


Abstract
Queuosine (Q) was discovered in the wobble position of a transfer RNA (tRNA) 47 years ago, yet the final biosynthetic enzyme responsible for Q-maturation, epoxyqueuosine (oQ) reductase (QueG), was only recently identified. QueG is a cobalamin (Cbl)-dependent, [4Fe-4S] cluster-containing protein that produces the hypermodified nucleoside Q in situ on four tRNAs. To understand how QueG is able to perform epoxide reduction, an unprecedented reaction for a Cbl-dependent enzyme, we have determined a series of high resolution structures of QueG from Bacillus subtilis. Our structure of QueG bound to a tRNATyr anticodon stem loop shows how this enzyme uses a HEAT-like domain to recognize the appropriate anticodons and position the hypermodified nucleoside into the enzyme active site. We find Q bound directly above the Cbl, consistent with a reaction mechanism that involves the formation of a covalent Cbl-tRNA intermediate. Using protein film electrochemistry, we show that two [4Fe-4S] clusters adjacent to the Cbl have redox potentials in the range expected for Cbl reduction, suggesting how Cbl can be activated for nucleophilic attack on oQ. Together, these structural and electrochemical data inform our understanding of Cbl dependent nucleic acid modification.
INTRODUCTION
To date, more than 100 post-transcriptional modifications of transfer RNA (tRNA) have been reported (1,2). These modifications appear to stabilize local tRNA structure and, when found in the wobble position of the anticodon, ensure control over specificity and/or efficiency of translation (3,4). One of the more impressive hypermodifications is represented by queuosine (Q) (Figure 1A and B) (5,6), an RNA nucleoside that was discovered 47 years ago in the course of sequencing tRNATyr from Escherichia coli (7–9). This unusual nucleoside and its analogs (Supplementary Figure S1) were subsequently found in the wobble position of tRNAAsp, tRNAAsn, tRNAHis and tRNATyr (G/Q-U-N anticodons) and shown to be present in a wide range of organisms from bacteria to man (8,10–12). Notably, archaea contain the structurally related hypermodified RNA nucleoside archaeosine (13), which is found within the D loop of archaeal tRNAs at position 15 rather than at the wobble position. Q is only biosynthesized by bacteria and is obtained by mammals from dietary sources or microbes in the gut (14). It is not essential under normal growth conditions in E. coli, but appears to confer selective advantage when a wild-type strain is grown in competition with a variant lacking Q (15). In mice, depletion of Q from dietary sources leads to neurological defects and death. More generally, hypomodified cellular RNA pools are correlated with cancer progression and viral infection (14).
Figure 1.

Queuosine biosynthesis and incorporation into four tRNAs. (A) Scheme for the conversion of GTP to Q. (B) Diagram of tRNA that includes the nucleotide sequence of the anticodon stem loop construct of tRNATyr that was used in this work. Incorporation of Q in the wobble position of tRNA enables decoding of substrate codons with either a corresponding U or C in the 3′ codon position.
The biosynthetic pathway for Q was recently elucidated (16–21) (Figure 1A) and the enzyme responsible for the final step identified and biochemically characterized (19,22). Epoxyqueuosine (oQ) reductase (QueG) catalyzes the reduction and dehydration of an oQ moiety that is already incorporated in the wobble position of tRNA (oQ-tRNA) to yield Q-tRNA (19). Surprisingly, QueG is not homologous to other tRNA modifying enzymes but instead is a cobalamin (Cbl, vitamin B12)/[4Fe-4S] protein (19) that is similar to CblC, an enzyme that catalyzes the reductive decyanation/dealkylation of Cbl derivatives (23,24). It is also similar to the reductive dehalogenase (RDH) family of enzymes (19,25–27), which degrade both environmental and human-introduced organohalides such as tetrachloroethene (28). Cbl-enzymes are generally referred to as having two flavors: ones that use a methylcobalamin (MeCbl) cofactor and catalyze methyl transfer reactions, and ones that use adenosylcobalamin (AdoCbl) and catalyze radical-mediated rearrangement reactions; yet QueG and RDHs do not appear to use either of these Cbl derivatives (29). Here, we present crystal structures of QueG from Bacillus subtilis, both alone and in complex with tRNA, in order to visualize how a Cbl-dependent enzyme can modify RNA.
MATERIALS AND METHODS
All crystallography experiments were performed under anaerobic conditions within either a 95–97% Ar and 3–5% H2 atmosphere in a Coy anaerobic chamber, or a purely N2 atmosphere within an MBraun anaerobic chamber. All materials and solutions used for purification were deoxygenated in the anaerobic chamber many days prior and were RNase-free when possible. Crystallization-grade reagents were purchased from Hampton Research and purged with Ar prior to use. Small aliquots of <0.1 ml were flash frozen and exposed to vacuum before being brought into the anaerobic chambers. RNase-free crystallization trays were generated aerobically, flash frozen, exposed to vacuum and allowed to gas exchange while covered with crystallization tape in an anaerobic chamber overnight prior to crystallization. QueG was purified and reconstituted under anaerobic conditions in a Coy anaerobic chamber containing a 95–97% N2 and 3–5% H2 atmosphere.
Detection of RNase activity in QueG protein samples
We were surprised to find no electron density for the anticodon bases of the unmodified tRNA anticodon stem loop (ACSL). This lack of density suggested to us that low levels of RNase contamination in the purified protein could have clipped the RNA. RNase contamination during protein purifications is a known issue (30) that may be exacerbated at the concentrations of protein/RNA used in crystallography. Whereas dilute assays may show little-to-no RNase contaminating activity, concentrated samples may contain high enough RNase levels to observe RNA degradation. Over the course of a crystallization experiment, this contaminating activity may result in total loss of the desired RNA sample. To test for contamination, total E. coli RNA samples, enriched for tRNA, were incubated with QueG (expressed and purified as detailed below) and monitored for RNase activity. QueG (150 μM) and RNA (70 μM) were mixed and incubated at 37°C for 3 h, with a control in which only QueG buffer was added to the RNA sample. Formamide-containing loading dye was added to each sample, and samples were then frozen and thawed prior to analysis by 8% PAGE/7M urea. Gels show that addition of the QueG protein sample to the RNA leads to substantial RNA degradation (Supplementary Figure S2A), suggesting the presence of a contaminating RNase. Murine RNase inhibitor (NEB), which inhibits RNases A, B and C by binding in a 1:1 stoichiometric ratio, was incubated with RNA/QueG mixtures at 4, 0.4, 0.04, 0.004 and 0.0004 units, at 37°C for 3 h. RNA degradation levels were analyzed by 7 M urea/8% PAGE and were found to be greatly diminished as a result of the RNase inhibitor addition (Supplementary Figure S2B). The observed decrease in RNase activity suggested that the observed RNA degradation was due to an RNase A-, B- or C-type contamination, with RNase A as a likely culprit. To minimize RNA degradation by A-, B- and C-type RNases, we began using RNase-free reagents (see below) during the protein preparation and took extra precautions in handling glassware. We also switched to expressing QueG in BL21(DE3)Δrna, which contains a deletion for the RNase I gene. The generation of BL21(DE3)Δrna has been described in Supplementary Data for reference (31). QueG produced and purified in this manner showed no detectable RNase activity when assayed as described above (data not shown).
Expression and purification of SeMET-labeled QueG and wild-type B. subtilis QueG
A construct of B. subtilis QueG in the pASK-IBA43plus vector (IBA), containing an N-terminal hexahistidine tag and a C-terminal Strep-tag II, was utilized for expression and purification (22). Selenomethionine (SeMET)-labeled QueG was obtained by growth in minimal media using a method to suppress methionine biosynthesis (32). Wild-type B. subtilis QueG was expressed recombinantly using two different E. coli cell lines. Initially, BL21(DE3) cells were used as previously described (22). After the discovery of RNase contamination of QueG samples, all disposable materials used were certified RNase-free. All non-disposable materials and glassware were treated with RNaseZap (Applied Biosystems) according to the manufacturer's protocol prior to use. RNase-free water was made by adding diethyl pyrocarbonate to MilliQ water to a final volume of 0.05% (v/v) followed by incubation overnight at room temperature, with subsequent autoclaving the next day. All buffers/solutions were then made using the RNase-free water. Additionally, the cell line was switched to BL21(DE3)Δrna, which contains a deletion for the RNase I gene (see Supplementary Data in reference (31)).
