

Abstract
Background
Graft-versus-host disease (GVHD) is a severe complication of hematopoietic stem cell transplantation. Current therapies to prevent alloreactive T cell activation largely cause generalized immunosuppression and may result in adverse drug, anti-leukemia and anti-pathogen responses. Recently, several immunomodulatory therapeutics have been developed that show efficacy in maintaining anti-leukemia responses while inhibiting GVHD in murine models. To analyze efficacy and better understand immunological tolerance, escape mechanisms, and side-effects of clinical reagents, testing of species-cross-reactive human agents in large animal GVHD models is critical.
Methods
We have previously developed and refined a NHP large animal GVHD model. However, this model is not readily amenable to semi-high throughput screening of candidate clinical reagents.
Results
Here, we report a novel, optimized NHP xenogeneic GVHD (xeno-GVHD) small animal model that recapitulates many aspects of NHP and human GVHD. This model was validated using a clinically available blocking, monovalent anti-CD28 antibody (FR104) whose effects in a human xeno-GVHD rodent model are known.
Conclusions
Since human-reactive reagents may not be fully cross-reactive or effective in vivo on NHP immune cells, this NHP xeno-GVHD model provides immunological insights and direct testing on NHP-induced GVHD prior to committing to the intensive NHP studies that are being increasingly used for detailed evaluation of new immune therapeutic strategies prior to human trials.
Introduction
Hematopoietic stem cell transplantation (HSCT) is a life-saving therapy that is limited by graft-versus-host disease (GVHD). Acute GVHD occurs in as many as 70% of transplant recipients, leading to high rates of post-HSCT morbidity and mortality1,2. Importantly, while current drug- (eg calcineurin inhibitors; methotrexate) and broadly-reactive antibody- (eg antithymocyte globulin) based immunosuppressive therapies dampen acute GVHD, they also may inhibit necessary immune functions (eg tumor killing and pathogen clearance) and can be antithetical to immune tolerance induction3,4.
New immunomodulatory therapies are being developed that show great promise in murine GVHD models 2,5. However, given that significant differences exist between murine and human immune systems, especially in regards to thymic output, CD4/8 ratio and peripheral memory T cell frequency, mouse experiments alone may not be sufficient to bridge the gap between preclinical experiments and clinical translation 6,7. In the fields of both solid organ 8-11 and now, HSCT 12-15, nonhuman primate (NHP) models have been critical to the translation of novel targeted therapies to the clinic. These models provide a critical platform for comprehensive analysis of the peripheral blood (PB) as well as GVHD target tissues to assess toxicity, efficacy, and gain unique biological insights into the immunobiology of novel interventions. However, NHP studies can present their own challenges, given the unknown in vivo potency of clinical reagents that have biological effects on NHP cells in vitro, the intensive management required for in vivo transplant experiments in NHP, and the high costs of animals and their supportive care. Moreover, because significant quantities of reagents are required to complete in vivo studies, this often incurs a high cost and/or complex materials transfer agreements with biopharma companies, especially for combinatorial strategies that may prove essential for achieving optimal efficacy, adding to the challenge of NHP studies. Furthermore, subsequent FDA requests for large animal testing prior to approving clinical studies may be better justified if there is evidence for in vivo efficacy in small animal models using the immune cells to be targeted in such large animal models. Conversely, requirements for large animal studies may not be fully justified or useful if testing in small animal models is found to be ineffective.
Given these issues, for immunomodulatory strategies that have not previously been tested in NHP models, an experimental pipeline that would allow preevaluation of their in vivo impact on NHP T cell biology prior to full-scale NHP allo-transplantation would represent a major advantage in the field. For this reason, we sought to develop such an approach. We first created and then optimized a novel NHP xeno-GVHD model amenable to a higher throughput screening rate and subsequent selection of antibodies, proteins, small molecule, drug and cell-based therapies that are most worthwhile to pursue in large animal NHP models. Guided by murine allo-transplantation models and human biomarkers that can predict desirable targets, we can now proceed with in vivo testing of human reagents progressing from small animal human xeno-GVHD to NHP xeno-GVHD to large animal NHP allo-transplantation to human clinical trials. This NHP xeno-GVHD model thus provides a novel platform for relatively high-throughput testing of immunomodulatory therapeutics for potential efficacy prior to commitment to full-scale in vivo NHP studies.
Materials and Methods
Animals
Female NOD/SCID/γc−/− (NSG) mice were purchased from The Jackson Laboratory (Bar Harbor, ME), and housed in a specific pathogen-free facility in micro-isolator cages. Mice were used at 8-12 weeks. Mouse protocols were approved by IACUC at the University of Minnesota. This study used specific pathogen-free, juvenile rhesus macaques (4.6 to 10.4 years of age) that were housed at the Yerkes National Primate Research Center and the Washington National Primate Research Center. All animals were treated in accordance with IACUC regulations.
NHP Peripheral blood mononuclear cell procurement and shipping
Peripheral blood mononuclear cell (PBMNC) were isolated from NHP leukapheresis products as previously described 16, but without G-CSF mobilization. In initial studies, cells were shipped overnight (at RT), and PBMNC isolated by Ficoll. In most experiments, cells were aliquoted at 50 ×106/ml in freezing medium (10% DMSO, 45%human AB serum, 45% complete media) and frozen in a rate-controlled freezer.
NHP small animal model of xenogeneic GVHD
NSG mice, irradiated with 50 or 200 cGray total body irradiation or left as unirradiated, were injected intravenously (IV) with NHP (Rhesus Macaque) peripheral blood mononuclear cells (PBMNC) at doses ranging from 20-30×106 cells. Mice were assessed for survival daily and weighed and clinically scored 17 3 times per week. Peripheral T cell expansion was assessed by flow cytometry as in 18, with mAbs to human CD4, CD8, CD45 that cross-react with NHP. FR104 (Effimune Inc.) is a monovalent humanized Fab’ antibody fragment antagonist of CD28 that was pegylated to prolong its half-life 19. FR104 or irrelevant IgG (Fab; Jackson Immunoresearch 015-000-007) was administered IP (100μg/mouse) on day 0 prior to NHP PBMNC injection, and 3 times weekly for 4 weeks.
