


Abstract
The major human pathogen Mycobacterium tuberculosis can survive in the host organism for decades without causing symptoms. A large cohort of Toxin–Antitoxin (TA) modules contribute to this persistence. Of these, 48 TA modules belong to the vapBC (virulence associated protein) gene family. VapC toxins are PIN domain endonucleases that, in enterobacteria, inhibit translation by site-specific cleavage of initiator tRNA. In contrast, VapC20 of M. tuberculosis inhibits translation by site-specific cleavage of the universally conserved Sarcin-Ricin loop (SRL) in 23S rRNA. Here we identify the cellular targets of 12 VapCs from M. tuberculosis by applying UV-crosslinking and deep sequencing. Remarkably, these VapCs are all endoribonucleases that cleave RNAs essential for decoding at the ribosomal A-site. Eleven VapCs cleave specific tRNAs while one exhibits SRL cleavage activity. These findings suggest that multiple vapBC modules contribute to the survival of M. tuberculosis in its human host by reducing the level of translation.
INTRODUCTION
The major human pathogen Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), currently infects more than 8 million and kills >1 million people per year (1). The prevalence of multi-drug resistant TB is increasing and is a cause for significant concern (2). M. tuberculosis can persist latently without symptoms for many years in human carriers (3). The molecular mechanisms underlying latency and slowed bacterial growth are still poorly understood but may involve multiple regulatory pathways. One of these depends on the stringent response and Toxin–Antitoxin (TA) modules (4).
All bacteria produce slow growing ‘persister cells’ that are tolerant to a broad spectrum of antibiotics (5,6). Recent research on Escherichia coli and Salmonella shows that persistence is controlled by the stringent response and TA modules. Such TA modules are almost ubiquitous in bacteria and are often present in perplexingly high numbers (7). Mycobacterium tuberculosis has at least 88 type II TAs (8,9), raising important questions concerning their biological function(s).
Type II TA modules encode two genes in an operon, a protein ‘toxin’ that inhibits cell growth and a protein ‘antitoxin’ that counteracts the inhibitory effect of the toxin by direct protein contact (10). The antitoxins are usually metabolically unstable while the toxins are stable. Thus, regulated proteolysis of a given antitoxin determines the activity of the cognate toxin. Evidence from E. coli K-12 and Salmonella indicates that TA modules are effector genes that induce persistence when activated and step-wise deletion of 10 type II TAs progressively reduced persistence (11). Correspondingly, inactivation of Lon, the protease that degrades all known type II antitoxins of E. coli K-12, strongly reduced persistence. Remarkably, the TAs of E. coli are induced stochastically by a mechanism that depends on (p)ppGpp, polyphosphate and Lon. In a population of rapidly growing cells, approximately 10−4 have a high level of (p)ppGpp that leads to accumulation of polyphosphate, which activates Lon to degrade antitoxins. Toxin activation then leads to growth arrest, multidrug tolerance, and persistence (12). Independent support for this model has come from several comprehensive studies of Salmonella enterica serovar Typhimurium. S. Typhimurium has up to 20 type II TAs and deletions of single TA loci showed reduced survival of S. Typhimurium within fibroblasts (13). Furthermore, deletion of single TA genes reduced persister cell formation and survival when the bacterium was grown inside macrophages (14). Consistently, multiple type II TAs have been found to be activated in S. Typhimurium when residing inside macrophages (15). Studies with M. tuberculosis also support a role for TAs in persistence and virulence (8,16,17).
Mycobacterium tuberculosis has a highly expanded repertoire of TA modules, with 48 representatives of the vapBC family (7,8) (Figure 1A). VapC toxins are PIN (pilT N-terminal) endoribonucleases containing three or four conserved acidic residues that coordinate Mg2+ ion(s) in the active site (18). In Eukaryotes, PIN domains have been identified in multidomain endonucleases involved in RNA metabolism, RNA quality control and rRNA maturation (19). In Prokaryotes, however, most PIN domain proteins belong to the highly abundant VapC toxin family that are present in staggering numbers in certain prokaryotic genomes, including M. tuberculosis (7–9).
Figure 1.

Known vapBC modules of Mycobacterium tuberculosis H37Rv, growth-inhibition by selected VapCs and outline of the CRAC analysis procedure. (A) Chromosomal location of 48 vapBC modules (7,8). Genes shown in blue are analysed here, gene shown in red was analysed previously (22). (B) Growth-inhibition tests of selected vapCs of M. tuberculosis in M. smegmatis strain MC2155 containing plasmid pMEND carrying a tetracycline inducible promoter and encoding C-terminally His-TEV-FLAG tagged (HTF) vapC genes. Strain MC2155 carrying vapC4 (Rv0595c) vapC11 (Rv1561), vapC15 (Rv2010), vapC25 (Rv0277c), vapC26 (Rv0582), vapC28 (Rv0609), vapC29 (Rv0617), vapC30 (Rv0624), vapC32 (Rv1114), vapC33 (Rv1242), vapC37 (Rv2103c) or vapC39 (Rv2530c) were grown overnight in LBT at 37°C. The optical density (OD600) was then adjusted to 0.5, and the bacterial cultures serially diluted (10-fold) and spotted (3 μl) on nutrient agar plates without or with inducer (20 ng/ml tetracycline). The plates were incubated 3 days at 37°C. (C) Outline of CRosslinking and Analysis of cDNA protocol (CRAC). (1) The HTF tagged VapC protein is UV cross-linked (UV-C) with target RNAs in vivo, the RNA–protein complexes are purified and the RNA trimmed by a cocktail of RNase A/T1 and barcoded linkers are ligated to the RNA. (2) cDNA is synthesized by reverse transcription using 3′-end specific primer and the library generated by PCR. (3) Finally, the DNA library is purified and deep sequenced using the Illumina platform. The sequencing output is aligned and analysed using the pyCRAC software package.
The molecular targets of most prokaryotic VapCs are unknown; however, the VapCs of S. Typhimurium, Shigella flexneri and Leptospira interrogans cleave initiator tRNA site-specifically in the anticodon loop, thereby inhibiting global translation (20,21). In contrast, VapC20 of M. tuberculosis inhibits translation via cleavage of the conserved Sarcin–Ricin loop (SRL) of 23S rRNA (22).
Here, we applied UV-induced RNA-protein crosslinking and analysis of cDNA by high throughput sequencing (CRAC) to identify transcriptome-wide targets of VapC paralogues in M. tuberculosis using M. smegmatis as a surrogate host organism. First, we identified the cellular targets of six different VapCs and showed that they all cleave tRNAs site-specifically within the anticodon loop. We then used phylogenetic analysis to identify the cellular targets of an additional six VapCs. Several of these VapCs were previously shown to have non-specific RNase activity in vitro (23–25). Strikingly, we show that all 12 VapCs catalyse site-specific cleavage of RNAs essential for protein synthesis.
MATERIALS AND METHODS
Strains, plasmids and growth conditions
Mycobacterium smegmatis strain MC2155 was routinely grown in LB medium (Difco) containing 0.1% Tween-80 (LBT) at 37°C. For UV-crosslinking experiments strain MC2155 was grown in M9 medium (Difco) containing 0.1% Tween-80 (M9T) with 0.1% Casein hydrolysate, 1 μg/ml thiamine and 0.2% glucose as carbon source at 37°C. When appropriate, 50 μg/ml kanamycin was added the medium to maintain the plasmid. Furthermore, when stated 20 ng/ml tetracycline was added to liquid or solid medium to induce transcription from tetracycline inducible promoters.