Both wild-type and SeMET-labeled QueG were purified as follows. Cells (∼20 g) were suspended in buffer containing 50 mM Tris-HCl (pH 7.6), 0.5 M NaCl, 20 mM imidazole, 0.5 mM dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride and disrupted using a Branson digital sonifier (45% amplitude). The lysate was centrifuged for 20 min at 18 000 ×g (4°C) to pellet insoluble lysate. The cleared lysate was loaded onto a 5 ml HisTrap HP column (GE Healthcare), which had been charged with NiSO4 and equilibrated with a buffer containing 50 mM Tris-HCl (pH 7.6), 0.5 M NaCl, 20 mM imidazole (buffer A). The column was then washed with 25 ml of buffer A, and QueG was eluted with 25 ml of buffer A containing 0.75 M imidazole. Fractions containing QueG were pooled based on analysis by SDS-PAGE and then buffer exchanged using a BioRad DG-10 column into 100 mM Tris-HCl (pH 8.0) and 150 mM NaCl (buffer B). The protein was subsequently loaded onto a 5 ml StrepTrap HP column (GE Healthcare) that had been equilibrated with buffer B. The column was then washed with 25 ml of buffer B and QueG was eluted with 25 ml of buffer B containing 2.5 mM d-desthiobiotin (Sigma Aldrich). Fractions containing QueG were again identified using SDS-PAGE and pooled. The protein was reconstituted with its two [4Fe-4S] clusters in a Coy anaerobic chamber as described previously (22). The reconstituted protein solution was buffer exchanged using a BioRad DG-10 column into 20 mM Tris-HCl (pH 8.0), 100 mM KCl and 2 mM DTT (buffer C), and concentrated to a final volume of less than 1 ml by centrifugation at 6000 ×g (4°C) using an Amicon Ultra-4 10 kDa cutoff concentrator. One hundred microliters of 20 mM hydroxocobalamin acetate salt (Sigma-Aldrich) was added to the resulting concentrated [4Fe-4S]-QueG, and after a 5 min incubation period at room temperature, the protein was loaded onto a column packed with Sephacryl S300 HR resin (GE Healthcare) equilibrated in buffer C, and eluted at a constant flow rate of 1 ml min−1 as previously described (22). Following SDS-PAGE, fractions were pooled and concentrated by centrifugation at 4000 ×g (4°C) using an Amicon Ultra-15 10 kDa cutoff concentrator. Protein concentration was assessed using the Bradford method. Protein aliquots were frozen in liquid N2 and stored at −80°C.
Protein film electrochemistry
Experiments were performed anaerobically in an MBraun Labmaster glovebox using a PGSTAT 12 potentiostat (EcoChemie) using the tagged B. subtilis QueG protein. A three-electrode configuration was used with a standard calomel reference electrode, a platinum wire counter electrode and a pyrolytic graphite edge (PGE) working electrode in a water-jacketed glass cell. The electrochemical cell was thermostated using a circulating water bath and the reference electrode was maintained at room temperature in the course of the experiments. Potentials reported are relative to the standard hydrogen electrode.
Baseline measurements were collected using the PGE electrode polished with 1 μM alumina, rinsed and placed into the cell containing a 10°C mixed buffer solution composed of 10 mM MES, CHES, TAPS, HEPES, at pH 8.0 with 200 mM NaCl. A 3 μl sample of 340 μM protein was applied directly to the polished PGE electrode surface, the protein sample was removed after 3 min, and the electrode was immediately placed back into the cell solution. Non-turnover electrochemical signals were analyzed by correction of the non-Faradaic component of the current from the raw data using the SOAS package (33).
Crystallization
Crystallization of tagged and SeMET-labeled B. subtilis QueG was performed in an MBraun chamber under a nitrogen environment. Initial crystallization conditions were obtained using a Mosquito pipetting robot (TTP Factory) within this chamber. Optimized crystallization conditions were obtained in the same anaerobic chamber by using sitting drop vapor diffusion method at 21°C, mixing 0.5 μl protein (150 μM SeMET-labeled QueG [6.5 mg ml−1], 20 mM Tris (pH 8.0), 100 mM KCl, 10 mM DTT) with 1.5 μl of reservoir solution (0.1 M acetate (pH 4.5), 0.4 M (NH4)H2PO4, 12% (w/v) PEG 3350) and equilibrating against 600 μl of reservoir solution. Optimal SeMET-labeled QueG crystals of 100 × 100 × 50 μm3 dimensions grew within two to three weeks and were transferred to a 25°C polyvinyl anaerobic chamber (Coy Lab) under 95% Argon/5% hydrogen atmosphere. Crystals were harvested, cryoprotected in reservoir solution supplemented with 20% (v/v) glycerol, and cryocooled in liquid N2 within the anaerobic chamber.
Crystals that generated structures of QueG bound to a cleaved ACSL (see Supplementary Table S2) were obtained through co-crystallization experiments that employed 150 μM [6.5 mg ml−1] of wild-type QueG in 20 mM Tris (pH 8.0), 100 mM KCl and 10 mM DTT, and 375 μM of a construct of the E. coli tRNATyr ACSL (5′-G-C-A-G-A-C-U-G-U-A-A-A-U-C-U-G-C-3′) (IDT) (Supplementary Figure S3) and the same crystallization conditions detailed above. These crystals were transferred to the Coy Lab anaerobic chamber and either cryoprotected with 20% (v/v) glycerol or 20% (v/v) PEG 400 and flash cooled in liquid N2 within the chamber.
Crystals that generated a structure of QueG bound to an intact ACSL were obtained in the MBraun anaerobic chamber through co-crystallization of 150 μM wild-type QueG (purified from BL21(DE3)Δrna cells and stored in 20 mM Tris (pH 8.0)), 100 mM KCl, 10 mM DTT, and 375 μM of oQ-modified substrate (5′-G-C-A-G-A-C-U-oQ-U-A-A-A-U-C-U-G-C-3′) that was generated as previously described (19). In addition to the methods mentioned above for minimizing the amount of RNase contamination in the enzyme samples, murine RNase inhibitor (0.006 units) was added to the co-crystallization experiment as a preventative measure to preserve the RNA substrate. The QueG-ACSL complex crystallized under the same conditions as detailed above and was cryoprotected with 20% PEG 400 and flash cooled in liquid N2 within the Coy Lab anaerobic chamber.