Flow cytometry and Antibodies
NHP-reactive antibodies against CD3 (AF700; 557917), CD4 (APC-CY7, 341105), CD8α (BV510; 560774), IFNγ (V450; 560371) were purchased from BD Pharmingen and IL-17 (PE; 12-7178-41) was from eBioscience; and Granzyme B (GzmB) (APC; MHGB05) was from Invitrogen.
To assess NHP T cell expansion and differentiation in the periphery, mice were bled on the indicated days, red blood cells were removed by ACK lysis. Samples were then stained with fluorochrome-conjugated antibodies to NHP T cell markers and a known number of counting beads added prior to flow analysis. For intracellular cytokine staining, PB or splenocytes were ACK lysed and stimulated for 4 hours ± PMA (2 pg/ml) and Ionomycin (1 μg/ml) in the presence of Brefeldin A (100 ng/ml) (all from Sigma). Cells were then stained for CD4 and cytokine (IL-17, and IFNγ) or GzmB using a standard intracellular staining kit (BioLegend). Acquisition was performed using an LSRII (BD Bioscience) and data were analyzed using FlowJo software (Tree Star Inc.).
Pathology data
Histopathology was performed on tissues (liver, lung, small intestine, colon, skin, spleen, and bone marrow) from mice at the time of sacrifice to evaluate GVHD by 1 of the co-authors (APM), blinded with regard to experimental design and cohort grouping. Cryosections (6 μm) were acetone-fixed and stained by hematoxylin and eosin. Coded tissues were assessed for GVHD using a 0 to 4+ scale 20.
Statistical analysis
Survival data were analyzed by Mantel-Cox test (Prism 5). Other data were analyzed by ANOVA or paired Student's t-test. Correlation was determined by Pearson correlation coefficient (Prism 5). Probability (P) values ≤0.05 were considered statistically significant.
Supplemental Information
Information on the animals used for this study, the collection of PBMNC from NHP, and on flow cytometry can be found in Supplemental Information.
Results
Establishing a new and exportable NHP small animal mouse model of xenogeneic GVHD
The primary variables contributing to GVHD severity in the human→murine xenogeneic GVHD model are cell dose and exposure to radiation 21,22, with increasing doses of either of these variables speeding the tempo and increasing the severity of GVHD, as also discussed below for the NHP→murine model. To determine whether an analogous model could be designed and operationalized for NHP, PBMNC were isolated from rhesus macaques, immune cells phenotyped (Table 1) and injected into nonirradiated or sub-lethally irradiated NSG immune deficient mice, as indicated. Rhesus apheresis units consisted of mean values of 33% T cells (CD3+CD20−; range 27 to 33%), 6% B cells (CD3−CD20+; range 4-8%), 10% NK cells (CD3−CD16+; range 6-12%) and 31% monocyte/macrophages (CD14+; range 16-47%), which is in agreement with previously reported frequencies (Table 1)23. After Ficoll, 22% of PBMNC were CD4+ (range 12 to 28%), 25% were CD8+ (range 18 to 29%), and 3% were CD4+8+ (range 2 to 4%), which is also consistent with age-matched animals (Table 2)23-25. Disease severity was monitored by weight loss, clinical GVHD score and survival.
Table 1. Cell types present in NHP apheresis products.
Leukocyte composition of apheresis products was determined by flow cytometry.
| Apheresis product | T cells | B cells | NK cells | Mø |
|---|---|---|---|---|
| %CD3+ | %CD20+ | %CD16+ | %CD14+ | |
| 1 | 32.8 | 8.1 | 6.0 | 15.7 |
| 2 | 39.6 | 5.4 | 11.7 | 29.0 |
| 3 | 26.6 | 4.2 | 11.8 | 46.7 |
| 4-5 | NA | NA | NA | NA |
| Avg. | 33.0 | 5.9 | 9.8 | 30.5 |
Table 2. T Cell subsets in NHP PBMNC.
Post thaw/wash, NHP PBMNC were phenotyped to determine subset composition.
| Aph. prod. | CD4+ | CD8+ | CD4+8+ | |||
|---|---|---|---|---|---|---|
| (Figure) | %Total | %T cells | %Total | %T cells | %Total | %T cells |
| 2 (1) | 27.0 | 46.1 | 27.4 | 46.8 | 4.2 | 7.2 |
| 2 (2) | 27.7 | 47.3 | 26.8 | 45.7 | 4.1 | 7.0 |
| 3 (3) | 12.4 | 40.1 | 18.4 | 53.0 | 2.2 | 7.0 |
| 4 (4-5) | 20.3 | 40.2 | 28.7 | 56.8 | 1.5 | 3.0 |
| Avg. ± SEM | 22 ± 4 | 43 ± 2 | 25 ±3 | 51 ±3 | 3 ± 1 | 6 ±1 |
To increase the logistical ease of the NHP xeno-GVHD model, we tested whether a frozen/thawed product could successfully induce xeno-GVHD in irradiated NSG recipients. Importantly, frozen/thawed PBMNC were as effective at inducing GVHD manifestations and lethality as fresh PBMNC, allowing this model to be more readily exported to other laboratories that may not have ready access to freshly apheresed NHP PBMNC (Figures 1A-C). Since results were similar with fresh vs. frozen/thawed NHP PBMNC, all subsequent experiments were performed with frozen/thawed cells.
Figure 1. Establishing a murine/NHP xenogeneic model of GVHD.
