

SUMMARY
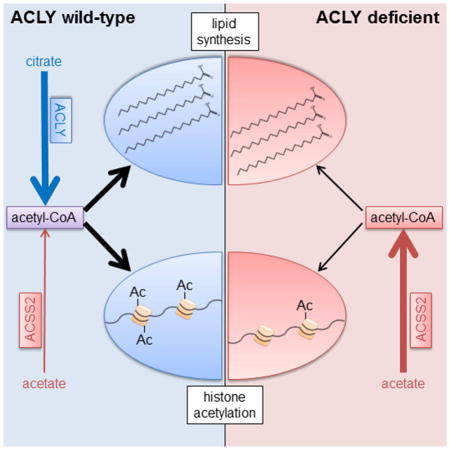
Mechanisms of metabolic flexibility enable cells to survive under stressful conditions and can thwart therapeutic responses. Acetyl-CoA plays central roles in energy production, lipid metabolism, and epigenomic modifications. Here we show that upon genetic deletion of Acly, the gene coding for ATP-citrate lyase (ACLY), cells remain viable and proliferate, although at an impaired rate. In the absence of ACLY, cells upregulate ACSS2 and utilize exogenous acetate to provide acetyl-CoA for de novo lipogenesis (DNL) and histone acetylation. A physiological level of acetate is sufficient for cell viability and abundant acetyl-CoA production, although histone acetylation levels remain low in ACLY-deficient cells unless supplemented with high levels of acetate. ACLY-deficient adipocytes accumulate lipid in vivo, exhibit increased acetyl-CoA and malonyl-CoA production from acetate, and display some differences in fatty acid content and synthesis. Together, these data indicate that engagement of acetate metabolism is a crucial, although partial, mechanism of compensation for ACLY deficiency.
eTOC BLURB
Zhao et al. demonstrate that ACLY deficiency causes upregulation of ACSS2 in proliferating cells in vitro and adipocytes in vivo. Acetate is needed for viability and is used for lipid synthesis and histone acetylation in the absence of ACLY. Proliferation is constrained in ACLY-deficient cells despite ACSS2 compensation.
INTRODUCTION
Acetyl-CoA is a central molecule in cell metabolism, signaling, and epigenetics. It serves crucial roles in energy production, macromolecular biosynthesis, and protein modification (Carrer and Wellen, 2015; Pietrocola et al., 2015). Within mitochondria, acetyl-CoA is generated from pyruvate by the pyruvate dehydrogenase complex (PDC), as well as from catabolism of fatty acids and amino acids. To enter the TCA cycle, acetyl-CoA condenses with oxaloacetate, producing citrate, a reaction catalyzed by citrate synthase. Transfer of acetyl-CoA from mitochondria to the cytosol and nucleus involves the export of citrate and its subsequent cleavage by ATP-citrate lyase (ACLY), generating acetyl-CoA and oxaloacetate. This acetyl-CoA is used for a number of important metabolic functions, including synthesis of fatty acids, cholesterol, and nucleotide sugars such as UDP-N-acetylglucosamine. Acetyl-CoA also serves as the acetyl-group donor for both lysine and N-terminal acetylation (Carrer and Wellen, 2015; Pietrocola et al., 2015). ACLY plays an important role in regulating histone acetylation levels in diverse mammalian cell types (Covarrubias et al., 2016; Donohoe et al., 2012; Lee et al., 2014; Wellen et al., 2009).
In addition to ACLY, nuclear-cytosolic acetyl-CoA is produced from acetate by acyl-CoA synthetase short chain family member 2 (ACSS2) (Luong et al., 2000). Recent studies have revealed an important role for this enzyme in hypoxia and in some cancers (Comerford et al., 2014; Gao et al., 2016; Kamphorst et al., 2014; Mashimo et al., 2014; Schug et al., 2015; Schug et al., 2016; Yoshii et al., 2009). Acetate can be produced intracellularly by histone deacetylase reactions or can be imported from the environment (Carrer and Wellen, 2015). Levels of acetate in circulating blood are rather low, ranging from 50–200 μM in humans, although acetate concentrations can increase substantially in certain conditions, such as following alcohol consumption, high fat feeding, or infection, or in specific locations, such as the portal vein (Balmer et al., 2016; Herrmann et al., 1985; Lundquist et al., 1962; Perry et al., 2016; Scheppach et al., 1991; Skutches et al., 1979; Tollinger et al., 1979). Acetate is also exported by cells under certain conditions, such as low intracellular pH (McBrian et al., 2013), and thus could potentially be made available for uptake by other cells in the immediate microenvironment. Two additional acetyl-CoA-producing enzymes, the PDC and carnitine acetyltransferase (CrAT), have been reported to be present in the nucleus and to contribute acetyl-CoA for histone acetylation (Madiraju et al., 2009; Sutendra et al., 2014). The PDC was shown to translocate from mitochondria to the nucleus under certain conditions, such as growth factor stimulation; within the nucleus, the complex is intact and retains the ability to convert pyruvate to acetyl-CoA (Sutendra et al., 2014). The relative contributions of each of these enzymes to the regulation of histone acetylation and lipid synthesis, as well as the mechanisms of metabolic flexibility between these enzymes, are poorly understood.