Data collection, structure solution and refinement
SeMET-labeled QueG crystallized with two molecules per asymmetric unit (Vm of 2.6 Å3 Da-1 and solvent content of 53%). These crystals diffracted to 1.75 Å resolution (Supplementary Table S1). Anomalous peak, inflection and remote data sets of SeMET-labeled QueG were collected at 0.9793 Å, 0.9795 Å and 0.9421 Å wavelengths at 100 K to phase the structure. Data were collected at beamline 24ID-C at the Advanced Photon Source (Argonne National Labs, Argonne, IL, USA) using a Pilatus 6M detector and minikappa to align Friedel Mates (34) and processed using HKL2000 (35). Sixteen initial heavy atom sites, corresponding to eight selenomethionines per molecule, were determined using SHELXC and given to AutoSol in the PHENIX suite of programs (36,37). A solution was found in AutoSol using the peak, inflection and remote data sets with a figure-of-merit of 0.57 to 2.5 Å resolution. Experimental maps at 2.5 Å resolution enabled direct building of one protein molecule and nicely revealed the locations of all cofactors (Cbl and two [4Fe-4S] clusters). Pure translational non-crystallographic symmetry (NCS) was used to generate the second protein molecule within the asymmetric unit. Iterative rounds of refinement in PHENIX (37) and model building in COOT (38) were carried out. Refinement was conducted against the full resolution of data using NCS restraints (simulated annealing, positional and B-factor refinement). The cobalamin parameter file for refinement was provided by O. Smart at Global Phasing, and the parameter file for iron–sulfur clusters was generated using phenix.elbow (39) and the high resolution structures of the HydE protein (40). NCS restraints were gradually released during refinement, and waters, glycerols and phosphate ions were added in the later rounds of refinement. The final model refined without NCS restraints was verified by simulated annealing composite omit maps calculated in CNS (41), and the model's geometry was checked and validated using ProCheck and MolProbity (42,43). Analysis of the calculated Ramachandran Plot statistics from ProCheck held 93.2% of residues in the most favored regions, with 6.5% in favored and 0.3% in disallowed (corresponding to Asp134 of the active site). The refined protein model contains residues 2–385 and 2–376 for molecules A and B, respectively. Glu125 has poor density. Less than 811 Å2 interface surface area is calculated between the two molecules using the PISA server (44) compared to a total surface area of 16 970 Å2, which is consistent with QueG being a monomer.
Data for the 2.10 Å resolution structure of QueG with a cleaved ACSL (Supplementary Table S2) were collected at beamline 24ID-C at a wavelength of 0.9792 Å and temperature of 100 K. The structure was solved by molecular replacement (MR) in PHASER (45) using the model of 1.75 Å resolution SeMET-labeled QueG minus ligands and water molecules. The MR solution yielded a final LLG value of 21 243 for the 2.1 Å resolution structure with four QueG molecules per asymmetric unit. Refinement and model building were carried out similarly to that described above. RNA and phosphates were built into the electron density and waters were added in the later rounds of refinement. The final model (Supplementary Table S2) refined without NCS restraints was verified by simulated annealing composite omit maps calculated in CNS and PHENIX (37,41), and the model's geometry was checked and validated using ProCheck and MolProbity (42,43). As in the SeMET-labeled QueG structure, the refined protein model contains residues 2–381, 2–381, 2–378 and 2–377 for molecules A, B, C and D, respectively, with residue Glu125 disordered in all protein molecules. Additionally, the side chain of Arg86 exhibits no electron density in molecules A and B and was omitted from the structure. Electron density for RNA enabled building of RNA interacting with QueG monomers A (13 nucleotides: G27–U33 and A38–C43) and B (11 nucleotides: A29–U33 and A37–G42); QueG monomers C and D do not bind RNA and have glycerol bound within the enzyme active site. Phosphate ions are observed in identical positions with respect to the SeMET-labeled QueG structure. Ramachandran statistics from ProCheck are as follows: 92.6% most favored; 7.1% favored; and 0.3% allowed.
Data for the 2.65 Å resolution structure of QueG with a cleaved ACSL (Supplementary Table S2) were collected at beamline 24ID-C (Supplementary Table S2) at a wavelength of 0.9795 Å and temperature of 100 K. This structure was solved by molecular replacement using the software PHASER (45) and the 1.75 Å resolution SeMET-labeled QueG structure, minus ligands and water molecules, as the search model. Two molecules of QueG were located with a final LLG value of 3668. Rounds of simulated annealing, positional and B-factor refinement were performed in PHENIX (37) using NCS restraints and parameter files described above. Refinement rounds were conducted iteratively with model building in COOT (38). NCS restraints were gradually released, and RNA, phosphates and water molecules were added in the later rounds of refinement. The final model (Supplementary Table S2) refined without NCS restraints was verified by simulated annealing composite omit maps calculated in CNS and PHENIX (37,41), and the model's geometry was checked and validated using ProCheck and MolProbity (42,43). The refined protein model contains residues 2–375 for molecule A, and 2–132 and 136–376 for molecule B. Notably, the Asp134-containing loop displays weaker electron density in a ‘flipped-out’ conformation in molecule A, and this loop is disordered in molecule B and could not be built into the model. Electron density for RNA nucleotides interacting with monomer B enabled refinement of A29–U33, although only three phosphate groups for the complementary bases are observed and built into the structure. A phosphate ion was also observed within the active site of both protein molecules A and B near their respective cobalamins, and crystallographic packing phosphates are observed in identical positions as in the SeMET-labeled QueG structure. Ramachandran statistics from ProCheck are as follows: 91.0% most favored; 8.7% favored; 0.2% allowed; and 0.2% disallowed (Asp134 in the ‘flipped out’ conformation).
Data for the structure of QueG with an intact ACSL (Supplementary Table S2) were collected on two different crystals at the Stanford Synchrotron Radiation Lightsource, beamline 12-2, at a wavelength of 0.9795 Å and temperature of 100 K on a Pilatus 6M detector. Data from two crystals were then merged to create a 92.8% complete data set at 2.65 Å resolution. The structure was solved to 2.65 Å resolution by MR in PHASER (45) using the 1.75 Å resolution SeMET-labeled QueG structure minus ligands and water molecules as an initial search probe. An LLG value of 3795 with two QueG molecules in the asymmetric unit was obtained. Iterative rounds of refinement in PHENIX (37) were paired with model building in COOT (38) as described above. Also as above, the RNA and waters were added in the later stages of refinement. The parameter file for queuosine and epoxyquouosine were generated using SMILES notation in the Grade Web Server (http://grade.globalphasing.org). The final model was verified by simulated annealing composite omit maps calculated in CNS and PHENIX (37,41), and the model geometry was checked and validated using ProCheck and MolProbity (42,43). An intact ACSL binds to QueG in molecule A only, and glycerol binds within the active site of molecule B. Phosphate ions are observed that are identical to the SeMET-labeled QueG structure. Both protein molecules contain residues 2–377, except that Glu125 is completely disordered and omitted from molecule B. The full RNA ACSL (17 nucleotides: G27–C43) is built and refined in the model. Ramachandran statistics from ProCheck are as follows: 92.9% most favored; 6.8% favored; and 0.3% allowed.
In this structure of QueG with an intact ACSL, the electron density within the active site is best fit as the product, queuosine (Supplementary Figure S4A). In particular, there is no electron density for the epoxide oxygen in the omit 2mFo-DFc electron density map (Supplementary Figure S4B). The presence of Q is consistent with the fact that the QueG:oQ ACSL complex is fully loaded and ready for catalysis; requiring only electrons. Upon X-ray exposure during data collection, it is possible that QueG is able to convert the oQ substrate into the product, queuosine. The refined product complex contains Q at full occupancy, with B-factor values comparable to other residues and cofactors within the active site (Supplementary Table S2).
Composite omit maps and simulated annealing omit maps for figures were generated using PHENIX and CNS (37,41). Figures were created using PyMOL, electrostatics were calculated using the APBS plugin (46), and the topology diagram was generated using TopDraw (47).
RESULTS
Structure of B. subtilis QueG reveals a modular fold with a base-off Cbl and two [4Fe-4S] clusters
Structural data were obtained from recombinant B. subtilis QueG that was reconstituted with hydroxocob(III)alamin and two [4Fe-4S] clusters and shown to be active in end-point assays examining conversion of oQ into Q (19). The crystal structure was determined to 1.75 Å resolution using SeMET-labeled QueG and the multiwavelength anomalous dispersion method (Supplementary Table S1). As was observed recently for a QueG homolog from Streptococcus thermophilus (27), QueG contains three functional domains that bind Cbl, the [4Fe-4S] clusters and tRNA, respectively (Figure 2 and Supplementary Figure S5). The Cbl-binding domain at the N-terminus (Asn2 – Ser189) adopts an α−β−α sandwich fold with a central, four-stranded antiparallel β-sheet and a peripheral β-hairpin. A ferredoxin-like domain (Fd-domain) (Ser190 – Leu256) follows that binds the two [4Fe-4S] clusters. The RNA-binding domain (Leu257 – Leu385) is at the C-terminus and consists of an antiparallel six-helical bundle with a small helix (α8b) inserted after the first helix (α8a). This antiparallel helical bundle resembles a HEAT domain, a scaffold that typically serves to mediate protein–protein or protein–RNA/DNA interaction (48), and rests on two extended loops from the Cbl-binding (Cbl-insert; Tyr77 – Asp104) and Fd-domain (Fd-insert; Ile215 – Gly239) domains, respectively.