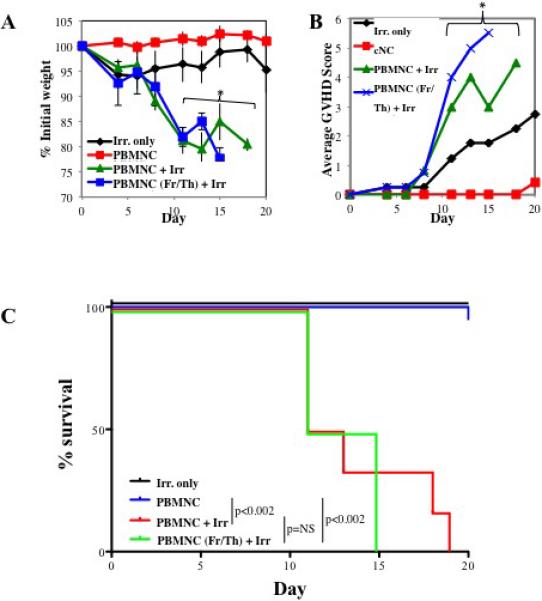
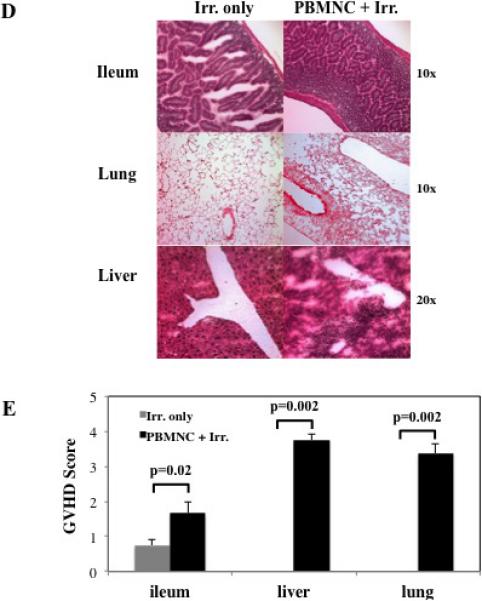
To determine whether NHP PBMNC could mediate a xeno-GVHD response, PBMNC were prepared from NHP leukapheresis products and were injected (30×106 cells) into NOD/Scid/γc−/− (NSG) ± 200 cGray total body irradiation. Control NSG mice receiving irradiation only were also monitored. Disease progression was monitored by weight loss (A), GVHD score (B) and survival (C). Representative example (D) and summary (E) of xenoGVHD histology of Ileum, liver and lung from irradiated animals with or without NHP PBMNC. (D and E). n=6 mice per group. * denotes days when average weight or clinical score for both fresh and frozen/thawed PBMNC + Irr. Resulted in a p<0.05 compared to Irr. or PBMNC only.
Although 30×106 PBMNC induced disease without prior recipient irradiation in the human xeno-GVHD model18,26, transfer of 30×106 NHP PBMNC alone in the absence of recipient irradiation resulted in no or minimal weight loss (Figure 1A) nor clinical GVHD symptoms (Figure 1B). Moreover, all recipients survived the 20-day observation period (Figure 1C). This is likely due to CD4+ T cell dose, as NHP apheresis products have a significantly lower mean percentage of CD4+ cells than human apheresis products (~23% vs. 46%, respectively; p <0.009), and we have observed an inverse correlation between the percentage of CD4+ T cells in the human PBMNC sample and median survival time (data not shown). However, 30×106 PBMNC induced a severe and fatal GVHD in NSG mice that were preirradiated at a sublethal dose of 200 cGy. These data suggested that, in addition to T cell dose, radiation-induced tissue injury and associated inflammation was required upon which superimposed xenogeneic NHP GVHD reactions then were sufficient to result in lethality. Alternatively, cytoreduction of host innate immune cells may have been required to induce tissue injury and proinflammatory cytokine release that may support expansion to a critical threshold of transferred NHP T cells to cause GVHD. Similar to the human xeno-GVHD model, transfer of NHP PBMNC in the setting of irradiation resulted in significant and quantifiable pathology in the liver, lung and GI tract (Figure 1D) readily distinguishable from radiation-injury without NHP PBMNC transfer (Figure 1E).
Decreasing the dose of radiation ameliorates radiation-induced pathology at the expense of xeno-GVHD lethality
Because preconditioning with 200 cGy alone caused pathology (Figures 1D,E), we compared disease severity in mice receiving a lower dose of radiation (50 vs. 200 cGray) with or without the infusion of 30×106 NHP PBMNC. Reducing the dose to 50 cGray effectively ameliorated radiation-induced pathology, including weight loss (days 3-10) and clinical scores (days 3-20) (Figure 2A and B, respectively). However, mice receiving 50 cGray + 30×106 PBMNC had significantly decreased weight loss and clinical GVHD scores in comparison to those receiving 200 cGray + 30×106 PBMNC, and no lethality was observed (Figure 2C). Thus, the optimized working model for NHP xeno-GVHD in our laboratory is 200 cGray preconditioning (given by x-ray) followed by the infusion of NHP PBMNC isolated from apheresis products. However, it should be noted that intermediate doses of radiation were not tested, thus, there may be an intermediate dose, not yet identified, that further reduces radiation-toxicity while still permitting GVHD.
Figure 2. Decreasing the dose of radiation ameliorates radiation-induced pathology, but doesn't support xenoGVHD.
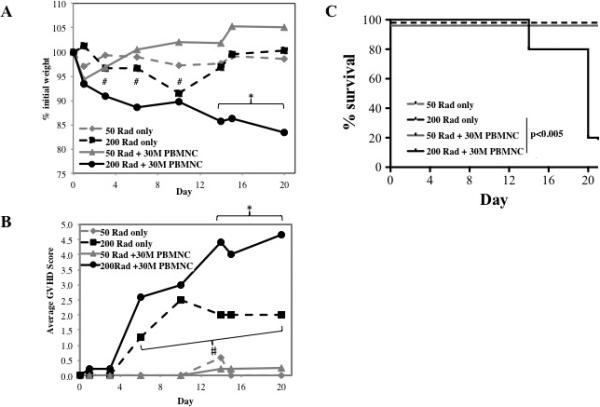
To determine the effect of radiation dose on xeno-GVHD, NSG mice were treated with either 50 or 200 cGray and administered PBS only or 30×106 PBMNC. Disease severity was measured by weight loss (A), clinical score (B) and survival (C). * denotes days when average weight or clinical score for 200rad + 30×106 PBMNC had p<0.05 compared to 200rad only. # denotes days when average weight or clinical score for 50rad only had p<0.05 compared to 200rad only. n= 5 mice per group.