Whole body loss of ACLY is early embryonic lethal, indicating that it serves non-redundant roles during development (Beigneux et al., 2004). Silencing or inhibition of ACLY suppresses the proliferation of many cancer cell lines and impairs tumor growth (Bauer et al., 2005; Hanai et al., 2012; Hatzivassiliou et al., 2005; Migita et al., 2008; Shah et al., 2016; Zaidi et al., 2012). Depending on context, ACLY silencing or inhibition can also promote senescence (Lee et al., 2015), induce differentiation (Hatzivassiliou et al., 2005), or suppress cancer stemness (Hanai et al., 2013), further pointing to its potential as a target for cancer therapy. Inhibition of ACLY in adult animals and humans is reasonably well tolerated and produces blood lipid-lowering effects (Ballantyne et al., 2013; Gutierrez et al., 2014; Pearce et al., 1998). Thus, there may be a therapeutic window for ACLY inhibition in treatment of cancer and/or metabolic diseases, although the extent to which cells could leverage other compensatory mechanisms upon reduced ACLY function is not clear.
In this study, we aimed to elucidate two questions: first, does use of glucose-derived carbon for fatty acid synthesis and histone acetylation require ACLY, and second, can cells compensate for ACLY deficiency and, if so, by which mechanisms or pathways? To address these questions, we generated a conditional mouse model of Acly deficiency (Aclyf/f mice), as well as immortalized mouse embryonic fibroblast cell lines (Aclyf/f MEFs). As a complement to these models, we used CRISPR-Cas9 genome editing to delete ACLY from human glioblastoma cells. ACLY deficiency in both MEFs and glioblastoma cells potently impaired proliferation and suppressed histone acetylation levels. Both lipid synthesis and histone acetylation from glucose-derived carbon were severely impaired in ACLY-deficient MEFs. Cells partially compensated for the absence of ACLY by upregulating ACSS2, and ACLY-deficient MEFs became dependent on exogenous acetate for viability. Acetate was used to supply acetyl-CoA for both lipid synthesis and histone acetylation, although global histone acetylation levels remained low unless cells were supplemented with high levels of acetate. ACSS2 upregulation in the absence of ACLY was also observed in vivo upon deletion of Acly from adipocytes in mice. AclyFAT−/− mice exhibited normal body weight and adipose tissue architecture, and production of acetyl-CoA and malonyl-CoA from acetate was enhanced in ACLY-deficient adipocytes. Upon D2O labeling of WT and AclyFAT−/− mice, we observed that de novo synthesized fatty acids were present in white adipose tissue (WAT) in both genotypes, although some differences between depots were apparent. Visceral (epididymal) WAT (VWAT) exhibited no significant differences between WT and AclyFAT−/− mice in quantities of de novo synthesized fatty acids, while synthesized saturated fatty acids were reduced in subcutaneous (inguinal) WAT (SWAT) of AclyFAT−/− mice. Histone acetylation levels were also significantly altered in AclyFAT−/− SWAT. Taken together, this study demonstrates that ACLY is required for glucose-dependent fatty acid synthesis and histone acetylation and that a major, albeit partial, compensatory mechanism for ACLY deficiency involves engagement of acetate metabolism.
RESULTS
Genetic deletion of Acly in cells is consistent with viability but impairs proliferation
To facilitate investigation of the role of ACLY in vitro and in vivo, we generated a conditional mouse model of Acly deficiency using a conventional Cre-lox strategy (Aclyf/f mice) (Fig. S1A). MEFs from Aclyf/f mice were immortalized (Aclyf/f MEFs). Acly was efficiently deleted from Aclyf/f MEFs upon administration of Cre recombinase (Fig. S1B). AclyΔ/Δ MEFs continued to proliferate, although more slowly than parental cells (Fig. S1C). Over time, however, these cells regained ACLY expression, indicating that deletion occurred in less than 100% of cells and that those that retained ACLY had a growth advantage over AclyΔ/Δ cells (Fig. S1B). To address this, we generated 3 clonal Acly knockout (KO) cell lines, designated PC7, PC8, and PC9 (Fig. 1A). ACSS2 was strikingly upregulated in these cell lines (Fig. 1A). Proliferation in the absence of ACLY was significantly slower in each of the KO cell lines than in the parental Aclyf/f cells (Fig. 1B). We also used CRISPR-Cas9 to delete ACLY from LN229 glioblastoma cells (Fig. 1C). ACSS2 levels were high at baseline in LN229 cells and only modestly increased with ACLY deletion (Fig. 1C). However, similar to the ACLY-deficient MEFs, ACLY-deficient LN229 clones exhibited a marked proliferative impairment (Fig. 1D).
Figure 1. Genetic deletion of Acly is consistent with cell viability but impairs proliferation.

A) Western blot of 3 clonal ACLY deficient (KO) cell lines (PC7, PC8, and PC9) generated from Aclyf/f MEFs. B) Proliferation curve of Aclyf/f and ACLY-KO MEFs over 5 days, mean +/− SEM of triplicate wells, statistical significance compared to Aclyf/f. C) Western blot verification of ACLY knockout by CRISPR-Cas9 in LN229 glioblastoma cells. D) Proliferation curve of LN229 and two ACLY-knockout clonal cell lines over 5 days, mean +/− SEM of triplicate wells, statistical significance compared to LN229. E) Western blot of nuclear and cytoplasmic fractions of Aclyf/f, PC9, and reconstituted ACLY-WT and ACLY-H760A PC9 cells. FASN and LMNA are cytoplasmic and nuclear markers, respectively. F) Proliferation curve of Aclyf/f MEF and PC9 lines compared to PC9 reconstituted with ACLY-WT or ACLY-H760A over 5 days, mean +/− SEM of triplicate wells, statistical significance compared to PC9. G) Western blot of ACLY and ACSS2 protein levels in Aclyf/f MEFs over 144 hours following administration of Cre recombinase. H) Western blot of ACLY and ACSS2 protein levels in Aclyf/f MEFs over 144 hours with pharmacological inhibition of ACLY (50 μM BMS-303141). For all panels: **, p<0.01; ***, p<0.001; ****, p<0.0001. See also Figure S1.