Plasmids
Construction of plasmids is described in Supplementary Information; Supplementary Table S1 contains a list of oligonucleotides used to construct plasmids and to detect RNAs in northern analysis.
Crosslinking and analysis of cDNA
Cultures (2 l) of M. smegmatis MC21555 containing plasmids pMEND::HTF, pMEND::vapC4::HTF, pMEND::vapC11::HTF, pMEND::vapC28::HTF, pMEND::vapC29::HTF, pMEND::vapC30::HTF, pMEND::vapC37::HTF, were grown exponentially in M9T at 37°C under constant aeration. At OD600 = 0.3–0.4 cultures were induced by addition of 20 ng/ml tetracycline. After 20 min of incubation cultures were irradiated with 1800 mJ of UV-C for 100 seconds (Van Remmen UV Techniek). The cells were subsequently harvested by centrifugation, washed in ice-cold phosphate buffered saline (PBS) containing 0.1% Tween-80 and snap frozen in liquid nitrogen. The pellets were stored at −80°C. The HTF tagged proteins were then purified, linkers ligated to crosslinked RNA, cDNA synthesized and DNA libraries generated as described in (26). The DNA libraries were then sequenced on the illumina MiSeq platform and the sequencing output analysed using the pyCRAC tool package (27).
Northern blotting analysis of tRNA and rRNA after VapC induction
M. smegmatis MC2155 containing plasmid pMEND-HTF with VapC of interest were grown exponentially in LBT containing kanamycin (50 μg/ml) at 37°C. At an OD600 of 0.3–0.4, vapC transcription was induced by the addition of tetracycline (20 ng/ml). A sample was collected before and after 120 min of incubation with inducer. Total RNA was purified using the FastRNA Blue Kit (MPbiomedicals) according to manufactures instructions. The cell samples were lysed by homogenization using the MagNA lyzer (Roche) twice at 6500 rpm for 40 s, separated by a 1 min rest on ice. After precipitation the RNA was dissolved in nuclease free water. Total RNA (2.5 μg) was denatured in Formamide loading buffer and separated on a 4.5% (rRNA) or 8% (tRNA) 8 M urea polyacrylamide gel in 1× TBE (100 mM Tris–borate and 2 mM ethylenediaminetetraacetic acid, EDTA). The RNA was then transferred to a Zeta-Probe membrane (Bio-Rad) by semi-dry electrophoretic transfer and the membrane pre-hybridized with SES1 buffer (0.25 M NaPO4 pH 7.2, 1 mM EDTA and 7% SDS) at 42°C for at least 30 min. Probe oligonucleotides (20 pmol) (see Supplementary Table S1) were labelled using 30μCi γ 32P-ATP and T4 Polynucleotide kinase (Fermentas) according to manufactures procedures and added to the membrane (due to the high primary sequence similarity between M. smegmatis and M. tuberculosis tRNAs, M. smegmatis tRNA probes could be used in most cases except for tRNA24Ser-GGA and tRNA28Ser-CGA). The tube was then incubated overnight at 29°C in a hybridization oven. After hybridization the membrane was washed 2–3 times in SES3 buffer (0.25 M NaPO4 pH 7.2, 1 mM EDTA and 5% SDS) at room temperature. The bands were visualized by phosphorimaging. The membrane could be stripped with repeated washes of 0.1% SDS at 85°C and subsequently re-probed.
Purification of VapC4-HTF, VapC11-HTF, VapC28-HTF and VapC37-HTF
VapC toxins were purified from Mycobacterium smegmatis using histidine affinity chromatography as we described previously (22). Mycobacterium smegmatis MC2155 containing plasmids pMEND::vapC4::HTF, pMEND::vapC11::HTF, pMEND::vapC28::HTF and pMEND::vapC37::HTF were grown exponentially in 1-l LBT at 37°C with shaking. At OD600 = 0.4, the culture was induced with tetracycline (20 ng/ml) for 1 h before cells were pelleted by centrifugation. The cells were then washed in ice-cold PBS containing 0.1% Tween-80 and lysed in 1 ml (v/w) lysis buffer (50 mM NaH2PO4, pH 8, 300 mM NaCl, 10 mM Imidazole, 0.1% NP-40, 5 mM β-mercaptothanol) by bead beating (2 × 6500 rpm for 40 s, MagNA lyzer cell homogenizer) using 0.5 ml 100 μM glass beads per 1.5 ml of cell suspension. The lysate was cleared by centrifugation at 14 krpm for 20 min and incubated overnight at 4°C with 0.5 ml equilibrated Ni-NTA resin (Qiagen). The resin was then loaded onto a gravity-flow column and washed with 40 column volume of wash buffer (50 mM NaH2PO4, pH 8, 300 mM NaCl, 20 mM Imidazole, 0.1% NP-40, 5 mM β-mercaptothanol). The column was then washed with four column volume wash buffer containing 40 mM imidazole and four column volume wash buffer containing 50 mM imidazole. The protein was eluted by incubating the column twice with elution buffer (50 mM NaH2PO4, pH 8, 300 mM NaCl, 500 mM Imidazole, 0.1% NP-40, 5 mM β-mercaptothanol). Elution fraction containing VapC toxin was dialysed for 6 h against PBS containing 1 mM DTT and overnight against PBS containing 50% glycerol and 1 mM DTT at 4°C.
Detection of in vitro RNA cleavage by Northern blotting analysis
M. smegmatis total RNA was purified using the FastRNA blue kit (see previous section). To ensure high quality, RNA was purified by an additional step of phenol/chloroform extraction. Total RNA from M. tuberculosis H37Rv was a generous gift from Douglas Young and Rachel Lai (National Institute for Medical Research, London and Imperial College London, UK). For the cleavage reaction 1 μg or 2.5 μg of total RNA was mixed with 1 μg or 2 μg of purified VapC4-HTF VapC11-HTF, VapC28-HTF or VapC37-HTF in cleavage buffer (final: 10 mM HEPES pH7.5, 15 mM KCl, 10 mM MgCl2, 1 mM DTT and 25% glycerol) and left to incubate at 37°C for 30 min. As a control VapC toxin was omitted in some reactions and Mg2+ chelated by the addition of 25 mM EDTA. The cleavage reactions were terminated by addition of FD-loading buffer and the RNA separated on an 8% Urea polyacrylamide gel buffered with 1× TBE. The RNA was transferred to a Zeta-Probe membrane and the RNA of interest detected using a radiolabelled probe as previously described.