Donor factors affect the incidence and severity of NHP xenogeneic GVHD in a small animal model system
Our overall goal was to develop an amenable, semi-high throughput screen for immunomodulatory therapeutics prior to commitment to in vivo NHP experiments. Therefore, it was important to optimize the number of NHP PBMNC, and to be able to titrate the degree of GVHD severity as most objectively measured by lethality. Given that 30×106 PBMNC plus radiation induced aggressive disease (median survival time of 13 days), we reasoned that this would represent a high bar for reagent efficacy, especially for GVHD therapy. We therefore compared doses of 30×106 vs. 20×106 PBMNC. The adoptive transfer of 20×106 PBMNC induced less severe disease than 30×106 PBMNC as assessed by overall survival and mean survival times (median survival of 33 vs. 14 days, p<0.003), mean weight loss and mean clinical GVHD scores (Figure 3A-C). Using a lower PBMNC cell number would be advantageous for screening since disease is less severe and hence more amenable to longitudinal analysis and detailed disease characterization in the setting of immunologic interventions.
Figure 3. Optimizing the murine/NHP xenogeneic model of GVHD.
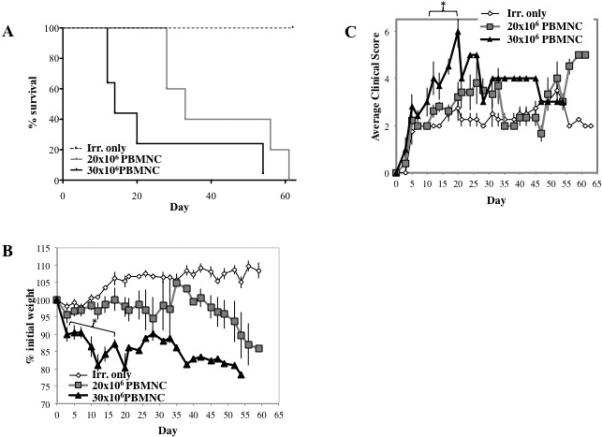
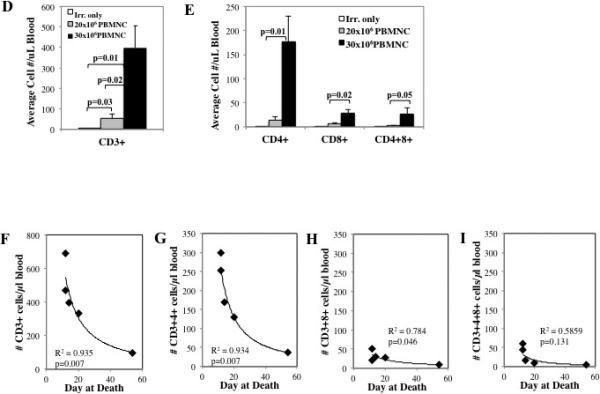
To determine the effect of cell dose on xeno-GVHD, NSG mice were irradiated (200 cGray) and administered PBS only, 20×106 PBMNC or 30×10 PBMNC. Disease severity was measured by survival (p<0.05 for 20×106 vs. 30×106 PBMNC) (A), weight loss (B) and GVHD score (C). To assess peripheral expansion of NHP T cells, mice were bled on day 10, and T cells were phenotyped and enumerated by flow cytometry. Quantitation of the total number of T cells (D) or T cell subsets (E) per μl blood on day 10 from mice receiving 20×106 or 30×106 NHP PBMNC (n=5 for 20×106 and 30×106 PBMNC) (n=5 for 20×106 and 30×106 PBMNC). Correlation between survival and total CD3+ T cells (F) or CD4+, CD8+ and CD4+8+ subsets (G-I, respectively) in blood on day 10. n=5 mice per group. * denotes days when p<0.05 for average weight and clinical score for both fresh and frozen/thawed PBMNC + Irr. compared to either Irr. or PBMNC only. * denotes days when average weight or clinical score for 30×106 PBMNC has p<0.05 compared to 20×106 PBMNC and Irr. only. R2 values were determined by Pearson correlation analysis.
To determine whether, as was seen with the human xeno-GVHD model 27, NHP T-cells were detectable in the PB of recipients posttransplant, animals were bled on day 10 and PBMNC stained for NHP CD3, CD4 and CD8 (Figure S1) and investigated for the impact of NHP cell dose on xeno-T cell accumulation in recipient PB. In addition to CD4 and CD8 single positive cells, NHP PB has previously been shown to contain a significant population of double positive T cells (ie CD4+8+) that have a resting memory phenotype and display both helper and cytotoxic functions 25,28,29. PB T cells were enumerated by flow cytometry using counting beads, and significantly more NHP CD3+ T cells were observed in mice receiving 30×106 PBMNC compared to 20×106 PBMNC (Figure 3D; P=0.02). While mice receiving 30×106 PBMNC predominantly accumulated significantly higher numbers of NHP CD4+ T cells, they also demonstrated increasing numbers of CD8+ and CD4+8+ T cells by Day 10 post-infusion compared to mice receiving 20×106 PBMNC (Figure 3E). The observation of predominant expansion of CD4+ cells in the NHP Xeno-GVHD model is notable, given that we have previously shown that peripheral CD4+ T cell numbers are inversely correlated with survival in the human model of xenoGVHD26. To determine whether peripheral T cell number also correlated with survival in the NHP xeno-GVHD model, we compared the number of CD3+, CD4+, CD8+ and CD4+8+ cells present on day 10 with survival for mice receiving 30×106 PBMNC (Figure 3F-I). Similar to what is observed in the human xeno-GVHD NSG model, lethality strongly correlated with total CD3+ and CD3+4+ T cells (R2=0.935 and 0.934, respectively; P=0.007 for each). A slight positive correlation was also observed for CD3+8+ T cells, although it considerably lower (R2=0.784; P=0.046), and no correlation was observed with CD3+4+8+ T cells.
