C., Sagrera A., Carrillo-de Santa-Pau E., Earl J., Marquez M., Vazquez M., Lapi E., Castro-Giner F., Beltran S., Bayes M., et al. Recurrent inactivation of stag2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 2013;45:1464–U221. doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asan Y, Xu H., Jiang C., Tyler-Smith Y., Xue T., Jiang J., Wang M., Wu X., Liu G., Tian J., et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011;12:R95. doi: 10.1186/gb-2011-12-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon S., Xuan Z., Makarov V., Ye K., Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.