Mapping of VapC4, VapC11, VapC28 and VapC37 cleavage site by primer extension analysis
The cleavage reactions were set up as previously described, using total RNA (4 μg) with or without 2 μg of purified VapC4-HTF, VapC11-HTF, VapC28-HTF or VapC37-HTF. The cleavage reactions were then incubated for 30 min at 37°C in cleavage buffer. The reaction was terminated by addition of 400 μl 100 mM Na-acetate pH 4.5 followed by phenol/chloroform extraction and ethanol precipitation. The precipitate was washed with 70% ethanol, air dried and dissolved in nuclease free dH2O. The reverse transcription was performed by setting up a hybridization reaction containing 0.2 pmol radiolabelled KW-MS-PXT-CysGCA-rv, KW-MS-PXT-TrpCCA-rv, KW-MS-PXT-SerTGA-S1-rv or KW-MS-PXT-LeuCAG-S-rv oligonucleotide and 1 μg of purified cleaved RNA in nuclease free dH2O. The oligonucleotide (4 pmol) was phosphorylated using 30 μCi γ 32P-ATP and T4 Polynucleotide kinase (Fermentas) and subsequently desalted using G-25 desalting columns (GE healthcare). The hybridization reactions were incubated at 80°C for 5 min and transferred to an icebath and left to incubate for 5 min. To the chilled reaction 1× FS buffer (Invitrogen), 10 mM DTT and 1 mM dNTP was added and the tube transferred to 54°C to incubate for 2 min. Then 20 U of Superscript III reverse transcriptase (Invitrogen) was added and the tubes left to incubate for 1 h. The reaction was terminated by addition of an equal volume of FD-loading buffer. The reactions were loaded onto a 10% polyacrylamide gel containing 8 M urea and 1× TBE. Along with the reverse transcription reactions a dideoxy sequencing ladder was loaded which had been made from a PCR template generated using oligonucleotides KW-MS-PXT-CysGCA-f and KW-MS-PXT-CysGCA-rv, KW-MS-PXT-TrpCCA-rv and KW-MS-PXT-TrpCCA-f, KW-MS-PXT-SerTGA-S1-rv and KW-MS-PXT-SerTGA-f or KW-MS-PXT-LeuCAG-S-rv and KW-MS-PXT-LeuCAG-f. After the cDNA had been separated, the gel was dried and the bands visualized by phosphorimaging.
RESULTS
Identification of cellular VapC targets using CRAC
We implemented CRAC to systematically identify the cellular targets of VapC RNases from M. tuberculosis, using M. smegmatis as a surrogate host organism. CRAC identifies RNAs that interact directly with a tagged bait protein in living cells and potentially detects both stable and relatively transient interactions (26,28). To apply the CRAC protocol, all 48 vapC genes of M. tuberculosis H37Rv (Figure 1A) were cloned into plasmid pMEND-HTF downstream of a tetracycline inducible promoter and in frame with a dual affinity tag that introduced a hexaHis, TEV protease cleavage site, and 3× FLAG tags at the C-termini (HTF-tag). Figure 1B shows the effect of transcriptional induction of selected vapC genes. Expression of VapC4, 11, 25, 26, 28, 29, 30, 33, 37 and 39 strongly inhibited cell growth, whereas expression of VapC15 and 32 had a more moderate effect (for the results of phenotypic testing of all 48 VapCs, see Supplementary Figure S1A). The pattern of growth inhibition was generally consistent with previous tests in which native VapCs were expressed in M. smegmatis (8,29). The only exception was VapC32 that did not significantly reduce cell growth in our experimental system (Figure 1B). Thus, we can conclude that the C-terminal HTF tag does not interfere with VapC-mediated inhibition of cell growth by the toxins selected in this study.
The majority of VapCs expressed in M. smegmatis showed weak or no inhibition of growth (Supplementary Figure S1A). Therefore, we determined the expression levels of a number of these VapCs (Supplementary Figure S1B). We found that in some cases, including VapC41, 43 and 44, no VapC expression was detectable 60 min after induction that could explain the lack of growth inhibition. However, for the majority of non-toxic VapCs that we tested, i.e. VapC6, 10, 12, 19, 31 and 47, protein expression was detectable, indicating that the proteins either are not functional (e.g. the cellular target could be missing in the heterologous host). Alternatively, their biological functions do not entail inhibition of cell growth.
Cultures of M. smegmatis MC2155 were pulsed with HTF-tagged VapC and were UV-irradiated to covalently crosslink VapCs to their target RNAs in vivo (Figure 1C). The covalently bound VapC-RNA complexes were then purified using M2 anti-FLAG resin, eluted by TEV-cleavage, and RNAs were trimmed to allow ‘footprinting’ of the protein interaction site. Trimmed VapC-RNA complexes were bound to Ni-NTA resin and barcoded linkers were ligated to the 5′ and 3′ termini. Linker-ligated RNA–protein complexes were eluted and size-selected using SDS-PAGE. Protein-RNA complexes of the appropriate molecular weight were extracted and protease digested. cDNAs were generated by RT-PCR, subjected to deep sequencing and reads were mapped to the M. smegmatis genome. VapC binding sites were initially identified using the pyCRAC software package to identify transcripts bound by each VapC. We used duplicate controls (pMEND::HTF) to assess the background of the assay. CRAC data from samples expressing different VapCs were obtained from single experiments. The results are shown as reads per million allowing us to visually compare for significance across samples.
In the following section, we describe the identification of the cellular targets of previously uncharacterized VapCs. We will refer to RNA interactions that were confirmed to confer RNA cleavage as ‘productive interactions’, and other interactions as ‘unproductive interactions’.
All six VapCs interact with a subset of tRNAs, SRP RNA and 23S rRNA
Analysis of CRAC data for VapC4, 11, 28, 29, 30 and 37 each revealed enrichment for tRNA5Thr-GGT, tRNA8fMet-CAT, tRNA12Val-CAC, tRNA37Asn-GTT and tRNA31Asp-GTC (Supplementary Figure S2A-E) (the tRNA nomenclature used is in accordance with GtRNAdb: http://gtrnadb.ucsc.edu and the reads have been plotted as per million). However, we were unable to demonstrate that any of these tRNAs were cleaved by the respective VapCs when expressed in M. smegmatis cells (Supplementary Figure S2H-L). The CRAC analysis also revealed enrichment for SRP RNA and 23S rRNA (Supplementary S2F and S2G). However, these interactions could not be confirmed as productive as no cleavage by any of the VapCs was observed (Supplementary Figure S2M and S2N). VapC28 and VapC29 did indeed showed higher enrichment with the 5′-end region of 23S rRNA (Supplementary Figure S2G) but we were not able to confirm cleavage in this region of 23S rRNA (Supplementary Figure S2O). As none of these RNA species were cleaved by VapC4, 11, 28, 29, 30 and 37 we categorize these interactions as unproductive and they were not studied further.
VapC4 specifically cleaves tRNA44Cys-GCA in M. smegmatis
Analysis of specific RNA interaction with VapC4 using CRAC revealed strong enrichment for RNA fragments mapping to tRNA44Cys-GCA (Figure 2A). The interaction with tRNA44Cys-GCA was productive as the full-length tRNA decreased upon induction of vapC4 simultaneous with the accumulation of a smaller cleavage product (compare lanes 1 and 2 with 3 and 4 in Figure 2B). We also observed weak cleavage of tRNA44Cys-GCA before induction of vapC4. This observation can be explained due to ‘leaky’ transcription initiation from the tetracycline inducible promoter as no cleavage is observed in the control. Induction of the other vapC toxins did not affect tRNA44Cys-GCA stability (lanes 5–14). The tRNAs; tRNA23Pro-CGG and tRNA32Phe-GAA were also specifically enriched by VapC4 (Supplementary Figure S3A and S3B, respectively) but none of these tRNA species were cleaved by VapC4 (compare lanes 1 and 2 in Supplementary Figure S3D and S3E, respectively). The other cysteine-accepting tRNA of M. smegmatis, annotated as tRNA40Cys-GCA, was not enriched by VapC4 in the analysis, and was not investigated further (Supplementary Figure S3C).