Phenotype of NHP T cells in the peripheral blood
The T cell phenotype of NHP cells present in blood on day 18 was determined by flow cytometry by staining for markers of naïve (CD45RA) and activated/memory cells (CD95) (Figure 4A and B). While CD4 and CD8 T cells isolated from age-matched primates contain a significant percentage of naïve cells (52±3% and 40±6% CD45RA+, respectively), ≤8% of any T cell subset (CD4+, CD8+ or CD4+8+) isolated from mice on day 18 expressed CD45RA. Conversely, whereas only 41±5% and 60±9% of CD4 and CD8+ T cells isolated from NHP express CD95 (Fas), a marker of memory NHP T cells 29, nearly all (≥99%) T cells isolated from mice expressed high levels of CD95. Memory NHP T cells (ie CD3+95+) can be further differentiated based upon co-expression of CD28 and CCR7 into effector memory (EM; CD28−CCR7−), transitional central memory (TCM; CD28+CCR7−) and central memory (CM; CD28+CCR7+) (Figure 4C). Memory CD4+ T cells isolated from age-matched primates are primarily CM or TCM (59±6% and 34±4%, respectively), and only 7±2% are EM. In contrast, CD8+ memory cells are primarily EM (44±5%), and only 39±4% and 17±2% are CM and TCM respectively. Consistent with these results, a significantly lower percentage of CD4+ T cells expressed the effector memory phenotype (CD28−CCR7−), which is associated with terminal differentiation, compared to CD8+ or CD4+8+ T cells (8±6% vs. 26±6% or 22±4%, respectively; P=0.001 and −0.024). Additionally, a higher percentage of CD4+ T cells demonstrated the central memory or transitional central memory phenotype compared to CD8+ (32% vs. 20% CD28+CCR7+ and 59% vs. 47% CD28+CCR7−) or CD4+8+ T cells (32% vs. 28% CD28+CCR7+ and 59% vs. 49% CD28+CCR7−). These data, in which fewer CD4+ cells displayed an effector memory phenotype, are consistent with what is observed in aging primates 24.
Figure 4. Phenotype of NHP T cells in the peripheral blood.
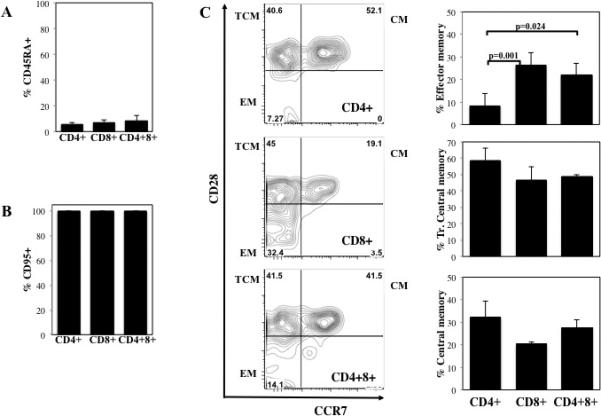
NOD/Scid/γc−/− mice receiving NHP PBMNC (30×106 cells) were bled on day 18. CD4+, CD8+ and CD4+8+ T cells present in day 18 peripheral blood were phenotyped for expression of markers of naïve (A; CD45RA) and activated/memory (B; CD95) T cells. (C) Day 18 memory T cells (CD3+95+) were further subsetted based upon CD28 and CCR7 expression into effector memory (CD28−CCR7−), transitional central memory (CD28+CCR7−) and central memory (CD28+CCR7+) T cells. n=6 mice per group.
Monovalent, pegylated, anti-human CD28 Fab’ antibody FR104 inhibits T cell expansion and xeno-GVHD in a NHP small animal model
To determine whether the NHP xenogeneic model of GVHD could be influenced by a human reagent that has shown efficacy in human xeno-GVHD models19, we tested an antagonistic monovalent, pegylated anti-CD28 Fab’ antibody (FR104) designed to selectively block CD28:B7 interactions 19. In prior renal allograft studies in NHPs, FR104, when combined with low doses of tacrolimus or with rapamycin, prevented acute rejection and alloantibody development, prolonging allograft survival30. Thus, FR104 represented a validated therapeutic, and, if this agent was ineffective in our NHP xeno-GVHD model, then the utility of such an approach as a semi-high throughput screen would be uncertain. Consistent with NHP renal allograft studies discussed above, FR104 treatment given from days 0-24 (Figure 5A) significantly decreased mortality in the NHP xeno-GVHD mouse model (Figure 5B). Whereas none of the animals treated with the control mAb survived past day 24, 80% (4 of 5) of mice receiving FR104 survived until d100 (Figure 5B; p ≤0.001). GVHD-associated weight loss and clinical scores were significantly reduced in FR104-treated mice compared to irrelevant IgG after day 17 (Figure 5C and D). These data indicated that this new NHP xeno-GVHD small animal model could have been used to successfully predict the usefulness of testing FR104 efficacy in NHP large animal allograft studies.
Figure 5. CD28 blockade with FR104 inhibits T cell expansion and effectively suppresses xeno-GVHD.
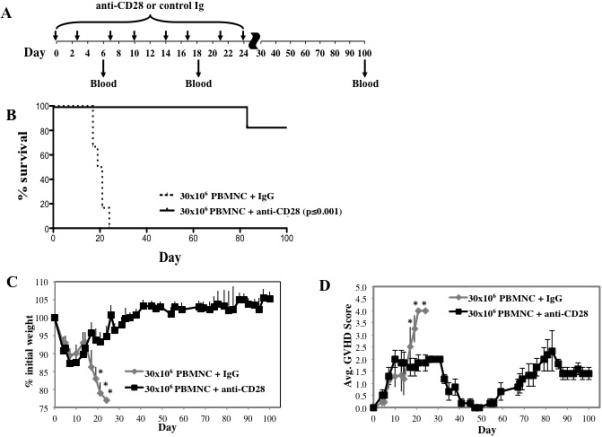
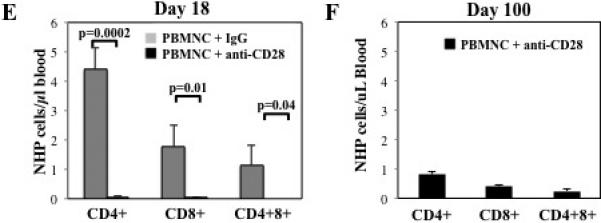
NOD/Scid/γc−/− mice receiving NHP PBMNC (30×106 cells) were treated with control IgG Fab’ antibody or anti–CD28 mAb (FR104) to assess potency for preventing xenogeneic GVHD. (A) Schematic showing the antibody dosing schedule: 100 μg per injection; injections starting on day 0, and 3 times weekly for 4 weeks. (B) Kaplan-Meier survival curves for mice receiving PBMNC + control IgG (dashed line) or + anti–CD28 mAb (solid line) (p<0.001). (C) Average weight (percentage of initial) for mice surviving on a given day for different groups of mice. *P < 0.05 for anti–CD28 treated mice from days 17 to 21. (D) Average GVHD score for mice surviving on a given day for different groups of mice. *P < 0.05 for anti–CD28 treated mice on days 19 and 21. GVHD severity was measured by enumerating CD4+, CD8+ and CD4+8+ T cell numbers in circulation on day 18 (E) and at the end of the experiment (d100) (F). n=6 mice per group.