Two of the ACLY-KO clones, PC7 and PC9, were reconstituted with wild type ACLY (ACLY-WT) or a catalytically inactive ACLY mutant (ACLY-H760A) (Fig. 1E and Fig. S1D). ACLY-WT but not ACLY-H760A significantly restored proliferation in the KO clones (Fig. 1F and Fig. S1E). Of note, despite comparable expression upon initial reconstitution (data not shown), ACLY-H760A failed to stably express as highly as ACLY-WT (Fig. S1D), further pointing to a strong selective advantage for cells expressing catalytically active ACLY. ACSS2 levels were elevated in both the nucleus and cytoplasm of ACLY-deficient cells, and this was reversed upon reconstitution of ACLY-WT (Fig. 1E).
We next inquired whether ACSS2 upregulation was induced by ACLY deletion or whether growing up ACLY-deficient clones selected for those that already had high ACSS2 expression. To test this, we examined the timing of ACSS2 upregulation upon loss of ACLY function. In Aclyf/f MEFs, ACSS2 was rapidly upregulated in parallel to loss of ACLY protein following Cre administration (Fig. 1G). Moreover, treatment of MEFs with an ACLY inhibitor (BMS-303141) led to increased ACSS2 within 96 hours (Fig. 1H). Thus, we conclude that the loss of ACLY activity induces ACSS2 upregulation.
ACLY deficient MEFs require use of exogenous acetate for viability
The amount of acetate in the serum used in these experiments was quantified by NMR. Undiluted calf serum (CS) contained ~800–900 μM acetate, while acetate was undetectable in dialyzed FBS (dFBS) (Fig. 2A and Supplemental Fig. 2A). Since acetate was also undetectable in DMEM, our standard culture conditions (DMEM + 10% CS) exposed cells to slightly less than 100 μM acetate. ACLY-deficient cells began to die when cultured in the absence of exogenous acetate (DMEM + 10% dFBS) (Fig. 2B, C, and D), and adding 100 μM acetate was sufficient to restore viability (Fig. 2C and E). No added proliferative benefit was gained by further increasing the amount of acetate supplemented (Fig. 2F). Additionally, reconstitution of ACLY-WT, but not ACLY-H760A, restored the ability of KO cells to grow in acetate-depleted conditions (Fig. 2B, E). To test whether acetyl-CoA production by ACSS2 was required for viability, we used CRISPR-Cas9 to delete Acss2 in Aclyf/f MEFs (Fig. S2B). Little to no difference in proliferation rate was observed upon Acss2 deletion when Acly was intact (Fig. S2C). However, subsequent deletion of Acly resulted in extensive toxicity (Fig. 2G and Fig. S2D), which was not observed in cells expressing Acss2, confirming that cells rely on ACSS2 for survival in the absence of ACLY.
Figure 2. ACLY-deficient MEFs require exogenous acetate for viability.

A) Acetate concentrations in DMEM, RPMI, 100% dialyzed fetal bovine serum (dFBS), and 100% calf serum (CS), mean +/− SEM of triplicate aliquots. See Fig. S2A for spectrum. B) Proliferation curve over 5 days of Aclyf/f, PC9, PC9-ACLY-WT, and PC9-ACLY-H760A cells in acetate-free conditions (DMEM + 10% dFBS + 10 mM glucose), mean +/− SEM of triplicate wells C) Image of ACLY-deficient PC9 cells cultured for 5 days in DMEM + 10% dFBS + 10 mM glucose, without (left), or with (right) 100 μM sodium acetate. D) Western blot of apoptotic markers cleaved PARP and cleaved caspase 3 (CASP3) in Aclyf/f and PC9 cells cultured in acetate-free conditions (DMEM + 10% dFBS + 10 mM glucose) for 4 (D4) or 5 (D5) days. E) Cell numbers following 5 days in culture in DMEM + 10% dFBS + 10 mM glucose alone (black) or supplemented with 100 μM sodium acetate (red) in Aclyf/f, PC9, PC9-ACLY-WT and PC9-ACLY-H760A cells, mean +/− SEM of triplicates, ***, p<0.001. Dotted line represents cell number at plating. F) Proliferation of PC9 cells over 5 days cultured in DMEM + 10% dFBS + 10 mM glucose, with 100 μM or 1 mM sodium acetate, mean +/− SEM of triplicate wells. G) Parental Aclyf/f MEFs and two clones of ACSS2-deficient Aclyf/f MEFs were administered Cre recombinase once (+) or twice (++) and proteins collected for Western blot after 2 days (+) and 2 weeks (++). See Fig. S2D for corresponding images. See also Figure S2.