Figure 2.

Structure and electrochemistry of QueG from Bacillus subtilis. (A) Ribbon depiction of QueG with the N-terminal Cbl-binding domain colored with blue helices and purple β-strands, the Fd-domain in gray and the RNA-binding domain in red. Inserts into the protein sequence that are described in text are indicated with arrows. (B) Cofactor binding in QueG. Cbl, iron sulfur clusters and glycerol (from crystallization buffer) are shown in sticks, colored as follows: carbon, yellow; oxygen, red; nitrogen, blue; cobalt, pink sphere; iron, ruby; sulfur, yellow; phosphorus, orange. His106 and Arg141 are in blue sticks. Dashed magenta lines indicate hydrogen bonds, whereas gray dashed lines are used to indicate important distances between the cofactors. (C) Cyclic voltammogram of the two [4Fe-4S] clusters in QueG. QueG was deposited on a PGE electrode, with the baseline response of the electrode shown as the dashed line. The baseline-corrected data are shown as the inset, with the fitting for the two one-electron centers displayed in dotted lines. Data were collected at 100 mV/s, pH 8, 200 mM NaCl and 10°C.
Consistent with electron paramagnetic resonance (EPR) measurements (22), 5-coordinate Cbl is bound to QueG with water as the upper axial ligand (Supplementary Figure S5C). The Cbl-binding domain of QueG is structurally similar to CblC (Supplementary Figure S6) and to RDH enzymes, PceA from Sulfurospirillum multivorans (49) (Supplementary Figure S7) and RdhA from Nitratireductor pacificus pht-3B (50). In all cases, Cbl is bound in an extended mode with the dimethylbenzimidazole displaced as the lower ligand to the cobalt (Figure 2 and Supplementary Figure S5C). Instead of a histidine serving as the lower ligand, as is often the case in Cbl-binding proteins (51,52), the lower axial position of the cobalt is vacant. In QueG, access of water or other small ligands to the lower axial position appears to be blocked by Arg141, which is positioned 4 Å below the corrin ring with its guanidino group parallel to the corrin ring. In CblC, a glutamine occupies this position (Supplementary Figure S6), and in PceA, the Cbl tail itself is rotated further under the corrin ring blocking access (Supplementary Figure S7).
The Fd-domain of QueG is similar to the ferredoxin from Clostridium acidurici (Supplementary Figure S8) (53) and to the Fd-domains of RDH enzymes (Supplementary Figure S7). The most notable differences are the presence of inserts in QueG, one in the Cbl-binding domain and one insert in the Fd-domain, which appear to be responsible for creating a platform on which the RNA-binding domain can reside (Supplementary Figure S5–S8). The [4Fe-4S] clusters of QueG are coordinated by a cysteine-rich motif, ‘Cys188-X2-Cys191-X2-Cys194-X3-Cys198-Xn-Cys214-Xm-Cys240-X2-Cys243-X3-Cys247’ (19). Cys198, Cys214, Cys240 and Cys243 bind the cluster proximal to Cbl (termed cluster A), and Cys188, Cys191, Cys194 and Cys247 bind a distal cluster (termed cluster B). Both clusters are solvent accessible, and cluster A is only 4.0 Å from a propionamide side chain of the corrin ring and 8.7 Å to the cobalt center, whereas cluster B is positioned 9.5 Å from cluster A (Figure 2B). This positioning is conserved in RDH enzymes.
Protein film electrochemistry reveals that the QueG fold stabilizes low-potential iron–sulfur clusters
The positioning of the [4Fe-4S] clusters in the structure suggests that they provide a route for electrons from the protein surface to the Cbl, indicating a role in Cbl reduction. However, Cbl has notoriously low redox potentials for the Co2+/Co1+ couple, (approximately −530 to −610 mV) (54), raising the question of whether the cluster potentials are low enough to reduce the Cbl to the supernucleophile, cob(I)alamin, state. Here, we used protein film electrochemistry (PFE) to investigate the redox potentials of the QueG clusters. PFE of QueG on a PGE electrode displays a single, reversible feature in the cyclic voltammogram that is very broad with a peak width at half-height of ∼150 mV (Figure 2C). This feature is much broader than that expected for a single one-electron center (86 mV at 10°C), and is instead consistent with two one-electron centers, such as two [4Fe-4S] clusters, with reduction potentials of -575 mV and −510 mV (versus SHE) at pH 8. These potentials are lower than the potential of approximately −480 mV reported for the two [4Fe-4S] clusters in one RDH, Dehalobacter restrictus tetrachloroethene reductase; however, these data for an RDH were merely an estimate, as a complete titration was not obtained (25). The lower potentials determined here for B. subtilis QueG are closer to what one would expect for clusters that are capable of reducing Cbl.
tRNA anticodon stem loop binds QueG with the wobble base inserted deep into active site cavity
To investigate how QueG interacts with substrate, three structures of QueG bound to RNA have been obtained (Supplementary Table S2). In each case, a 17-mer tRNA ACSL construct was used that is a known substrate of QueG and displays the nucleotide sequence of E. coli tRNATyr (Figure 1B and Supplementary Figure S3) (19,22). Although the substrate specificity of QueG has not yet been assessed quantitatively, it is known to process oQ to Q in both this 17-mer ACSL construct and in bulk RNA samples from a QueG knockout strain (19).
A 2.1 Å resolution structure of QueG with RNA has four molecules in the asymmetric unit, two of which have electron density for RNA. However, in neither molecule is the ACSL intact (Figure 3A and B). These structural data confirmed our expectation that the C-terminal HEAT-like domain would serve to mediate enzyme:nucleotide interactions, as we find nucleotides of the ACSL (G-27 to U-33) nicely ordered against the HEAT-like domain, with the protein making direct hydrogen-bonds to the phosphates of the phosphodiester backbone (Figure 3A and B and Supplementary Figure S9A). The antisense strand (A-38 to C-43) is also present in the structure, with contacts between these nucleotides and protein involving crystal packing exclusively (Supplementary Figure S9B). The anticodon loop is completely disordered and is likely cleaved (see Materials and Methods). Another structure, which is at lower resolution (2.65 Å), also contains cleaved RNA. These two structures of QueG bound to a cleaved ACSL are very similar to one another, except for the fact that a loop near the Cbl has an altered position in the 2.65 Å resolution structure (Supplementary Figure S10A). Both are also very similar to the QueG structure without RNA (Supplementary Figure S10B).
Figure 3.