To assess the effect of FR104 administration on peripheral T cell numbers in NHP xeno-GVHD recipients, mice were bled on days 18 and 100 and the number and relative frequency of CD4+, CD8+ or CD4+8+ cells was determined. The overall numbers of CD4+, CD8+ and CD4+8+ T cells in the blood of FR104-treated animals were each reduced 50-100 fold compared to controls (Figure 5E). FR104's effect on peripheral T cell numbers was durable, and very few NHP T-cells were found at the end of the experiment (day 100), 76 days after the last dose of antibody (Figure 5F).
To determine whether FR104 had an effect on functional T cell differentiation to Teffector cells, mice were sacrificed on day 10 and splenocytes stimulated for 4 hours with PMA and Ionomycin in the presence of Brefeldin A, and then stained for IFNγ, IL-17, GzmB and T cell antigens. Similar to PB, FR104 treatment decreased the overall number of CD4+, CD8+ or CD4+8+ T cells (≥25-fold) in the spleen (Figure 6A). Compared to mice injected with control IgG, FR104-treatment significantly decreased the percentage and absolute number of CD4+, CD8+ or CD4+8+ T cells that secreted IFNγ (20 vs. 5%, 69 vs. 34% and 53 vs. 24%; and ~200,000 vs. ~2,000, 33,000 vs. 500 and 35,000 vs. 500, respectively) (Figure 6B). Compared to control IgG treated animals, FR104-treatment also reduced both the frequency and absolute numbers of IL-17 producing splenic CD4+ and CD8+ T cells and lowered the absolute number of CD4+8+ T-cells producing IL-17 (Figure 6C). FR104 decreased the percentage of splenic CD8+ cells secreting GzmB 2.5-fold and the total number of CD8+GzmB+ cells 50-fold (Figure 6D). These results indicate that FR104 is able to both reduce cell number as well as inhibit differentiation into effector cells. In preliminary unpublished studies in NHP, CD28 restrains T cell proliferation and upregulation of granzyme B, resulting in preservation of the naïve T cell phenotype with concomitant control of Teffector/memory differentiation.
Figure 6. CD28 blockade with FR104 inhibits T cell expansion and effector differentiation.
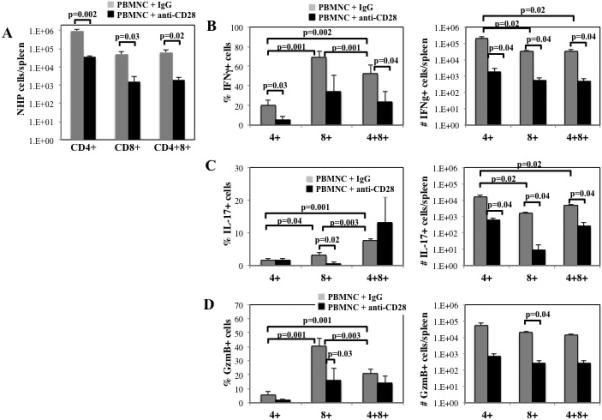
To assess the effects of FR104 on NHP T cell expansion and differentiation in secondary lymphoid organs, spleens were harvested on day 10 from mice receiving PBMNC + control IgG or FR104. Splenocytes were isolated, stimulated with phorbol PMA/Ionomycin, and cytokine expressing cells determined by intracellular cytokine staining. (A) Average number of CD4+, CD8+ and CD4+8+ T cells (±SEM) present in spleen. Percentage and average number of CD4+, CD8+ and CD4+8+ T cells producing IFNγ (B), IL-17 (C) or GzmB (D). n=4 mice per group for elective sac on day 10. p-values as indicated.
These results demonstrate that the NHP xeno-GVHD model is able to quantitate multiple clinical immunologic facets of GVHD, including target organ pathology, weight loss, external clinical score, as well as T cell differentiation and expansion in PB and secondary lymphoid organs. Finally, this model has predictive value, as therapeutics (ie FR104) with known efficacy in ameliorating NHP renal allograft rejection 30 and NHP/murine xenogeneic GVHD 19.
Discussion
Here we describe the creation of a NHP xeno-GVHD model that can be used to prioritize novel therapeutics for full evaluation in a large animal NHP allogeneic GVHD model. This model creates significant value-added compared to both human xeno-GVHD systems and large animal NHP allo-transplant models: Compared to human xeno-GVHD, the NHP xeno-GVHD model permits direct evaluation of the impact of new immunomodulatory strategies on NHP T cells prior to full-scale NHP allo-transplant studies. In contrast to NHP allo-transplant studies, the NHP small animal xeno-model enables a higher-throughput, cost-effective pipeline through which to evaluate novel immunomodulatory strategies. It also provides a platform for assessing the impact of these agents on T-cell differentiation, proliferation and activation profiles, as well as on clinical disease. This platform can also be used to determine the relative efficacy of therapeutics such as FR104 as prophylaxis vs. treatment of ongoing disease.