Physiological levels of acetate support lipid synthesis in the absence of ACLY
ACLY deficiency did not alter rates of glucose or glutamine consumption, although lactate and glutamate production were elevated (Fig. 3A). To confirm the requirement for ACLY for glucose-dependent fatty acid synthesis and test the use of acetate, we set up parallel stable isotope tracer experiments in which Aclyf/f, PC9, PC9-ACLY-WT, and PC9-ACLY-H760A cells were incubated for 48 hours either with [U-13C]glucose (10 mM) and unlabeled acetate (100 μM) or [1,2-13C]acetate (100 μM) and unlabeled glucose (10 mM) (Fig. 3B). In ACLY proficient cells, palmitate was strongly labeled from glucose-derived carbon, as expected. In PC9 ACLY-KO cells, labeling of palmitate from 13C-glucose was nearly abolished; this could be restored by reconstitution of ACLY-WT, but not ACLY-H760A (Fig. 3C). Conversely, a marked increase in use of acetate for fatty acid synthesis was observed in PC9 and PC9-ACLY-H760A cells (Fig. 3D). We also examined the use of glucose and acetate carbon for synthesis of HMG-CoA, an intermediate in the mevalonate pathway and ketone body synthesis. Again, parental and PC9-ACLY-WT cells used glucose-derived carbon for HMG-CoA synthesis (Fig. 3E). In the absence of ACLY, glucose carbon use for HMG-CoA synthesis was extremely limited (Fig. 3E), and instead, acetate was used (Fig. 3F). Total levels of HMG-CoA trended slightly lower in the PC9 cells, though this difference was not statistically significant (Fig. 3G). The data thus show that in MEFs, glucose-dependent synthesis of fatty acids and HMG-CoA is nearly completely dependent on ACLY, and a physiological level of acetate can support lipid synthesis in its absence.
Figure 3. Acetate supports lipid synthesis in the absence of ACLY.

A) Measurements of glucose consumption and lactate production (left) and glutamine consumption and glutamate production (right), normalized to cell volume (cell number X mean cell volume), mean +/− SEM of triplicate wells, **, p<0.01; ***, p<0.001. Experiment was performed in glucose-free DMEM + 10% dFBS + 10 mM glucose + 100 μM sodium acetate. B) Experimental design for heavy isotope labeling of fatty acids using [U-13C]glucose, with unlabeled acetate present (left) and [1,2-13C]acetate, with unlabeled glucose present (right). C) Isotopologue distribution of palmitate after 48 hours labeling in 10 mM [U-13C]glucose in Aclyf/f, PC9, PC9-ACLY-WT, PC9-ACLY-H760A MEFs (top). Expressed as percent enrichment of palmitate (bottom), mean +/− SD of triplicates. D) Isotopologues of palmitate after 48 hours labeling in 100 μM [1,2-13C]acetate in Aclyf/f, PC9, PC9-ACLY-WT, PC9-Acly H760A MEFs (top). Expressed as percent enrichment of palmitate (bottom), mean +/− SD of triplicates. E) Isotopologues of HMG-CoA upon 6 hours labeling in 10 mM [U-13C]glucose (100 μM unlabeled acetate present) in Aclyf/f and PC9 MEFs, mean +/− SEM of triplicates. F) Isotopologues of HMG-CoA upon 6 hours labeling in 100 μM [1,2-13C]acetate (10 mM unlabeled glucose present) in Aclyf/f and PC9 MEFs, mean +/− SEM of triplicates. G) Total HMG-CoA quantitation in cells cultured in DMEM + 10% dFBS + 10 mM glucose + 100 μM sodium acetate (unlabeled), mean +/− SEM of triplicates.
ACLY is the primary supplier of acetyl-CoA for maintaining global histone acetylation
Histone acetylation is another major fate of nuclear-cytosolic acetyl-CoA. Consistent with previous data using RNAi-mediated ACLY silencing (Lee et al., 2014; Wellen et al., 2009), global levels of histone acetylation were strikingly reduced upon genetic deletion of Acly, despite increased ACSS2. Moreover, although 100 μM acetate was sufficient to restore survival in dFBS-cultured KO cells, it failed to rescue histone acetylation levels. Incubating cells with a high level of acetate (1 mM), however, markedly increased histone acetylation levels in KO cells (Fig. 4A). Reciprocally, histone acetylation levels were low in WT MEFs when cultured in 1 mM glucose and increased with greater glucose concentrations. In KO cells, histone acetylation levels were low at all concentrations of glucose tested, up to 25 mM (Fig. S3A). Reconstitution of PC9 cells with ACLY-WT but not ACLY-H760A restored histone acetylation levels to those in the parental cells (Fig. 4A).
Figure 4. ACLY is required for sustaining histone acetylation levels, despite ACSS2 compensation.

A) Western blot of acetylated histones extracted from Aclyf/f, PC9, PC9-ACLY-WT, PC9-ACLY-H760A MEFs cultured in complete (DMEM + 10%CS), dFBS (DMEM + 10%dFBS), +100 μM Acetate (DMEM + 10%dFBS + 100 μM sodium acetate), and +1 mM Acetate (DMEM + 10%dFBS + 1 mM sodium acetate) for 48 hours. B–D) Fraction of histone H3-K14, -K18, and -K23 acetylation (m+2) derived from 10 mM [U-13C]glucose, with unlabeled 100 μM acetate present (B), 100 μM [1,2-13C]acetate, with 10 mM unlabeled glucose present (C), or 1 mM [1,2-13C]acetate with 10 mM unlabeled glucose present (D), mean +/− SEM of triplicate samples. Labeling was for 24 hours (see also Fig. S3B for experimental design). E–G) Overall percentage of H3K23 acetylated in each cell line (y-axis), as well as the relative fraction of this acetylation incorporated from a labeled source (red): 10 mM [U-13C]glucose (E), 100 μM [1,2-13C]acetate (F), 1 mM [1,2-13C]acetate (G) or unlabeled sources (black), mean +/− SEM of triplicate samples. The same dataset is represented in parts B–D and E–G. See Figs. S3C–E for overall percentage data on K14 and K18 acetylation. See also Figures S3 and S4.