Figure 2.

VapC4 cleaves tRNA44Cys-GCA in M. smegmatis. (A) Enrichment of tRNA44Cys-GCA fragments by six different VapCs (VapC4, 11, 28, 29, 30 and 37) analysed by CRAC. M. smegmatis strain MC2155 containing plasmid pMEND::HTF was used as a Control. The X-axis indicates nucleotide position relative to gene start site and Y-axis shows number of reads per million. (B) Northern analysis of tRNA44Cys-GCA cleavage. M. smegmatis containing plasmid pMEND::HTF with six different vapC as in (A) were grown exponentially in LBT at 37°C. At an OD600 of 0.3–0.4, the vapC gene was induced by the addition of tetracycline (20 ng/ml). Total RNA was purified from samples taken before (indicated by 0) and after 120 min of induction (indicated by 120). Total RNA (2.5 μg) was separated on a polyacrylamide gel (8%) containing urea (8 M) and blotted onto a membrane and the tRNAs of interest detected using radiolabelled oligonucleotides detecting tRNA44Cys-GCA. Full-length tRNA44Cys-GCA is indicated and the cleavage product is indicated with an arrow. (C) in vitro cleavage of M. smegmatis tRNA44Cys-GCA by purified VapC4-HTF (addition of protein is indicated with a +) detected by Northern analysis as described in Materials and Methods. A reaction with EDTA was included as a control for dependency on Mg2+. FL indicates the position of the full-length tRNA and the cleavage product is indicated by an arrow. (D) Cleavage site mapping of tRNA44Cys-GCA by primer extension analysis in the absence (indicated by –) and presence (indicated by +) of VapC4-HTF as described in Materials and Methods, using the 32P-labelled oligonucleotide MS-PXT-Cys-GCA-rv. An asterisk indicates the position of a putative modification in the tRNA. (E) Visualization of the VapC4-mediated in vitro cleavage sites on the secondary structure of tRNA44Cys-GCA. The base positions of the cleavage sites are indicated in the anticodon loop.
To confirm that the observed cleavage of tRNA44Cys-GCA was direct, cleavage was investigated in vitro by Northern analysis using purified VapC4-HTF in a reaction containing total RNA from M. smegmatis. Indeed we observed cleavage of tRNA44Cys-GCA only in the presence of VapC4 (Figure 2C). Consistent with the VapC PIN domain coordinating Mg2+ in the active site, addition of EDTA to the reaction abolished cleavage. Mapping of the cleavage site in tRNA44Cys-GCA by primer extension analysis revealed cleavage in the anticodon loop between bases C34↓A35, A35↓A36, A36↓A37 and A37↓G38 (arrows indicate cleavages between numbered nucleotides), with the A36↓A37 site being the dominant cleavage site (Figure 2D and E). The reverse transcriptase weakly terminated at this site before addition of VapC4, probably due to modification of the corresponding base.
VapC4 has recently been suggested to cleave tRNA2Ala-TGC, tRNA24Ser-GGA, tRNA26Ser-GCT of M. tuberculosis H37Rv in vitro (30). We did observe weak enrichment of tRNA24Ser-GGA tRNA26Ser-GCT by VapC4 (Supplementary Figure S4E and S4F). However, we did not observe any cleavage of these tRNA species in vivo (compare lane 1 and 2, Supplementary Figure S3F-H). We also did not observe cleavage of tRNA24Ser-GGA or tRNA26Ser-GCT in vitro with M. smegmatis RNA (Supplementary Figure S3I and S3J) or in vitro with M. tuberculosis H37Rv RNA (Supplementary Figure S3K and S3L).
VapC11 cleaves tRNA3Leu-CAG, tRNA13Leu-GAG and tRNA10Gln-CTG
CRAC analysis showed enrichment for RNA fragments derived from tRNA3Leu-CAG, tRNA13Leu-GAG and tRNA10Gln-CTG by VapC11 (Figure 3A–C). These interactions were productive since tRNA cleavage products accumulated upon vapC11 induction in all three cases (compare lanes 1 and 2 with 5 and 6 in Figure 3D–F). None of the other VapCs tested showed productive interactions with these tRNA species (lanes 3, 4 and 7–14). The positive interaction with tRNA3Leu-CAG seemed to be strongest as a cleavage product could be detected even before induction of vapC11 (lane 5), probably reflecting leakiness of the tetracycline-inducible promoter driving vapC11 transcription.
Figure 3.

VapC11 cleaves tRNA3Leu-CAG, tRNA10Leu-GAG and tRNA13Gln-CTG in M. smegmatis. Enrichment of (A) tRNA3Leu-CAG, (B) tRNA10Leu-GAG and (C) tRNA13Gln-CTG by six different VapCs analysed by CRAC as in Figure 2(A). In vivo cleavage by six VapCs was analysed as in Figure 2(B) and the tRNAs of interest detected using radiolabelled oligonucleotides complementary to (D) tRNA3Leu-CAG. (E) tRNA13Leu-GAG. (F) tRNA10Gln-CTG. The tRNAs were analysed before (indicated by 0) and after 120 min of induction (indicated by 120) in M. smegmatis cells. Cleavage products are indicated with arrows. (G) in vitro cleavage assay of tRNA3Leu-CAG without (indicated by –) or with (indicated by +) purified VapC11-HTF detected by Northern analysis as in Figure 2(C). A reaction with EDTA was included as a control for dependency on Mg2+. (H) Cleavage site mapping by primer extension analysis was performed as in Figure 2(D), using the 32P-labeled oligonucleotide MS-PXT-LeuCAG-S-rv. An asterisk indicates the position of a putative modification in the tRNA. (I) Visualization of the VapC11-mediated in vitro cleavage sites on the secondary structure of tRNA3Leu-CAG. The base positions of the cleavage sites are indicated in the anticodon loop.
To determine whether the activity of VapC11 was direct, we assayed cleavage of tRNA3Leu-CAG in vitro. Purified VapC11-HTF was incubated with total RNA of M. smegmatis. Northern analysis confirmed that tRNA3Leu-CAG was cleaved by VapC11, with accumulation of a discrete cleavage product (Figure 3G). Cleavage was inactivated by the addition of EDTA, demonstrating that the cleavage activity was Mg2+ dependant. Primer extension analysis revealed that VapC11 cleaved tRNA3Leu-CAG in the anticodon loop between nucleotide pairs G37↓U38, and U38↓G39, respectively (Figure 3H and I). Like tRNA44Cys-GCA cleavage by VapC4 we observe termination of the reverse transcriptase at these sites before addition of VapC11 indicating that the sites are putative sites of modification.