It is important to note that this small animal NHP xeno-GVHD model is intended to compliment and not replace an NHP large-animal model of GVHD. For example, some therapeutics may have different pharmacokinetics/pharacodynamics in the xeno-GVHD model compared to large animal studies, and thus may not correctly mimic combinatorial therapies or be fully useful for testing drugs for effects on NHP cells in vivo. Additionally, toxicities that occur as a result of conditioning regimen intensity and radiation repair mechanisms, especially of the GI tract, are expected to be different in immunodeficient NSG mice compared to NHP. Furthermore, pathogenic antibodies or cytokine therapeutics may not be uniformly cross-reactive between rodent and NHP cells and thus safety read-outs may not be valid in the xeno-GVHD model. Further, drug-based GVHD prophylaxis and treatment regimens that can be combined with new agents being tested are better simulated in large animal models guided by sequential pharmacokinetic and pharmacodynamics monitoring. Thus, when logistically (biologically, financially and reagent availability) feasible, large animal models serve a valuable purpose for predicting outcomes in the clinic that may avoid circumstances in which rodent studies have not well translated into the clinic 7. As such, adding a NHP xeno-GVHD screen to the small animal human xeno- and large animal NHP allo-transplantation pipeline will allow a first-pass examination of the efficiency of NHP targeted agents. These examples highlight the importance of the full translational pipeline, with the NHP Xeno-GVHD model feeding into the fully allogeneic NHP transplant models, which will be required to most closely parallel the human clinical setting.
Given the limitations discussed above of any xeno-model (either NHP or human), for both HSCT and organ grafting, pharmaceutical and biotech companies, along with the FDA, often wish to have in vivo NHP data before embarking on a clinical trial for both safety and efficacy reasons. Therefore, we envision a 2- or 3- stage approach to drug development in which candidates successfully identified in rodent allogeneic GVHD studies that would benefit from large animal NHP studies, are studied in human xeno-GVHD (stage 1) then the NHP xeno-GVHD small animal model (stage 2) followed by large animal NHP allogeneic GVHD models (stage 3). Stage 1 may also be accomplished by examining the biological properties of human reagents on human compared to NHP cells in vitro and then using the latter 2 stages to determine their in vivo efficacy, toxicity and detailed clinical and laboratory immunological assessment as part of the decision making process toward clinical trial development. This approach would be especially useful for PB and tissue kinetics studies, tolerance, long-term outcomes, and combined therapies for both studies of GVHD and solid organ transplantation.
In summary, this new NHP xeno-GVHD model recapitulates many aspects of NHP and human GVHD, resulting in an effective platform for high-throughput testing of immunomodulatory therapeutics for their effects on T cell differentiation and activation and clinical xeno-GVHD. Despite the inherent limitations of all preclinical models, the creation of this NHP small animal xeno-GVHD model offers a critical intermediary evaluation opportunity for new therapeutics to significantly stream-line the translational pipeline for approaches requiring or benefitting from large animal studies. Further, our results in the NHP xeno-GVHD model provide a strong rationale for a full exploration of monovalent pegylated anti-CD28 in the in vivo NHP large animal GVHD model 14,31, allowing a comprehensive evaluation on the immunology of engraftment and GVHD as well as a determination of the impact of mono- and combinatorial therapies on GVHD prevention and treatment.
Supplementary Material
Acknowledgments
We gratefully acknowledge Lauren Li (posthumously), Nara Paulson and the staff at Yerkes National Primate Research Center for veterinary and animal husbandry assistance.
Funding:
This work was supported by the Yerkes National Primate Research Center Base Grant, #RR00165, as well as NIH 2U19 AI051731, NIH 1R01 HL095791 and by a Burroughs Wellcome Fund Career Award (LSK). This work was also supported in part by NIH P30 CA77598 utilizing the shared resource Flow Cytometry Core from the Masonic Cancer Center, University of Minnesota.
Abbreviations
- GVHD
Graft-versus-host disease
- HSCT
hematopoietic stem cell transplantation
- xeno-GVHD
xenogeneic GVHD
- NHP
nonhuman primate
- PB
peripheral blood
- IV
intravenous
- PBMNC
peripheral blood mononuclear cells
- GzmB
Granzyme B
- NSG
NOD/SCID/γc−/−
- Tem
effector/memory T cells
- Trans. CM
Transitional central memory
- Tcm
Central memory T cells
Footnotes
Authorship:
Contributions: V.T., A.M.L. performed the experiments, interpreted the data, and assisted with the paper. C.L., M.J.R., K.S., A.G. performed the experiments, interpreted the data. B.V. reviewed and discussed the data. K.L.H., A.P.M, J.T., L.S.K., and B.R.B designed the research, interpreted the data, and wrote the paper.
Conflict of Interest Statement:
B.V. is a shareholder of Effimune, a company developing CD28 antagonists. Other authors declare no competing financial interests.