To determine the respective use of glucose- and acetate-derived carbon for histone acetylation in each of the MEF cell lines, we conducted stable isotope tracer experiments under 3 conditions: 1) [U-13C]glucose (10 mM) and unlabeled acetate (100 μM), 2) physiological [1,2-13C]acetate (100 μM) and unlabeled glucose (10 mM), or 3) high [1,2-13C]acetate (1 mM) and unlabeled glucose (10 mM) (Fig. S3B). In condition 1, histone acetyl groups were strongly labeled from 13C-glucose in Aclyf/f and PC9-ACLY-WT cells (Fig. 4B and E and Fig. S3C). In PC9 and PC9-ACLY-H760A cells, labeling of histone acetyl groups from glucose carbon was severely compromised (Fig. 4B and E and Fig. S3C). Moreover, aligning with Western blot data, total levels of histone acetylation were lower in cells lacking functional ACLY (Fig. 4E). The data thus indicate that ACLY is required for the majority of glucose-dependent histone acetylation. In cells lacking functional ACLY (PC9 and PC9-ACLY-H760A), 100 μM acetate contributed carbon to histone acetylation with ~40–60% of acetyl groups derived from acetate after 24 hours labeling (Fig. 4C), but total acetylation remained low (Fig. 4F and Fig. S3D). In 1 mM 13C-acetate, total histone acetylation levels rose (Fig. 4G and Fig. S3E), consistent with Western blot data, and acetate carbon constituted the majority of histone acetyl groups (Fig. 4D). These data indicate that ACLY is the dominant supplier of acetyl-CoA for histone acetylation in standard, nutrient-rich conditions, and that in its absence, cells can use acetate to supply acetyl-CoA for histone acetylation, although high exogenous acetate availability is needed to bring histone acetylation up to levels matching ACLY proficient cells. Of note, high acetate did not produce a corresponding rescue of proliferation (Fig. 2F). Thus, while ACLY-deficient cells exhibit both slower proliferation and lower histone acetylation levels, histone acetylation can be raised with high acetate without restoration of normal rates of proliferation, supporting the notion that metabolism regulates histone acetylation at least partially independently of proliferation.
We previously defined acetyl-CoA-responsive gene sets in LN229 glioblastoma cells (Lee et al., 2014). Cell cycle and DNA-replication-related genes were enriched among those genes that were suppressed in low glucose and increased by both glucose and acetate, although only glucose impacted doubling time (Lee et al., 2014). As observed in MEFs, ACLY deletion in LN229 cells abolished glucose-dependent regulation of global histone acetylation (Supplemental Fig. 4A). Acetate supplementation increased histone acetylation in ACLY null LN229 cells in a dose-dependent manner (Supplemental Fig. 4A). Consistently, the ability of glucose to promote expression of proliferation-related genes (E2F2, MCM10, and SKP2) was potently inhibited in ACLY-deficient cells. Expression of these genes exhibited dose-dependent rescue by acetate (Fig. S4B), correlating with global histone acetylation levels, despite lack of a proliferation rescue (Fig. S4C). In addition, we were surprised to find that whole cell acetyl-CoA levels were minimally impacted in ACLY-KO as compared to WT LN299 cells in high glucose conditions (Fig. S4D).
Acetyl-CoA levels are maintained by acetate in ACLY-deficient cells
In prior studies, global histone acetylation levels have tracked closely with cellular acetyl-CoA levels (Cai et al., 2011; Cluntun et al., 2015; Lee et al., 2014). It was therefore unexpected to find these uncoupled in ACLY-KO LN229 cells (Fig. S4D). We further explored this in ACLY-KO MEFs, and found that acetyl-CoA levels were significantly higher in the KO cells than in the WT Aclyf/f cells when cultured in 10 mM glucose and 100 μM acetate (Fig. 5A). These data suggested either that mitochondrial acetyl-CoA, which is inaccessible for histone acetylation (Takahashi et al., 2006), is elevated in ACLY-KO cells, or that ACSS2 compensation allows plentiful nuclear-cytosolic acetyl-CoA production from acetate, but that this acetate-derived acetyl-CoA is used less effectively than glucose-derived acetyl-CoA for histone acetylation. We reasoned that mitochondrial and extra-mitochondrial acetyl-CoA pools in ACLY KO cells could be distinguished based on whether whole cell acetyl-CoA is derived from glucose or from acetate (Fig. 5B). This is because in the absence of ACLY, glucose carbon does not meaningfully contribute to nuclear-cytosolic acetyl-CoA, as determined by its minimal use for either lipid synthesis or histone acetylation (Figs. 3 and 4). Within mitochondria, both glucose (via PDC) and acetate (via mitochondrial acetyl-CoA synthetases) can be used to generate acetyl-CoA for citrate synthesis. However, as assessed by enrichment of citrate and malate, acetate contributes minimally to mitochondrial metabolism in both WT and KO cells, while glucose is oxidized in both cell lines under these conditions (albeit to a somewhat lesser extent in KO cells) (Fig. 5C and D and Fig. S5A and B). These data suggest that in ACLY-KO cells, any glucose-derived acetyl-CoA is mitochondrial, whereas acetate-derived acetyl-CoA is predominantly nuclear-cytosolic (Fig. 5B). Thus measuring the contribution of glucose and acetate to whole cell acetyl-CoA should allow us to distinguish if the increase in acetyl-CoA in ACLY-KO MEFs reflects elevated mitochondrial or extra-mitochondrial acetyl-CoA.
Figure 5. Acetyl-CoA pools are sustained by acetate in the absence of ACLY.