VapC28 and VapC30 both cleave tRNA25Ser-TGA and tRNA28Ser-CGA
VapC28 and VapC30 specifically enriched for RNA fragments derived from tRNA25Ser-TGA, tRNA28Ser-CGA (Compare lanes 1 and 2 with 7 and 8 and 11 and 12 of Figure 4A and B). Induction of vapC28 and vapC30 showed reduced amounts of full-length tRNA25Ser-TGA and tRNA28Ser-CGA. However, stable, discrete cleavage products were not detected (compare lane 1–2 with 7–8 and 11–12 Figure 4C and D). Expression of the other VapCs analysed by CRAC did not affect the stability of tRNA25Ser-TGA and tRNA28Ser-CGA. CRAC analysis also revealed tRNA43Gly-GCC, tRNA17Lys-CTT and tRNA2Ala-TGC to be enriched by VapC28 and VapC30 (Supplementary Figure S4A-C). However, none of these tRNAs were detectably affected by induction of vapC28 and vapC30 and these interactions were categorized as unproductive (Supplementary Figure S4H-J, compare lane 1 and 2). Furthermore, CRAC analysis revealed enrichment of tRNA16His-GTG, tRNA24Ser-GGA, tRNA26Ser-GCT and tRNA22Thr-TGT by VapC30 alone (Supplementary Figure S4D–G). These enrichments were also categorized as unproductive as no changes in stabilities were observed after induction of VapC30 (compare lanes 1 and 2 in Supplementary Figure S4K–N).
Figure 4.

VapC28 and VapC30 cleave tRNA25Ser-TGA and tRNA28Ser-CGA of M. smegmatis. Enrichment of tRNA fragments from (A) tRNA25Ser-TGA and (B) tRNA28Ser-CGA by six VapCs was identified by CRAC as described before. Detection of in vivo cleavage by VapCs were investigated as in Figure 2(B) before (indicated by 0) and after 120 minutes of induction (indicated by 120). The tRNA of interest was detected using radiolabelled oligonucleotides complementary to (C) tRNA25Ser-TGA and (D) tRNA28Ser-CGA. (E) Cleavage in vitro of tRNA25Ser-TGA was determined as Figure 2(C) in the absence (indicated by –) and in the presence (indicated by +) of purified VapC28-HTF. The cleavage product was detected by Northern analysis using a radiolabelled probe specific to tRNA25Ser-TGA. (F) The in vitro cleavage sites in tRNA25Ser-TGA were mapped as in Figure 2(D) using radiolabelled oligonucleotide MS-PXT-SerTGA-S1-rv in the reverse transcription reaction of VapC28 in vitro cleaved RNA from M. smegmatis. Absence or presence of VapC28-HTF is indicated by a – or +, respectively and the mapped cleavage site position is indicated with an arrow. An asterisk indicates the position of a putative modification in the RNA. (G) Visualization of the cleavage site in tRNA25Ser-TGA. The base position and cleavage site are indicated in the anticodon loop.
The activity of VapC28 was confirmed by in vitro cleavage. Thus, VapC28 cleaved tRNA25Ser-TGA of M. smegmatis (Figure 4E) and a discrete cleavage product was detected, suggesting that the absence of clear cleavage product in vivo reflected their rapid degradation. Addition of EDTA abolished cleavage, consistent with the requirement for Mg2+ in the reaction. The cleavage site of VapC28 in tRNA25Ser-TGA was mapped to nucleotide pairs G36↓A37 in the anticodon loop (Figure 4F and G). At A38, we observed strong termination of reverse transcriptase before the addition of VapC28, probably reflecting base modification at this position.
VapC29 and VapC37 cleave tRNA7Trp-CCA
CRAC with VapC29 and VapC37 as baits identified interactions with RNA fragments derived from tRNA7Trp-CCA (Figure 5A). In addition, VapC29 also specifically enriched for tRNA4Tyr-GTA-derived fragments while VapC37 specifically enriched for tRNA42Leu-CAA-derived fragments (Supplementary Figure S5A and S5B). Northern analysis revealed productive interactions between tRNA7Trp-CCA and VapC29 and VapC37 in M. smegmatis cells and two discrete cleavage products accumulated upon induction (compare lanes 1 and 2 with lanes 9 and 10 and 13 and 14 of Figure 5B). None of the other VapCs analysed by CRAC was observed to affect the stability of this tRNA (lanes 3–8 and 11–12). Neither tRNA4Tyr-GTA nor tRNA42Leu-CAA were cleaved by VapC29 and VapC37, respectively, and these interactions were therefore categorized as unproductive (compare lanes 1 and 2 in Supplementary Figure S5C and S5D). Incubation of purified VapC37 with total RNA of M. smegmatis confirmed direct cleavage of tRNA7Trp-CCA that was Mg2+ dependent (Figure 5C). We only observe one cleavage product in vitro, indicating that the two cleavage products observed in vivo is a result of cellular RNases similar to that observed for tRNA25Ser-TGA and tRNA28Ser-CGA. The VapC37 cleavage sites in tRNA7Trp-CCA were mapped to bases A36↓A37, A37↓A38 and A38↓A39, with the A37↓A38 cleavage site being the most prominent (Figure 5D and E). Similar to the other tRNAs mapped by primer extension analysis we observe termination of reverse transcription reaction in this region before addition of VapC37, which indicates a putative site of modification in the tRNA at the cleavage site.
Figure 5.

VapC29 and VapC37 cleave tRNA7Trp-CCA of M. smegmatis. (A) Enrichment of tRNA fragments from tRNA7Trp-CCA by six VapCs was identified by CRAC as described previously. (B) Cleavage of tRNA7Trp-CCA in M. smegmatis cells expressing VapCs was detected by Northern analysis as described before. Samples taken before and after 120 min of induction are indicated by 0 and 120, respectively and cleavage products are indicated with arrows. (C) In vitro cleavage of tRNA7Trp-CCA without (indicated by –) and with (indicated by +) purified VapC37-HTF was performed as in Figure 2(C) and cleavage detected using a radiolabelled probe specific to tRNA7Trp-CCA. (D) The in vitro cleavage site in tRNA7Trp-CCA was mapped as in Figure 2(D) using radiolabelled DNA oligonucleotide MS-PXT-TrpCCA-rv in the reverse transcription reaction and total RNA of M. smegmatis treated without (indicated by –) or with (indicated by +) VapC37-HTF. Arrows indicate cleavage sites and an asterisk indicates the position of a putative modification in the tRNA. (E) Visualization of the cleavage sites in tRNA7Trp-CCA. Base position and cleavage sites are indicated in the anticodon loop.
VapC4, VapC11, VapC28 and VapC37 cleave orthologous tRNAs in M. tuberculosis
The above-described in vitro cleavage reactions were performed using total RNA from M. smegmatis as the substrate. Even though the tRNAs of M. smegmatis are almost identical to the orthologous tRNAs of M. tuberculosis there are nucleotide differences that potentially could affect VapC recognition and cleavage (Supplementary Figure S6A–G). The VapC cleavage reactions were therefore also analysed using total RNA of M. tuberculosis H37Rv. Consistent with our results in M. smegmatis, VapC4 cleaved tRNA21Cys-GCA (Figure 6A, lanes 2 and 3), VapC11 specifically cleaved tRNA3Leu-CAG (Figure 6B, lane 2), VapC28 specifically cleaved tRNA25Ser-TGA and tRNA28Ser-CGA (Figure 6C and D, lane 5) and VapC37 specifically cleaved tRNA7Trp-CCA (Figure 6E, lane 8). The cleavages were tRNA-specific and all cleavages were also inhibited by the EDTA addition, indicating Mg2+ dependence (Figure 6B–E). These results confirmed that heterologously expressed VapCs have identical cleavage specificities in M. smegmatis, validating this organism as a useful surrogate host for the analysis of VapC toxins from M. tuberculosis.