References
- 1.Holtan SG, Pasquini M, Weisdorf DJ. Acute graft-versus-host disease: a bench-to-bedside update. Blood. 2014;124(3):363–373. doi: 10.1182/blood-2014-01-514786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDonald-Hyman C, Turka LA, Blazar BR. Advances and challenges in immunotherapy for solid organ and hematopoietic stem cell transplantation. Sci Transl Med. 2015;7(280):280rv282. doi: 10.1126/scitranslmed.aaa6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flowers ME, Martin PJ. How we treat chronic graft-versus-host disease. Blood. 2015;125(4):606–615. doi: 10.1182/blood-2014-08-551994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Socie G, Ritz J. Current issues in chronic graft-versus-host disease. Blood. 2014;124(3):374–384. doi: 10.1182/blood-2014-01-514752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin Oncol. 2014;11(9):536–547. doi: 10.1038/nrclinonc.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Socie G, Blazar BR. Acute graft-versus-host disease: from the bench to the bedside. Blood. 2009;114(20):4327–4336. doi: 10.1182/blood-2009-06-204669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeiser R, Blazar BR. Preclinical models of acute and chronic graft-versus-host disease: how predictive are they for a successful clinical translation? Blood. 2016 doi: 10.1182/blood-2016-02-699082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badell IR, Russell MC, Thompson PW, et al. LFA-1-specific therapy prolongs allograft survival in rhesus macaques. J Clin Invest. 2010;120(12):4520–4531. doi: 10.1172/JCI43895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larsen CP, Pearson TC, Adams AB, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5(3):443–453. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 10.Lowe M, Badell IR, Thompson P, et al. A novel monoclonal antibody to CD40 prolongs islet allograft survival. Am J Transplant. 2012;12(8):2079–2087. doi: 10.1111/j.1600-6143.2012.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weaver TA, Charafeddine AH, Agarwal A, et al. Alefacept promotes co-stimulation blockade based allograft survival in nonhuman primates. Nat Med. 2009;15(7):746–749. doi: 10.1038/nm.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaliyaperumal S, Watkins B, Sharma P, et al. CD8-predominant T-cell CNS infiltration accompanies GVHD in primates and is improved with immunoprophylaxis. Blood. 2014;123(12):1967–1969. doi: 10.1182/blood-2014-01-547612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koura DT, Horan JT, Langston AA, et al. In vivo T cell costimulation blockade with abatacept for acute graft-versus-host disease prevention: a first-in-disease trial. Biol Blood Marrow Transplant. 2013;19(11):1638–1649. doi: 10.1016/j.bbmt.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Miller WP, Srinivasan S, Panoskaltsis-Mortari A, et al. GVHD after haploidentical transplantation: a novel, MHC-defined rhesus macaque model identifies CD28− CD8+ T cells as a reservoir of breakthrough T-cell proliferation during costimulation blockade and sirolimus-based immunosuppression. Blood. 2010;116(24):5403–5418. doi: 10.1182/blood-2010-06-289272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh K, Stempora L, Harvey RD, et al. Superiority of rapamycin over tacrolimus in preserving nonhuman primate Treg half-life and phenotype after adoptive transfer. Am J Transplant. 2014;14(12):2691–2703. doi: 10.1111/ajt.12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kean LS, Adams AB, Strobert E, et al. Induction of chimerism in rhesus macaques through stem cell transplant and costimulation blockade-based immunosuppression. Am J Transplant. 2007;7(2):320–335. doi: 10.1111/j.1600-6143.2006.01622.x. [DOI] [PubMed] [Google Scholar]
- 17.Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88(8):3230–3239. [PubMed] [Google Scholar]
- 18.Hippen KL, Merkel SC, Schirm DK, et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Science Translational Medicine. 2011;3(83):83ra41. doi: 10.1126/scitranslmed.3001809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poirier N, Mary C, Dilek N, et al. Preclinical efficacy and immunological safety of FR104, an antagonist anti-CD28 monovalent Fab' antibody. Am J Transplant. 2012;12(10):2630–2640. doi: 10.1111/j.1600-6143.2012.04164.x. [DOI] [PubMed] [Google Scholar]
- 20.Blazar BR, Taylor PA, McElmurry R, et al. Engraftment of severe combined immune deficient mice receiving allogeneic bone marrow via In utero or postnatal transfer. Blood. 1998;92(10):3949–3959. [PubMed] [Google Scholar]
- 21.Ali N, Flutter B, Sanchez Rodriguez R, et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS One. 2012;7(8):e44219. doi: 10.1371/journal.pone.0044219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King MA, Covassin L, Brehm MA, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157(1):104–118. doi: 10.1111/j.1365-2249.2009.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nam KH, Akari H, Terao K, Itagaki S, Yoshikawa Y. Age-related changes in major lymphocyte subsets in cynomolgus monkeys. Exp Anim. 1998;47(3):159–166. doi: 10.1538/expanim.47.159. [DOI] [PubMed] [Google Scholar]
- 24.Lee WW, Nam KH, Terao K, Akari H, Yoshikawa Y. Age-related increase of peripheral CD4+ CD8+ double-positive T lymphocytes in cynomolgus monkeys: longitudinal study in relation to thymic involution. Immunology. 2003;109(2):217–225. doi: 10.1046/j.1365-2567.2003.01646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akari H, Terao K, Murayama Y, Nam KH, Yoshikawa Y. Peripheral blood CD4+CD8+ lymphocytes in cynomolgus monkeys are of resting memory T lineage. Int Immunol. 1997;9(4):591–597. doi: 10.1093/intimm/9.4.591. [DOI] [PubMed] [Google Scholar]
- 26.Hippen KL, Harker-Murray P, Porter SB, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood. 2008;112(7):2847–2857. doi: 10.1182/blood-2008-01-132951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Rijn RS, Simonetti ER, Hagenbeek A, et al. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2−/− gammac−/− double-mutant mice. Blood. 2003;102(7):2522–2531. doi: 10.1182/blood-2002-10-3241. [DOI] [PubMed] [Google Scholar]
- 28.Zuckermann FA. Extrathymic CD4/CD8 double positive T cells. Vet Immunol Immunopathol. 1999;72(1-2):55–66. doi: 10.1016/s0165-2427(99)00118-x. [DOI] [PubMed] [Google Scholar]
- 29.Messaoudi I, Estep R, Robinson B, Wong SW. Nonhuman primate models of human immunology. Antioxid Redox Signal. 2011;14(2):261–273. doi: 10.1089/ars.2010.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poirier N, Dilek N, Mary C, et al. FR104, an antagonist anti-CD28 monovalent fab' antibody, prevents alloimmunization and allows calcineurin inhibitor minimization in nonhuman primate renal allograft. Am J Transplant. 2015;15(1):88–100. doi: 10.1111/ajt.12964. [DOI] [PubMed] [Google Scholar]
- 31.Furlan SN WB, Tkachev V, Flynn R, Cooley S, Ramakrishnan S, Singh K, Giver C, Hamby K, Stempora L, Garrett A, Chen J, Betz K, Ziegler CG, Tharp G, Bosinger SE, Promislow D, Miller J, Waller E, Blazar B, Kean L. Defining the Molecular Signature of Acute GVHD: Transcriptome Analysis Reveals Aurora Kinase A as a Novel Hippen, K.L. Targetable Pathway for Disease Prevention. Science Translational Medicine. 2016 doi: 10.1126/scitranslmed.aad3231. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.