A) Relative whole-cell acetyl-CoA levels in Aclyf/f and PC9 MEFs cultured in glucose-free DMEM + 10% dFBS + 10 mM glucose + 100 μM sodium acetate for 6 hours, normalized to cellular volume, mean +/− SEM of triplicates. B) Schematic of acetyl-CoA production from glucose and acetate with (top) or without (bottom) ACLY. C) Isotopologue distribution of citrate after 6 hours incubation with 10 mM [U-13C]glucose, with 100 μM unlabeled acetate present (black) or 100 μM [1,2-13C]acetate with 10 mM unlabeled glucose present (red), in Aclyf/f (top) or PC9 (bottom) MEFs, mean +/− SEM of triplicates. D) Isotopologue distribution of malate in the same conditions as panel C. E–G) m+2 acetyl-CoA (E), malonyl-CoA (F), or succinyl-CoA (G) following 6 hours labeling in 10 mM [U-13C]glucose (with 100 μM unlabeled acetate present) or 100 μM [1,2-13C]acetate (with 10 mM unlabeled glucose present), mean +/− SEM of triplicates. For E, F, and G all statistical comparisons are to Aclyf/f. For all panels: *, p<0.05; **, p<0.01; ***, p<0.001. See also Figure S5.
We therefore incubated cells with [U-13C]-glucose (10 mM) and 100 μM unlabeled acetate, or reciprocally, [1,2-13C]-acetate (100 μM) and 10 mM unlabeled glucose. In WT (Aclyf/f) cells, as expected, acetyl-CoA, malonyl-CoA, and succinyl-CoA were more strongly enriched from glucose than acetate (Fig. 5E–G). Interestingly, despite minimal labeling of malonyl-CoA from acetate in WT cells (consistent with palmitate enrichment in Fig. 3D), ~20% of the acetyl-CoA pool was enriched from 13C-acetate (Fig. 5E and F), further hinting at differential partitioning of acetate- and glucose-derived acetyl-CoA. In contrast, in the PC9 ACLY-KO cells, acetyl-CoA was minimally labeled from glucose and ~80% of the acetyl-CoA pool was labeled from acetate after 6 hours (Fig. 5E). Malonyl-CoA, but not succinyl-CoA, was also strongly enriched from 13C-acetate in PC9 cells (Fig. 5F and G). In sum, these data indicate that acetate is the major source of acetyl-CoA in the absence of ACLY, and it appears to predominantly supply the extra-mitochondrial pool.
A second implication of these data is that at least in KO cells, the mitochondrial acetyl-CoA pool is likely quite low in comparison to the extra-mitochondrial pool, since acetyl-CoA is minimally labeled from glucose-derived carbon. A large difference in relative acetyl-CoA pool size can explain the apparently paradoxical finding that in KO cells citrate is labeled from glucose, despite minimal acetyl-CoA enrichment (Fig. 5C and E). This interpretation is consistent with findings from a recent study of the mitochondrial metabolome, which found that matrix acetyl-CoA levels are very low unless complex I is inhibited, which increases the NADH/NAD ratio, reducing the activity of citrate synthase (Chen et al., 2016). Notably, another implication of this result is that a much larger nuclear-cytosolic acetyl-CoA pool in cultured cells would explain why whole cell acetyl-CoA measurements in ACLY proficient cells correlate closely with histone acetylation levels (Cluntun et al., 2015; Lee et al., 2014).
Together, these data indicate that acetate carbon is used to supply acetyl-CoA for nuclear and cytosolic processes in the absence of ACLY. Nevertheless, histone acetylation levels remain low in the absence of ACLY unless a high level of acetate is supplied, and proliferation remains constrained even in the presence of high acetate. Thus ACSS2 is a key, but partial, mechanism of compensation for ACLY deficiency.
ACSS2 is upregulated in vivo upon deletion of Acly from adipocytes
Finally, we sought to determine whether ACSS2 is upregulated upon loss of ACLY in vivo. Glucose uptake and glucose-dependent lipid synthesis in adipocytes is closely associated with insulin sensitivity and systemic metabolic homeostasis (Herman and Kahn, 2006; Herman et al., 2012). Moreover, our prior work implicated ACLY in regulating histone acetylation levels and expression of key genes in glucose metabolism, such as Glut4, in 3T3-L1 adipocytes (Wellen et al., 2009). To interrogate the role of adipocyte ACLY in vivo, we bred Aclyf/f mice to Adiponectin-Cre transgenic mice, which express Cre specifically in adipocytes (Lee et al., 2013). ACSS2 was upregulated in SWAT and VWAT upon deletion of Acly (Fig. 6A and B). In VWAT, ACSS2 upregulation was more apparent at the protein than the mRNA level (Fig. 6A and B). FASN protein levels were also elevated in the absence of ACLY, particularly in SWAT (Fig. 6A). Lipid droplets formed normally in AclyFAT−/− adipocytes; in VWAT, adipocytes were larger than in WT mice, while in SWAT, adipocyte lipid droplet size was comparable between genotypes (Fig. 6C). Body weight was indistinguishable between WT and AclyFat−/− mice fed a regular chow diet (Fig. 6D). However, overall gene expression patterns were altered, with lower expression of adipocyte-specific genes such as Glut4 in the AclyFAT−/− mice (Fig. 6E).
Figure 6. ACSS2 is upregulated in vivo upon deletion of Acly from adipocytes.

A) Western blot of liver, SWAT, and VWAT from Aclyf/f and AclyFAT−/− mice. B) mRNA expression of Acly and Acss2 in SWAT (left) and VWAT (right) from Aclyf/f and AclyFAT−/− mice, mean +/− SEM. C) Representative SWAT and VWAT histology from male 16-week-old Aclyf/f and AclyFAT−/− mice, scale bars indicate 100 μm. D) Body weight of male Aclyf/f (n=9) and AclyFAT−/− (n=8) mice, mean +/− SD. E) Expression of adipocyte genes in SWAT (left) and VWAT (right) from Aclyf/f (n=8) and AclyFAT−/− (n=7) mice, mean +/− SEM. For all panels: *, p<0.05; **, p<0.01.