Figure 6.
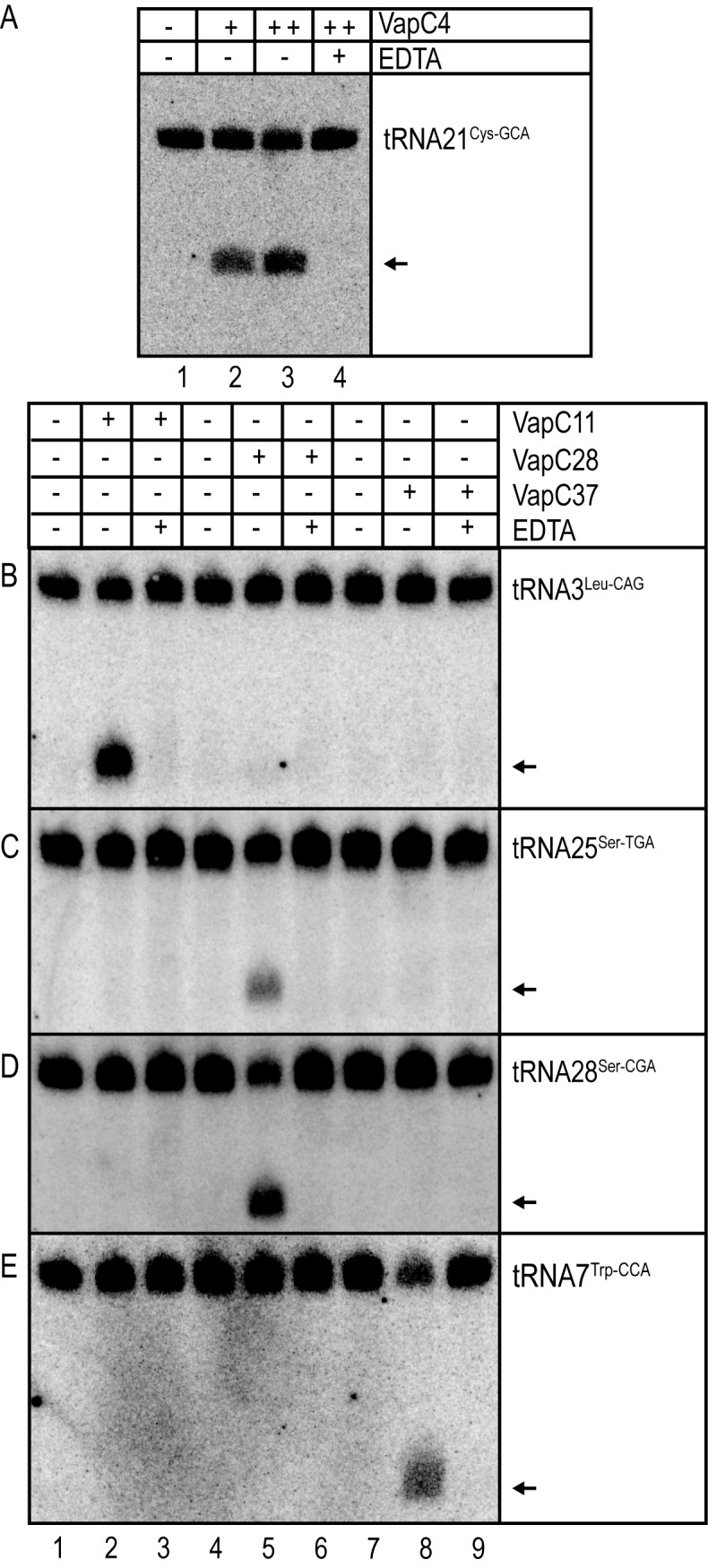
VapC4, VapC11, VapC28 and VapC37 cleave orthologous tRNAs in M. tuberculosis. In vitro cleavage of M. tuberculosis H37Rv tRNAs by purified VapC4, 11, 28 and 37 detected by Northern analysis was performed as described in Materials and Methods. Addition of toxins and EDTA are indicated by (+) and arrows indicate cleavage products. For reactions containing toxin (+) indicated addition of 1μg of toxin, whereas (++) indicated addition of 2μg. The membrane was incubated with tRNA specific DNA probe hybridizing to (A) tRNA21Cys-GCA. (B) tRNA3Leu-CAG. (C) tRNA25Ser-TGA. (D) tRNA28Ser-CGA. (E) tRNA7Trp-CCA of M. tuberculosis H37Rv.
Phylogeny can predict novel VapC targets
The identification of novel VapC targets is challenging and in the past required the application of laborious ‘trial-and-error’ approaches. Since M. tuberculosis has at least 48 different VapCs (Figure 1A), we tested the possibility that the targets we had identified could provide insights into the targets of related VapC toxins using a phylogenetic approach. From the phylogenetic tree shown in Supplementary Figure S1C, we selected six VapCs that group together with one of the VapCs with a known target, identified here or in our previous analysis (22), and tested whether these VapCs cleaved the same targets. The following examples showed that this approach was effective.
VapC15 and VapC32 cleave tRNA3Leu-CAG. VapC15 and VapC32 group together with VapC11 in the phylogram (Figure 7E and Supplementary Figure S1C). Therefore, we asked if these two VapCs also cleave tRNA3Leu-CAG, the tRNA that was cleaved most efficiently by VapC11. Indeed tRNA3Leu-CAG was cleaved upon induction of vapC15 and vapC32 (Figure 7A, lanes 3 - 6). These cleavages were relatively weak compared to those of VapC11, consistent with the lower toxicity of these VapCs in M. smegmatis (Figure 1B). VapC11 was also found to have productive interactions with tRNA13Leu-GAG and tRNA10Gln-CTG, but these tRNA species were not cleaved by VapC15 or VapC32 (Supplementary Figure S7A and S7B).
VapC25, 33 and 39 cleave tRNA7Trp-CCA. VapC25, 33 and 39 group together with VapC29 and 37 in the phylogram (Figure 7E and Supplementary Figure S1C) and induction of these toxins in M. smegmatis inhibited cell growth (Figure 1B). Notably, induction of vapC25, 33 or 39 resulted in cleavage products identical to those of VapC29 and 37 (Figure 7B lanes 3 - 8).
VapC26 cleaves 23S rRNA in the Sarcin–Ricin Loop (SRL). VapC26 groups with VapC20 in the phylogram (Figure 7E and Supplementary Figure S1C) and inhibited growth of M. smegmatis very efficiently (Figure 1B). Since VapC20 cleaves 23S rRNA in the SRL loop (22), we tested if VapC26 also cleaves 23S rRNA. Indeed, induction of vapC26 resulted in a 23S rRNA cleavage pattern identical to that of VapC20 showing that the two VapCs have identical targets (Figure 7C, lanes 3–6).
Figure 7.