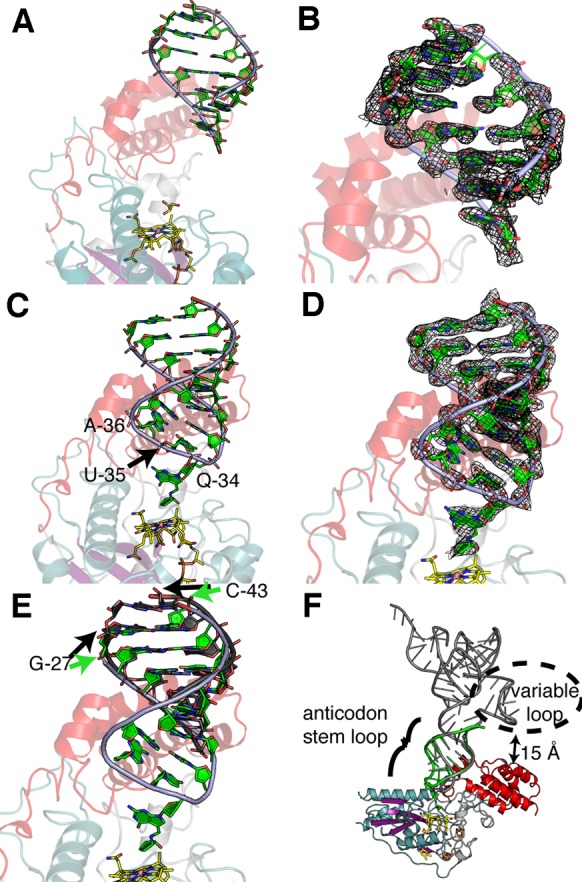
RNA binding to QueG. (A) A 2.10 Å resolution structure of QueG (colored as in Figure 2A) co-crystallized with an unmodified ACSL shows RNA (green backbone with green bases) bound to the C-terminal HEAT-like domain. RNA is apparently cleaved; Q-34 to A-37 of the ACSL are not observed. (B) SigmaA-weighted composite omit (2mFo-DFc) electron density for the RNA shown in panel A, displayed at 1σ level. (C) A 2.65 Å resolution structure of QueG bound to an intact ACSL. (D) SigmaA-weighted composite omit (2mFo-DFc) electron density for the RNA shown in panel C, displayed at 1σ level. (E) Bases of the intact ACSL (green) are shifted by one base pair toward the active site compared to structure of QueG with cleaved ACSL (dark gray). Black arrows point to bases in the cleaved RNA structure (from panel A) and green arrows point to bases from intact RNA structure (from panel C). (F) Predicted binding of full length tRNA, modeled by superposition of bacterial tRNATyr (PDB accession code 3UZ6), shown in gray, with the modified and intact ACSL (green) bound to QueG.
The third structure was obtained by co-crystallization of QueG with an oQ-modified ACSL under conditions that minimized RNase contamination (see Materials and Methods), and the ACSL is intact (Figure 3C and D). Again, little movement of the protein is observed, even compared to the structure without any RNA substrate bound (Supplementary Figure S10C). This result is in contrast with many other Cbl-dependent enzymes for which large-scale conformational changes upon substrate binding are a hallmark (51). In terms of RNA binding, this structure shows that the tRNA ACSL is sufficiently large to bury all of the highly positively charged surface on QueG (Supplementary Figure S9C), although modeling suggests that a full-length tRNATyr molecule could make additional contacts with smaller patches of positively charged surface (Supplementary Figure S9D). Comparison of partial and intact ACSL-bound QueG structures shows contacts to the same region of the QueG RNA-binding domain (Figure 3A–D). However, bases of the intact ACSL are shifted by one base pair toward the active site (from black arrow to green arrow position in Figure 3E). Interestingly, although the substrate, oQ-modified ACSL, was added to the co-crystallizations, electron density suggests turnover occurred, yielding a structure that is largely of the Q-containing product tRNA (Supplementary Figure S4A and B).
These structural data further show the interactions of the ACSL with QueG that position oQ/Q in the active site (Figure 4A and B, and Supplementary Table S3). Given that QueG acts on four tRNAs, with only positions oQ-34 and U-35 conserved, and A, U, G or C occupying position 36 (Supplementary Figure S3), we only expected to find specific interactions between QueG and the anticodon at positions 34 and 35, and that is exactly what we observe. In particular, the carbonyl at atom C4 of U-35 acts as a hydrogen bond acceptor with Arg295 and Lys222 from the QueG RNA-binding domain and the Fd-domain insertion sequence, respectively (Figure 4A and Supplementary Figure S11A and B). Cytosine at this position would be less favorable than uracil, with a hydrogen bond donating amino group replacing the hydrogen bond accepting carbonyl at C4, whereas purine bases would be too large to occupy this binding site (Supplementary Figure S11C). As expected, RNA base A-36 does not interact directly with the protein, explaining how all four bases can be accommodated at this position. Instead of contacting protein, A-36 appears to interact with U-35 of the ACSL by making a Hoogsteen hydrogen-bonding interaction from its exocyclic amine with the C2 carbonyl of U-35 (2.9 Å), and a non-hydrogen-bonding close contact (2.9 Å) between the N7 nitrogen of A-36 and the C2 carbonyl of U-35 (Figure 4A). Notably in a fully modified bacterial tRNATyr, A-37 would be modified with both a prenylation and methylthiolation to yield ms2i6A-37, and we find a glycerol molecule occupying this space left empty by the lack of these modifications (Supplementary Figure S11A).
Figure 4.

ACSL conformation and QueG interactions. (A) Close-up view of U-35 and A-36 of the QUN anticodon bound to QueG. Hydrogen-bonding and close van der Waals interactions are displayed as dashed cyan and black lines, respectively. (B) Zoom-in of Q binding within QueG with van der Waal radii displayed as spheres and protein residues colored by domain. Dashed lines are defined in panel A. (C) Stereoview of the non-canonical ACSL conformation observed in the QueG co-crystal structure. (D) Stereoview of the ACSL in classic ‘U-turn’ from a full tRNATyr molecule bound within the ribosome from Thermus thermophilus (PDB accession code 4V8D), which contains the ms2i6A-37 modification (represented with purple colored carbons). (E) Surface cut-away representation of QueG active site showing shape complementarity for the extended Q-base. (F) Two conformations of the Asp134-loop could facilitate the requisite ACSL rearrangements and the insertion of oQ into active site: an ‘open-loop’ conformation as observed in the 2.65 Å resolution QueG structure with a cleaved ACSL (carbons in green) and a ‘closed-loop’ conformation as observed in the intact ACSL QueG structure (carbons in yellow).
Q itself buries considerable surface area (240 Å2) as it extends into the QueG active site cavity (Figure 4B and E). The deazaguanine moiety of Q-34 is at the top of the active site, sandwiched between Trp294 of the RNA-binding domain and Thr135 of the Cbl-binding domain, and making a water-mediated hydrogen bond to Tyr238 of the Fd-domain (Figure 4B). The aminomethyl–cyclopentenediol moiety of Q transverses deeper into the active site and is rotated such that its amino group is 2.5 Å from the carbonyl of the deazaguanine base, allowing for the possibility of a favorable intramolecular hydrogen-bonding interaction (Figure 4B and Supplementary Figure S12C). The cyclopentenediol group is bound at the bottom of the active site, ∼4 Å from the Cbl cobalt. Although Q makes few hydrogen bonds to QueG, specificity for the oQ base versus an unmodified one can be explained by the depth of the active site cavity; an unmodified base would be too far from the Cbl for any reaction to occur (∼10 Å) and the differential in binding surface of the Q- versus G-base in this site (240 Å2 versus 124 Å2) would result in a weaker interaction of QueG with a tRNA containing an unmodified G in its anticodon (Figure 4E). When substrate is bound to QueG, both the upper axial water ligand to the Cbl and the crystallographic glycerol molecule are displaced (Supplementary Figure S12A–C). Refinement of the Q-base places the double bond of the cyclopentenediol 3.0 Å from Asp134, whereas one of the two cyclopentenediol hydroxyls is in hydrogen-bonding distance of Gln220 (2.9 Å) and the other is packed against Phe49 (Figure 4B). Assuming the substrate oQ binds similarly, Gln220 likely orients the ring structure for catalysis, whereas Asp134 would be in position to interact with the epoxy moiety, functioning perhaps as a catalytic acid (see Discussion).