Adipocyte acetyl-CoA and lipid metabolism is altered in the absence of ACLY
These data suggested that acetate metabolism might at least partially compensate for ACLY deficiency in adipocytes in vivo. Similar to that observed in MEFs, acetyl-CoA levels were higher in both VWAT and SWAT from AclyFAT−/− as compared to WT mice, while liver acetyl-CoA levels were slightly reduced (Fig. 7A). To test whether AclyFAT−/− adipocytes supply acetyl-CoA and dependent biosynthetic processes using acetate, we isolated primary visceral adipocytes and tested acetate uptake. Acetyl-CoA, as well as malonyl-CoA and HMG-CoA, indeed were more enriched from [1,2-13C]-acetate in primary adipocytes from AclyFAT−/− mice as compared to those from WT mice (Fig. 7B–D).
Figure 7. ACLY-deficient adipose tissue exhibits depot-specific alterations in DNL and histone acetylation.

A) Acetyl-CoA abundance in SWAT, VWAT, and liver in 11-week-old Aclyf/f (n=6) and AclyFAT−/− (n=7) mice. B–D) Primary mature adipocytes were isolated from 12-16 week old Aclyf/f (n=5) and AclyFAT−/− (n=3) mice and labeled with 100 μM [1,2-13C]acetate (with 5 mM unlabeled glucose present). Acetyl-CoA (B), malonyl-CoA (C), and HMG-CoA (D) enrichment from acetate was analyzed, mean +/− SEM. E–G) Relative quantities of fatty acids synthesized de novo in SWAT (E), VWAT (F), and liver (G) of Aclyf/f (n=6) and AclyFAT−/− (n=8) mice, mean +/− SEM. H–J) Overall histone H3 acetylation levels in 11-week-old SWAT (H), VWAT (I), and liver (J) of Aclyf/f (n=6) and AclyFAT−/− (n=7) mice, mean +/− SEM. For all panels, *, p<0.05; **, p<0.01. See also Figure S6.
We next investigated the extent to which de novo synthesized fatty acids were present in adipose tissue in the absence of ACLY. To capture rates of DNL in vivo, D2O was administered to mice via a bolus injection and subsequent addition to drinking water for 3 weeks. At the conclusion of labeling VWAT, SWAT, and liver were collected and total (saponified) fatty acids from each were analyzed by GC-MS. Plasma D2O enrichment was confirmed to be equivalent between genotypes (Fig. S6A). In both VWAT and SWAT, abundance of the saturated fatty acids palmitic acid (C16:0) and stearic acid (C18:0) was significantly reduced (Fig. S6B and C). Conversely, monounsaturated fatty acids, oleic acid (C18:1n9) and palmitoleic acid (C16:1n7), as well as the essential fatty acid linoleic acid (C18:2n6), were elevated in SWAT from AclyFAT−/− mice (Fig. S6B). A slight reduction in palmitic acid was also observed in liver (Fig. S6D). Fractional enrichment of fatty acids was not significantly different in VWAT between genotypes, although SWAT exhibited a moderate reduction in palmitic acid fractional synthesis (Fig. S6E and F). Fractional synthesis was not different between genotypes in the liver, except for a small reduction for palmitoleic acid (Fig. S6G).
The relative quantities of de novo synthesized fatty acids present in each tissue were calculated using plasma D2O enrichment, fatty acid labeling, and abundance. Of note, DNL-derived fatty acids present in WAT may be synthesized in adipocytes or produced in the liver and transported to fat. In SWAT of AclyFAT−/− mice, total de novo synthesized palmitic and stearic acid were significantly reduced (Fig. 7E). In contrast, no significant differences in the quantities of DNL-generated fatty acids were detected between AclyFAT−/− and Aclyf/f mice in VWAT (Fig. 7F). Liver DNL was largely unchanged by adipocyte ACLY deficiency, although a slight reduction in palmitic acid synthesis was observed (Fig. 7G). Since DNL-derived fatty acids were reduced in SWAT of AclyFAT−/− mice, this depot may maintain lipid droplet size through greater storage of diet-derived fatty acids, as suggested by elevated levels of linoleic acid (Fig. S6B).
Histone acetylation levels were also analyzed. Despite ACSS2 upregulation and elevated acetyl-CoA levels, H3K9ac and H3K23ac were significantly lower, and H3K18ac trended lower, in SWAT of AclyFAT−/− mice (Fig. 7H). Interestingly, this difference was not observed in VWAT, suggesting that acetate compensation for ACLY deficiency may be more complete in this depot or that other factors are dominant in determining histone acetylation levels (Fig. 7I). No differences in histone H3 acetylation were detected in liver (Fig. 7J). Altogether, the data suggest that in vivo, adipocytes lacking ACLY partially compensate by engaging acetate metabolism.
DISCUSSION
The findings of this study demonstrate that ACLY is required for the vast majority of glucose-dependent fatty acid synthesis and histone acetylation under standard culture conditions, and that ACSS2 upregulation and use of acetate carbon is a major mechanism of compensation for ACLY deficiency. Additionally, despite ACSS2 upregulation and higher acetyl-CoA levels, ACLY deficiency results in lower overall histone acetylation levels, slower proliferation, and altered gene expression patterns. The data suggest that ACLY and ACSS2 likely play distinct roles in the regulation of histone acetylation and gene expression, but also indicate that the potential for metabolic compensation from acetate should be considered if ACLY is pursued as a therapeutic target.