Phylogeny can predict cellular VapC targets. (A) Detection of in vivo cleavage of tRNA3Leu-CAG by VapC15 and VapC32 by Northern analysis was performed as in Figure 3 (D); VapC expression was induced from M. smegmatis MC2155 carrying plasmids pMEND::vapC15::HTF or pMEND::vapC32::HTF. (B) in vivo cleavage of tRNATrp-CCA by VapC25, VapC33 and VapC39 was detected by Northern analysis performed as in Figure 5 (B). Toxin expression was induced from plasmid pMEND::vapC25::HTF, pMEND::vapC33::HTF and pMEND::vapC39::HTF. (C) Cleavage of 23S rRNA detected by Northern analysis was performed on RNA purified from M. smegmatis strain MC2155 containing plasmids pMEND::vapC20::HTF and pMEND::vapC26::HTF. Cell samples were collected before (indicated by 0) and after 120min of induction (indicated by 120) and analysed as described in Materials and Methods. The arrow indicates the 276 nucleotide cleavage product of 23S rRNA generated by VapC20 and VapC26, as mapped previously (22). The full-length 23S rRNA (3164 nt) is also indicated with an arrow. (D) Region of the decoding center of 70S ribosome of Thermus thermophilus in complex with mRNA and the ternary complex of EF-Tu•Thr-tRNAThr•GDP (pdb entry: 4V5G from (31)). The Figure highlights that VapCs of M. tuberculosis cleave RNAs essential for decoding during translation. The Sarcin - Ricin Loop (SRL), which is cleaved by VapC20 and 26, is shown in orange. The tRNA anticodon loop, which is cleaved by VapC4, 11, 15, 25, 28, 29, 30, 32, 33 37 and 39, is also marked with a box. Bases cleaved are coloured red. Major parts of the ribosome structures were omitted for clarity. (E) Phylogenetic tree of VapCs of M. tuberculosis strain H37Rv with known targets was based on a multiple sequence alignment made using the ClustalX v2.0 software; amino acid sequences were retrieved from www.ncbi.nlm.nih.gov. VapCMYSH: VapC from Shigella flexneri 2a plasmid pMYSH6000 (21). VapCLepto: VapC of Leptospira interrogans (20). The tRNAs in the green, magenta, blue and light blue boxes have identical names in M. smegmatis MC2155 and M. tuberculosis H37Rv except tRNA13Leu-CAG and tRNA10Gln-CTG of M. smegmatis that are denoted tRNA15Leu-CAG and tRNA32Gln-CTG in M. tuberculosis H37Rv. tRNA44Cys-GCA in M. smegmatis is named tRNA21Cys-GCA in M. tuberculosis H37Rv. The sequences of the orthologous tRNAs studied here are shown in Supplementary Figure S6.
VapCs are highly target-specific
The above results indicated that VapCs grouping together phylogenetically exhibit identical RNA cleavage specificities (Figure 7E and Supplementary Figure S1C). Therefore, we analysed whether VapCs from one phylogenetic subgroup would cleave RNAs targeted by a different VapC subgroup. As seen from both in vivo (see overview in Supplementary Figure S8A–G) and in vitro cleavage assays (see overview in Supplementary Figure S9A-D) we observed no cross-reactivity and conclude that the VapCs analysed are highly specific endoribonucleases.
DISCUSSION
Using CRAC in combination with phylogenetic analysis and in vivo and in vitro RNA cleavage assays, we identified the cellular targets of 12 novel VapCs from M. tuberculosis H37Rv. UV-crosslinking and high throughput sequencing revealed binding sites that were both productive (cleaved) and apparently unproductive (Supplementary Figure S2). Functional tests identified site-specific tRNA cleavage sites for six VapCs. Using a phylogenetic approach, we exploited this novel information to identify specific targets for six additional VapCs. Since the target of VapC20 was already known (22), collectively we now know the targets of thirteen of the forty-eight VapCs of M. tuberculosis (Figure 7E and Supplementary Figure S1C). Remarkably, all these thirteen VapCs degrade RNAs that are essential for translation: eleven VapCs cleave tRNAs while two VapCs cleave the SRL of 23S rRNA. These RNA cleavages all inactivate RNAs essential for mRNA decoding at the A-site during translation (Figure 7D), thereby explaining the strong growth inhibition resulting from their overexpression. Together with previous reports, our data show that VapCs from different bacterial domains (actinomycetes, spirochaetes and enterobacteria) inhibit translation by highly related mechanisms, indicating conserved biological function(s) (20,22). Moreover, phylogenetic analyses determined that evolutionarily related VapC proteins show identical cleavage specificities, indicating a degree of functional redundancy. Within the VapC11/15/32 clade, VapC11 additionally cleaved tRNA13Leu-GAG and tRNA10Gln-CTG (Figure 7E). However, the cleavages of these tRNAs by VapC11 were less efficient (Figure 3E and F) and may represent an artefact of ectopic expression of VapC11 caused by the high sequence similarity of the anticodon loops (Supplementary Figure S6B–D).
Cruz et al. recently reported cleavage of tRNA2Ala-TGC, tRNA24Ser-GGA, tRNA26Ser-GCT by VapC4 (30). In our study we did not observe enrichment of tRNA2Ala-TGC by VapC4 (Supplementary Figure S4C) and, consistently, we did not observe cleavage of this tRNA when VapC4 was induced in M. smegmatis cells (Supplementary Figure S3F). We did indeed observe weak enrichment of RNA fragments from tRNA24Ser-GGA and tRNA26Ser-GCT by CRAC with VapC4 as the bait (Supplementary Figure S4E and S4F); however, no cleavage of tRNA24Ser-GGA or tRNA26Ser-GCT by VapC4 was observed in vivo (Supplementary Figure S3G and S3H) or in vitro (Supplementary Figure S3I-L). We note that the reported cleavage of tRNA2Ala-TGC, tRNA24Ser-GGA, and tRNA26Ser-GCT by VapC4 in vitro was only seen after highly extended incubation times only (3-12hr; (30)), raising the possibility that these tRNAs represent low-affinity targets of VapC4. Using UV-crosslinking and Northern analysis we found that VapC4 actually interacts with, and cleaves tRNA44Cys-GCA in vivo. We used a much shorter incubation time (30 min) and exclusively observed cleavage of tRNA44Cys-GCA of M. smegmatis and the orthologous tRNA21Cys-GCA of M. tuberculosis H37Rv.
In enterobacteria, TA gene modules encoding translational inhibitors promote increased survival following exposure to antibiotics or stressful conditions within macrophages (11–15,32). The biological function(s) of the multitude of vapBC modules encoded by M. tuberculosis has not yet been established. However, it is possible that cleavage of RNAs essential for translation similarly confers protection by inhibiting cell growth, as proposed previously by other groups (8,16,17,33). Consistent with this proposal, E. coli rapidly responds to oxidative stress by downregulating translation via tRNA degradation (34). It is also possible that the vapBC genes can prevent bacteriophages from spreading in cell populations by inducing so-called ‘abortive infection’ that has been described for type III TA modules also encoding inhibitors of translation (35,36).
The M. tuberculosis genome contains an unusually large expansion of TA modules that present a unique challenge for understanding their contribution to bacterial persistence in this significant human pathogen. Here we described a new methodology to identify the cellular targets of PIN-domain RNases in vivo and demonstrated that related clades of VapC toxins have similar substrate specificities. To our knowledge this study represents the most comprehensive analysis of VapC targets to date and greatly expands our understanding of VapC targets in M. tuberculosis.