QueG-bound ACSL has twists but no U-turns
The QueG bound-ACSL has undergone considerable remodeling and does not display the canonical ‘U-turn’ structure, i.e. the sharp reversal of the RNA backbone that is commonly associated with the positioning of the three bases of the anticodon for codon:anticodon base-pairing during protein synthesis (55) (Figure 4C and D). In a classic U-turn, as observed in the ribosome-bound tRNATyr structure (56), the anticodon bases G-34–U-35–A-36 are on one side of the turn, stacked with each other, and bases U-33, C-32 and A-31 are on the other side of the turn, also stacked. U-33 stabilizes this almost 180° reversal in direction by hydrogen bonding across the turn to a backbone phosphate. Similar to the tRNATyr-ribosome complex, our structure shows bases U-33, C-32 and A-31 stacked upon each other. However, all the anticodon bases (Q-34, A-36, U-35) and A-37 have altered positions and interactions, with the net result being that U-33 now stabilizes a broad rather than tight turn that positions Q-34 in the active site (Figure 4C and D). If we consider other enzymes that modify tRNA at position 34 for which there are co-crystal structures (57–60), we find that the ACSL is commonly splayed open rather than adopting the U-turn configuration (Supplementary Figure S13). Although most ACSLs have undergone a fairly large conformational change, none of the conformations resemble that found in QueG, and none truly resemble each other. It is impressive how many different loop conformations are possible, which is a feature of RNA structure that is likely essential for specificity of protein–RNA and RNA–RNA recognition.
The tight fit of the Q base into the deep active site cavity raises the question of how the modified base gets in there. The QueG active site cavity is designed to bind a broad anticodon turn with the Q base already splayed out and not the U-turn configuration of the ACSL (Figure 4). This active site design suggests that the ACSL conformational change should occur prior to QueG binding. However, chemical logic suggests that protein binding should facilitate the requisite ACSL conformational change, making a case for protein binding as the first step. Here, the various crystallographic snapshots provide insight. The 2.10 Å resolution structure of QueG with a cleaved ACSL shows that RNA can form non-specific interactions with QueG even in the absence of any specific anticodon loop-protein interactions, suggesting that the first step could be non-specific RNA–protein interactions made by protein residues from the HEAT domain that are outside of the active site. This non-specific binding of the ACSL to QueG would also position the anticodon loop near the enzyme active site (Figure 3). The 2.65 Å resolution structure of QueG with a cleaved ACSL shows that a conformationally flexible loop (the Asp134-loop) can flip out to generate an open active site (Figure 4F and Supplementary Figure S14A–C). Such an open active site could accommodate a U-turn configuration of the ACSL, which would need to undergo a conformational change prior to, or along with, the movement of the Asp134-loop back into the active site to assume the catalytic conformation of the protein that we observe in the structure of QueG with an intact ACSL. Importantly, the sequence of the anticodon would need to be correct for this active site closure to occur, both to satisfy the hydrogen bonding requirements described above and for the packing considerations described above and here. Thus, this set of structures predicts that non-specific interactions between the RNA and enzyme occur first, facilitating conformational changes of both the ACSL and one protein loop to afford a specific and tight complex between the enzyme active site and the oQ base. In this way, the splayed out base serves two purposes: ensuring specificity and also allowing for the appropriate juxtaposition of cofactor and substrate to afford catalysis.
DISCUSSION
Although the Q modification was discovered 47 years ago (7), its biosynthetic pathway was not fully established until recently (16–21), and though Cbl was proposed to be involved (26), the role that it played and the form of Cbl involved were not known. The structures and electrochemistry presented here provide new insight into the role of Cbl as well as the mechanism of oQ reduction.
We find that the ACSL binds QueG within a protein cavity located at the interface of the Cbl-binding, Fd- and RNA-binding domains, such that Q-34 is positioned directly above the Cbl. This juxtaposition of cofactor and product is consistent with a direct role of the cofactor in product formation. In particular, we propose (Figure 5) that the nucleophilic attack of cob(I)alamin on oQ leads to formation of a cob(III)alamin-substrate intermediate adduct, which dissociates upon reduction and protonation to form cob(II)alamin and the product Q. Such chemistry has precedence; cob(I)alamin is known to react with epoxides in solution, forming stable β-hydroxyalkylcobalamin adducts (61,62). Although the distance that we observe between the Q base and Cbl (4 Å) is appropriate for a product complex, we would expect the substrate oQ to be closer to the Cbl (∼3.5 Å) for the nucleophilic attack of cob(I)alamin and that covalent attachment would require an even closer distance (∼2.3 Å). Modeling shows that a small rotation around the substrate aminomethyl linker and translation of the nucleoside down toward the Cbl can generate these expected distances without any major change in the protein structure or loss of hydrogen bonding contacts to protein (Supplementary Figure S15). Thus, a covalent mechanism appears consistent with the active site architecture.
Figure 5.

A proposed mechanism. Based on QueG structures as well as mutagenesis data (22), Asp134 is predicted to serve as the general acid catalyst.
Our structural work has captured both the initial state of the enzyme without substrate and the final state with product Q bound (Figure 5). From EPR, we know that substrate-free QueG is in the cob(II)alamin state, with g- and A-values supporting a 5-coordinate Cbl in which the fifth coordination position is occupied by a water molecule (22). Our structure shows 5-coordinate Cbl with water as the upper axial ligand and no lower ligand. For nucleophilic attack on an epoxide, Cbl would need to be reduced by one electron to the +1 state (Figure 5), and the structure shows two [4Fe-4S] clusters positioned ideally for electron transfer to the Cbl at 8–9 Å apart. Although reduction of 5-coordinate Cbl is challenging for biological reductants due to cob(I)alamin's low midpoint potential (approximately −530 to −610 mV) (54), PFE presented here shows that these clusters are also low in potential (−575 mV and −510 mV) and thus well suited for this function. Arg141 is observed to block access to the lower face Cbl, which may preorganize the cofactor for reduction to a square-planar cob(I)alamin species, the preferred geometry for cob(I)alamin (54). Expression of an Arg141Ala QueG variant in an E. coli ΔqueG knockout strain significantly reduces the Q-content of the strain compared to wild-type, suggesting that limiting access to the lower face of Cbl is important for QueG activity (22). The upper water ligand observed in the initial structure would either dissociate upon substrate binding or be released upon reduction of the Cbl. A number of solvent binding sites are available near the cofactor that could provide a route to or from the Cbl cobalt (Supplementary Figure S12). Alternatively, movement of the Asp134-loop, as observed in the 2.65 Å resolution structure of QueG with a cleaved ACSL, could be responsible for releasing water molecules as substrate binds.
A potential caveat with this mechanistic proposal (Figure 5) is that β-hydroxyalkylCbls, such as the one proposed to form in this scheme, tend to be relatively stable. However, model chemistry indicates that protonation can decrease the stability of such adducts and facilitate Co-C bond cleavage and product release. Most relevant to QueG, β-hydroxy-substituted Cbl model systems are known to undergo acid-catalyzed Co-C bond cleavage (63). For QueG, protonation of the epoxide oxygen, either before or after ring opening, would be expected to facilitate Co-C bond heterolysis, releasing the product. Our structure shows that Asp134 is ideally located to protonate the epoxide oxygen. Consistent with such a function, an Asp134Ala variant in a ΔqueG knockout strain dramatically decreases Q production (22). Additionally or alternatively to acid catalyzed Co-C cleavage of an alkylCbl intermediate, a 1-electron reduction of alkylCbl is known to destabilize the C-Co bond in solution (54).
Overall, the mechanistic proposal in Figure 5 requires two protons and two electrons to reduce the epoxide oxygen to water. As mentioned above, Asp134 is positioned such that it could carry out both the protonation of the epoxide oxygen, facilitating the ring opening and the protonation of the resulting hydroxide, facilitating adduct release from Cbl and loss of water. Importantly, Asp134 hydrogen bonds to His106, which is near the surface of the protein, and could provide a conduit for protons into a closed active site. As with Asp134, mutation of His106 to Ala substantially affects Q production in a ΔqueG knockout strain, indicating that it plays an important catalytic role (22). Given that a neutral, rather than negatively charged, Asp134 should make it easier to reduce the Cbl, protonation of Asp134 and electron transfer to Cbl may be correlated, if not specifically coupled. As mentioned above, the vehicle for electron delivery to Cbl is immediately obvious from the structure, which shows two [4Fe-4S] clusters adjacent to Cbl. The importance of these clusters to QueG activity was confirmed through mutagenesis of the Cys coordinating ligands to Ala, all but one of which results in complete loss of Q production in vivo (22).
Data from these and other (27,49,50) structural and biochemical studies of QueG and RDHs are serving to expand the known reactivity of the Cbl cofactor and of Cbl-dependent enzymes. They also underscore the importance and prevalence of a previously under-appreciated third class of Cbl-dependent enzymes, ones that do not use MeCbl or AdoCbl in their chemistry (29). In terms of defining this class, it does appear that the lack of an upper ligand is functionally important to allow for a Cbl with a free upper position to form covalent adducts with substrate, as we propose here, and/or facilitate product release as has been suggested for some members of the RDH family (49,50). Thus, the relevant forms of Cbl are: MeCbl, AdoCbl, and open-Cbl. Cbl, often referred to as Nature's most beautiful cofactor (64), continues to offer up more incredible chemistry as we continue to discover its secrets.
Supplementary Material
Acknowledgments
The authors would like to thank Prof. Uttam L. RajBhandary for assistance in preparing the tRNA for crystallization.
Footnotes
Present addresses:
Daniel P. Dowling, Department of Chemistry, University of Massachusetts Boston, Boston, MA 02125, USA.
Zachary D. Miles, Scripps Institution of Oceanography, University of California-San Diego, La Jolla, CA 92037, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U.S. National Institutes of Health [GM72623 to V.B. with Administrative Supplement GM72623 S01 to V.B. for the collaboration between V.B. and C.L.D., GM120283 to S.J.E., and GM17151 to Prof. Uttam L. RajBhandary supported C.K.]; Career Award in Biomedical Sciences from the Burroughs Wellcome Fund [to V.B.]; National Science Foundation [MCB 1122977 to S.J.E.]; C.L.D. is a Howard Hughes Medical Institute Investigator; this work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health [P41 GM103403]; The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant [S10 RR029205]; These beamlines are located at the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357; Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515; DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences [to The SSRL Structural Molecular Biology Program including P41GM103393]; The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Funding for open access charge: HHMI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Grosjean H. Nucleic acids are not boring long polymers of only four types of nucleotides: A guided tour. In: Grosjean H, editor. Enzyme-RNA substrate recognition in RNA-modifying enzymes. Austin: Landes Bioscience; 2009. pp. 1–18. [Google Scholar]
- 2.Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A.P., Fabris D., Agris P.F. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006;34:721–733. doi: 10.1093/nar/gkj471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackman J.E., Alfonzo J.D. Transfer RNA modifications: Nature's combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA. 2013;4:35–48. doi: 10.1002/wrna.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasai H., Nishimura S., Oppenheimer N.J., Liehr J.G., Mccloskey J.A. Structure of the modified nucleoside Q isolated from Escherichia coli transfer ribonucleic acid. 7-(4, 5-cis-Dihydroxy-1-cyclopenten-3-ylaminomethyl)-7-deazaguanosine. Biochemistry. 1975;14:4198–4208. doi: 10.1021/bi00690a008. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama S., Miyazawa T., Iitaka Y. Three-dimensional structure of hyper-modified nucleoside Q located in the wobbling position of tRNA. Nature. 1979;282:107–109. doi: 10.1038/282107a0. [DOI] [PubMed] [Google Scholar]
- 7.RajBhandary U.L., Chang S.H., Harada F., Kimura F., Nishimura S. E. coli tyrosine transfer RNA-primary sequence and direct evidence for base pairing between terminal sequences. Fed. Proc. Fed. Am. Soc. Exp. Biol. 1969;28:409. [Google Scholar]
- 8.Harada F., Nishimura S. Possible anticodon sequences of tRNAHis, tRNAAsn, and tRNAAsp from Escherichia coli B. Universal presence of nucleoside Q in the first position of the anticondons of these transfer ribonucleic acids. Biochemistry. 1972;11:301–308. doi: 10.1021/bi00752a024. [DOI] [PubMed] [Google Scholar]
- 9.Goodman H.M., Abelson J., Landy A., Brenner S., Smith J.D. Amber suppression: a nucleotide change in the anticodon of a tyrosine transfer RNA. Nature. 1968;217:1019–1024. doi: 10.1038/2171019a0. [DOI] [PubMed] [Google Scholar]
- 10.Katze J.R., Basile B., McCloskey J.A. Queuine, a modified base incorporated posttranscriptionally into eukaryotic transfer RNA: Wide distribution in nature. Science. 1982;216:55–56. doi: 10.1126/science.7063869. [DOI] [PubMed] [Google Scholar]
- 11.Okada N., Shindo-Okada N., Nishimura S. Isolation of mammalian tRNAAsp and tRNATyr by lectin-sepharose affinity column chromatography. Nucleic Acids Res. 1977;4:415–423. doi: 10.1093/nar/4.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada N., Nishimura S. Enzymatic synthesis of Q* nucleoside containing mannose in the anticodon of tRNA: isolation of a novel mannosyltransferase from a cell-free extract of rat liver. Nucleic Acids Res. 1977;4:2931–2937. doi: 10.1093/nar/4.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregson J.M., Crain P.F., Edmonds C.G., Gupta R., Hashizume T., Phillipson D.W., McCloskey J.A. Structure of the archaeal transfer RNA nucleoside G*-15 (2-amino-4, 7-dihydro- 4-oxo-7-β-D-ribofuranosyl-1H-pyrrolo[2, 3-d]pyrimidine-5-carboximidamide (archaeosine)) J. Biol. Chem. 1993;268:10076–10086. [PubMed] [Google Scholar]
- 14.Vinayak M., Pathak C. Queuosine modification of tRNA: Its divergent role in cellular machinery. Biosci. Rep. 2010;30:135–148. doi: 10.1042/BSR20090057. [DOI] [PubMed] [Google Scholar]
- 15.Noguchi S., Nishimura Y., Hirota Y., Nishimura S. Isolation and characterization of an Escherichia coli mutant lacking tRNA-guanine transglycosylase. J. Biol. Chem. 1982;257:6544–6550. [PubMed] [Google Scholar]
- 16.McCarty R.M., Bandarian V. Biosynthesis of pyrrolopyrimidines. Bioorg. Chem. 2012;43:15–25. doi: 10.1016/j.bioorg.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slany R.K., Bösl M., Crain P.F., Kersten H. A new function of S-adenosylmethionine: the ribosyl moiety of AdoMet is the precursor of the cyclopentenediol moiety of the tRNA wobble base queuine. Biochemistry. 1993;32:7811–7817. doi: 10.1021/bi00081a028. [DOI] [PubMed] [Google Scholar]
- 18.McCarty R.M., Somogyi A., Lin G., Jacobsen N.E., Bandarian V. The deazapurine biosynthetic pathway revealed: In vitro enzymatic synthesis of PreQ0 from guanosine 5′-triphosphate in four steps. Biochemistry. 2009;48:3847–3852. doi: 10.1021/bi900400e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miles Z.D., McCarty R.M., Molnar G., Bandarian V. Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7368–7372. doi: 10.1073/pnas.1018636108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarty R.M., Bandarian V. Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem. Biol. 2008;15:790–798. doi: 10.1016/j.chembiol.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miles Z.D., Roberts S.A., McCarty R.M., Bandarian V. Biochemical and structural studies of 6-carboxy-5, 6, 7, 8-tetrahydropterin synthase reveal the molecular basis of catalytic promiscuity within the tunnel fold superfamily. J. Biol. Chem. 2014;289:23641–23652. doi: 10.1074/jbc.M114.555680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miles Z.D., Myers W.K., Kincannon W.M., Britt R.D., Bandarian V. Biochemical and spectroscopic studies of epoxyqueuosine reductase: a novel iron–sulfur cluster- and cobalamin-containing protein involved in the biosynthesis of queuosine. Biochemistry. 2015;54:4927–4935. doi: 10.1021/acs.biochem.5b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koutmos M., Gherasim C., Smith J.L., Banerjee R. Structural basis of multifunctionality in a vitamin B12-processing enzyme. J. Biol. Chem. 2011;286:29780–29787. doi: 10.1074/jbc.M111.261370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Froese D.S., Krojer T., Wu X., Shrestha R., Kiyani W., von Delft F., Gravel R.A., Oppermann U., Yue W.W. Structure of MMACHC reveals an arginine-rich pocket and a domain-swapped dimer for its B12 processing function. Biochemistry. 2012;51:5083–5090. doi: 10.1021/bi300150y. [DOI] [PubMed] [Google Scholar]