Supplementary Material
Acknowledgments
We thank Douglas Young and Rachel Lai (National Institute for Medical Research, London and Imperial College London, UK) for the donation of purified M. tuberculosis RNA and Ditlev E. Brodersen for critical comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Danish Natural Research Foundation [DNRF120; to K.G.]; Novo Nordisk Foundation (to K.G.); European Research Council Advanced Investigator Grant PERSIST [294517 to K.G.]. Funding for open access charge: Danmarks grundforskningsfond.
Conflict of interest statement. None declared.
REFERENCES
- 1.Zumla A., Raviglione M., Hafner R., von Reyn C.F. Tuberculosis. N. Engl. J. Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 2.Zumla A., Nahid P., Cole S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 3.Lillebaek T., Dirksen A., Baess I., Strunge B., Thomsen V.O., Andersen A.B. Molecular evidence of endogenous reactivation of Mycobacterium tuberculosis after 33 years of latent infection. J. Infect. Dis. 2002;185:401–404. doi: 10.1086/338342. [DOI] [PubMed] [Google Scholar]
- 4.Dutta N.K., Karakousis P.C. Latent tuberculosis infection: myths, models, and molecular mechanisms. Microbiol. Mol. Biol. Rev. 2014;78:343–371. doi: 10.1128/MMBR.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maisonneuve E., Gerdes K. Molecular mechanisms underlying bacterial persisters. Cell. 2014;157:539–548. doi: 10.1016/j.cell.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 6.Bigger J.W. Treatment of staphyloccal infections with penicillin by intermittent sterilisation. Lancet. 1944;2:497–500. [Google Scholar]
- 7.Pandey D.P., Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramage H.R., Connolly L.E., Cox J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jørgensen M.G., Pandey D.P., Jaskolska M., Gerdes K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 2009;191:1191–1199. doi: 10.1128/JB.01013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerdes K., Christensen S.K., Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 11.Maisonneuve E., Shakespeare L.J., Jørgensen M.G., Gerdes K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Maisonneuve E., Castro-Camargo M., Gerdes K. Gpp Controls Bacterial Persistence by Stochastic Induction of Toxin-Antitoxin Activity. Cell. 2013;154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 13.Lobato-Marquez D., Moreno-Cordoba I., Figueroa V., Diaz-Orejas R., Garcia-del Portillo F. Distinct type I and type II toxin-antitoxin modules control Salmonella lifestyle inside eukaryotic cells. Sci. Rep. 2015;5:9374. doi: 10.1038/srep09374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helaine S., Cheverton A.M., Watson K.G., Faure L.M., Matthews S.A., Holden D.W. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science. 2014;343:204–208. doi: 10.1126/science.1244705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva-Herzog E., McDonald E.M., Crooks A.L., Detweiler C.S. Physiologic stresses reveal a Salmonella persister state and TA family toxins modulate tolerance to these stresses. PLoS One. 2015;10:e0141343. doi: 10.1371/journal.pone.0141343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tiwari P., Arora G., Singh M., Kidwai S., Narayan O.P., Singh R. MazF ribonucleases promote Mycobacterium tuberculosis drug tolerance and virulence in guinea pigs. Nat. Commun. 2015;6:6059. doi: 10.1038/ncomms7059. [DOI] [PubMed] [Google Scholar]
- 17.Keren I., Minami S., Rubin E., Lewis K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio. 2011;2:e00100–00111. doi: 10.1128/mBio.00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fatica A., Tollervey D., Dlakic M. PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. RNA. 2004;10:1698–1701. doi: 10.1261/rna.7123504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houseley J., Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 20.Lopes A.P., Lopes L.M., Fraga T.R., Chura-Chambi R.M., Sanson A.L., Cheng E., Nakajima E., Morganti L., Martins E.A. VapC from the leptospiral VapBC toxin-antitoxin module displays ribonuclease activity on the initiator tRNA. PLoS One. 2014;9:e101678. doi: 10.1371/journal.pone.0101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winther K.S., Gerdes K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7403–7407. doi: 10.1073/pnas.1019587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winther K.S., Brodersen D.E., Brown A.K., Gerdes K. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat. Commun. 2013;4:2796. doi: 10.1038/ncomms3796. [DOI] [PubMed] [Google Scholar]
- 23.Das U., Pogenberg V., Subhramanyam U.K., Wilmanns M., Gourinath S., Srinivasan A. Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J. Struct. Biol. 2014;188:249–258. doi: 10.1016/j.jsb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Lee I.G., Lee S.J., Chae S., Lee K.Y., Kim J.H., Lee B.J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015;43:7624–7637. doi: 10.1093/nar/gkv689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenzie J.L., Duyvestyn J.M., Smith T., Bendak K., Mackay J., Cursons R., Cook G.M., Arcus V.L. Determination of ribonuclease sequence-specificity using Pentaprobes and mass spectrometry. RNA. 2012;18:1267–1278. doi: 10.1261/rna.031229.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tree J.J., Granneman S., McAteer S.P., Tollervey D., Gally D.L. Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol. Cell. 2014;55:199–213. doi: 10.1016/j.molcel.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Webb S., Hector R.D., Kudla G., Granneman S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biol. 2014;15:R8. doi: 10.1186/gb-2014-15-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granneman S., Kudla G., Petfalski E., Tollervey D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9613–9618. doi: 10.1073/pnas.0901997106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahidjo B.A., Kuhnert D., McKenzie J.L., Machowski E.E., Gordhan B.G., Arcus V., Abrahams G.L., Mizrahi V. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One. 2011;6:e21738. doi: 10.1371/journal.pone.0021738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruz J.W., Sharp J.D., Hoffer E.D., Maehigashi T., Vvedenskaya I.O., Konkimalla A., Husson R.N., Nickels B.E., Dunham C.M., Woychik N.A. Growth-regulating Mycobacterium tuberculosis VapC-mt4 toxin is an isoacceptor-specific tRNase. Nat. Commun. 2015;6:7480. doi: 10.1038/ncomms8480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmeing T.M., Voorhees R.M., Kelley A.C., Gao Y.G., Murphy F.V.t., Weir J.R., Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah D., Zhang Z., Khodursky A., Kaldalu N., Kurg K., Lewis K. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 2006;6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korch S.B., Contreras H., Clark-Curtiss J.E. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J. Bacteriol. 2009;191:1618–1630. doi: 10.1128/JB.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong J., Xiao C., Gu W., Du G., Sun X., He Q.Y., Zhang G. Transfer RNAs mediate the rapid adaptation of Escherichia coli to oxidative stress. PLoS Genet. 2015;11:e1005302. doi: 10.1371/journal.pgen.1005302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blower T.R., Pei X.Y., Short F.L., Fineran P.C., Humphreys D.P., Luisi B.F., Salmond G.P. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat. Struct. Mol. Biol. 2011;18:185–190. doi: 10.1038/nsmb.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blower T.R., Short F.L., Rao F., Mizuguchi K., Pei X.Y., Fineran P.C., Luisi B.F., Salmond G.P. Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012;40:6158–6173. doi: 10.1093/nar/gks231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.