- 25.Schumacher W., Holliger C., Zehnder A.J.B., Hagen W.R. Redox chemistry of cobalamin and iron-sulfur cofactors in the tetrachloroethene reductase of Dehalobacter restrictus. FEBS Lett. 1997;409:421–425. doi: 10.1016/s0014-5793(97)00520-6. [DOI] [PubMed] [Google Scholar]
- 26.Frey B., McCloskey J., Kersten W., Kersten H. New function of vitamin B12: Cobamide-dependent reduction of epoxyqueuosine to queuosine in tRNAs of Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1988;170:2078–2082. doi: 10.1128/jb.170.5.2078-2082.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Payne K.A., Fisher K., Sjuts H., Dunstan M.S., Bellina B., Johannissen L., Barran P., Hay S., Rigby S.E.J., Leys D. Epoxyqueuosine reductase structure suggests a mechanism for cobalamin-dependent tRNA modification. J. Biol. Chem. 2015;290:27572–27581. doi: 10.1074/jbc.M115.685693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holliger C., Hahn D., Harmsen H., Ludwig W., Schumacher W., Tindall B., Vazquez F., Weiss N., Zehnder A.J.B. Dehalobacter restrictus gen. nov. and sp. nov., a strictly anaerobic bacterium that reductively dechlorinates tetra-and trichloroethene in an anaerobic respiration. Arch. Microbiol. 1998;169:313–321. doi: 10.1007/s002030050577. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee R., Ragsdale S.W. The many faces of vitamin B12: Catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 30.Kurylo C.M., Alexander N., Dass R.A., Parks M.M., Altman R.A., Vincent C.T., Mason C.E., Blanchard S.C. Genome sequence and analysis of Escherichia coli MRE600, a colicinogenic, nonmotile strain that lacks RNase I and the type I methyltransferase, EcoKI. Genome Biol. Evol. 2016;8:742–752. doi: 10.1093/gbe/evw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacob A.I., Köhrer C., Davies B.W., RajBhandary U.L., Walker G.C. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell. 2013;49:427–438. doi: 10.1016/j.molcel.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 33.Fourmond V., Hoke K., Heering H.A., Baffert C., Leroux F., Bertrand P., Léger C. SOAS: A free program to analyze electrochemical data and other one-dimensional signals. Bioelectrochem. 2009;76:141–147. doi: 10.1016/j.bioelechem.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Brockhauser S., Ravelli R.B.G., McCarthy A.A. The use of a mini-κ goniometer head in macromolecular crystallography diffraction experiments. Acta Crystallogr. D. Biol. Crystallogr. 2013;69:1241–1251. doi: 10.1107/S0907444913003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Terwilliger T.C., Adams P.D., Read R.J., McCoy A.J., Moriarty N.W., Grosse-Kunstleve R.W., Afonine P. V, Zwart P.H., Hung L.W. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D. Biol. Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams P.D., Afonine P. V, Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P., Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Moriarty N.W., Grosse-Kunstleve R.W., Adams P.D. Electronic ligand builder and optimization workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicolet Y., Amara P., Mouesca J.-M., Fontecilla-Camps J.C. Unexpected electron transfer mechanism upon AdoMet cleavage in radical SAM proteins. Proc. Natl. Acad. Sci. U.S.A. 2009;106:14867–14871. doi: 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brunger A.T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 43.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 45.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bond C.S. TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
- 48.Andrade M.A., Petosa C., O'Donoghue S.I., Müller C.W., Bork P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- 49.Bommer M., Kunze C., Fesseler J., Schubert T., Diekert G., Dobbek H. Structural basis for organohalide respiration. Science. 2014;86:3698–3716. doi: 10.1126/science.1258118. [DOI] [PubMed] [Google Scholar]
- 50.Payne K.A.P., Quezada C.P., Fisher K., Dunstan M.S., Collins F.A., Sjuts H., Levy C., Hay S., Rigby S.E.J., Leys D. Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature. 2015;517:513–516. doi: 10.1038/nature13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dowling D.P., Croft A.K., Drennan C.L. Radical use of Rossmann and TIM barrel architectures for controlling coenzyme B12 chemistry. Annu. Rev. Biophys. 2012;41:403–427. doi: 10.1146/annurev-biophys-050511-102225. [DOI] [PubMed] [Google Scholar]
- 52.Drennan C.L., Huang S., Drummond J.T., Matthews R.G., Ludwig M.L. How a protein binds B12: a 3.0 Å X-ray structure of B12-binding domains of methionine synthase. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 53.Duée E.D., Fanchon E., Vicat J., Sieker L.C., Meyer J., Moulis J.M. Refined crystal structure of the 2[4Fe-4S] ferredoxin from Clostridium acidurici at 1.84 Å resolution. J. Mol. Biol. 1994;243:683–695. doi: 10.1016/0022-2836(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 54.Lexa D., Saveant J.M. The electrochemistry of vitamin B12. Acc. Chem. Res. 1983;16:235–243. [Google Scholar]
- 55.Cate J.H., Yusupov M.M., Yusupova G.Z., Earnest T.N., Noller H.F. X-ray crystal structures of 70S ribosome functional complexes. Science. 1999;285:2095–2104. doi: 10.1126/science.285.5436.2095. [DOI] [PubMed] [Google Scholar]
- 56.Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–259. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- 57.Losey H.C., Ruthenburg A.J., Verdine G.L. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 2006;13:153–159. doi: 10.1038/nsmb1047. [DOI] [PubMed] [Google Scholar]
- 58.Numata T., Ikeuchi Y., Fukai S., Suzuki T., Nureki O. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature. 2006;442:419–424. doi: 10.1038/nature04896. [DOI] [PubMed] [Google Scholar]
- 59.Xie W., Liu X., Huang R.H. Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nat. Struct. Biol. 2003;10:781–788. doi: 10.1038/nsb976. [DOI] [PubMed] [Google Scholar]
- 60.Nakanishi K., Bonnefond L., Kimura S., Suzuki T., Ishitani R., Nureki O. Structural basis for translational fidelity ensured by transfer RNA lysidine synthetase. Nature. 2009;461:1144–1148. doi: 10.1038/nature08474. [DOI] [PubMed] [Google Scholar]
- 61.Haglund J., Silvari V., Esmans E., Törnqvist M. Cobalamin as an analytical tool for analysis of oxirane metabolites of 1, 3-butadiene: development and validation of the method. J. Chromatogr. A. 2006;1119:246–250. doi: 10.1016/j.chroma.2005.11.069. [DOI] [PubMed] [Google Scholar]
- 62.Motwani H. V., Fred C., Haglund J., Golding B.T., Törnqvist M. Cob(I)alamin for trapping butadiene epoxides in metabolism with rat S9 and for determining associated kinetic parameters. Chem. Res. Toxicol. 2009;22:1509–1516. doi: 10.1021/tx900088w. [DOI] [PubMed] [Google Scholar]
- 63.Schrauzer G.N., Windgassen R.J. On hydroxyalkyl-cobaloximes and the mechanism of a cobamide-dependent diol dehydrase. J. Am. Chem. Soc. 1967;89:143–147. [Google Scholar]
- 64.Stubbe J. Binding site revealed of nature's most beautiful cofactor. Science. 1994;266:1663–1664. doi: 10.1126/science.7992049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.