From a clinical perspective, prior study of PET imaging in human hepatocellular carcinoma patients using 11C-acetate and 18F-fluorodeoxyglucose (FDG) revealed a dichotomy between acetate and glucose uptake. Patient tumors, or regions within tumors, with high 11C-acetate uptake demonstrated low 18F-FDG uptake and vice versa. Moreover, tumors with high 18F-FDG uptake were more proliferative (Yun et al., 2009). These data support the concept that mammalian cells, cancer cells in particular, possess an intrinsic flexibility in their ability to acquire acetyl-CoA from different sources to adjust to changing metabolic environments in vivo. Further elucidation of the mechanisms connecting ACLY and ACSS2, as well as the differential phenotypes observed downstream of their activity, could point towards synthetic lethal strategies for cancer therapy or improved tumor imaging protocols.
In considering the roles of these enzymes in normal physiology, given the importance of GLUT4-dependent glucose uptake and glucose-dependent fatty acid synthesis for systemic metabolic homeostasis (Herman and Kahn, 2006; Herman et al., 2012), deletion of Acly in adipocytes results in a surprisingly mild phenotype, with no overt metabolic dysfunction observed for mixed background mice on a regular chow diet. Nevertheless, larger adipocytes and reduced expression of genes, such as Glut4, observed in this model are also characteristic of obesity and are associated with poorer metabolic function. This suggests that AclyFAT−/− mice may be more susceptible to metabolic dysfunction when nutritionally stressed, for example with high fructose feeding. Another interesting question is whether these mice will exhibit exacerbated metabolic phenotypes under conditions that alter acetate availability in the bloodstream, such as ethanol consumption or antibiotic treatment.
The differential impact of ACLY on SWAT and VWAT also warrants further investigation. It is not clear why SWAT, but not VWAT, exhibits reduced histone acetylation and de novo fatty acid synthesis, despite evidence for compensatory mechanisms such as FASN upregulation. One possible explanation relates to overall greater fraction of fatty acids that are de novo synthesized in SWAT as compared to VWAT (Fig. S6E and F), placing a greater demand for acetyl-CoA. Potentially, in a tissue with a lower DNL rate, acetate may be more readily able to compensate in both DNL and histone acetylation. Distribution of fatty acids in AclyFAT−/− WAT depots is also altered; SWAT in particular exhibits increased levels of monounsaturated and essential fatty acids (Fig. S6B). Palmitoleate, which has been implicated as an insulin-sensitizing lipokine (Cao et al., 2008), is elevated in ACLY-deficient SWAT, raising questions about how altered levels of bioactive lipid species in the absence of ACLY may influence metabolic phenotypes. More mechanistic work is also clearly needed to elucidate the relationship between ACLY and gene regulation. The relationship between global histone acetylation and gene expression is not entirely consistent between VWAT and SWAT, possibly reflecting gene regulatory mechanisms that are specific to ACLY.
A noteworthy observation in this study is that acetyl-CoA and histone acetylation levels appear to become uncoupled in the absence of ACLY, suggesting that acetate-derived acetyl-CoA may not be efficiently used for histone acetylation. Several possible mechanisms could account for this. First, it may be that in MEFs, an insufficient amount of ACSS2 is present in the nucleus to efficiently drive histone acetylation. ACSS2 has been found to localize prominently to the nucleus in some conditions (Chen et al., 2015; Comerford et al., 2014; Xu et al., 2014), and thus investigation of whether acetate more readily contributes to overall histone acetylation levels in these contexts will be informative. However, potentially arguing against this possibility, hypoxia promotes ACSS2 nuclear localization (Xu et al., 2014); yet, while acetate does regulate histone acetylation in hypoxic cells, a high level of acetate (~2.5 mM) is required (Gao et al., 2016). A second possibility is that within the nucleus, acetyl-CoA producing enzymes are channeled, compartmentalized into niches, or sequestered with particular binding partners. Through such a mechanism, acetylation of specific proteins may be regulated not only by the relevant acetyltransferase, but also by a specific acetyl-CoA producing enzyme. Consistent with this possibility, acetylation of HIF2α was shown to be exclusively dependent on ACSS2 as a source of acetyl-CoA (Chen et al., 2015; Xu et al., 2014). A third possibility is that ACLY-deficient conditions may result in altered KAT or HDAC activity. Finally, a fourth possibility is that lower use of acetyl-CoA for histone acetylation could be a feature of slow proliferation in the absence of ACLY (i.e. secondary to the proliferation defect). However, prior findings that histone acetylation is sensitive to glucose availability over a range that did not impact proliferation (Lee et al., 2014), that the TCA cycle (which supplies ACLY substrate citrate) and mitochondrial membrane potential have distinct and separate roles in regulating histone acetylation and proliferation, respectively (Martinez-Reyes et al., 2016), as well as data in the current manuscript showing that histone acetylation can be boosted by high acetate without a corresponding rescue of proliferation, argue against this as a sole explanation. Nevertheless, elucidation of the mechanisms that constrain proliferation in the absence of ACLY could help to definitively address this.
Investigating these possibilities will illuminate whether cells possess mechanisms to differentially detect ACLY-generated versus ACSS2-generated acetyl-CoA, as well as define the functional relationship between histone acetylation levels and cellular functions and phenotypes. Since ACLY dominates in nutrient and oxygen replete conditions, while ACSS2 becomes important in nutrient- and oxygen-poor conditions (Gao et al., 2016; Schug et al., 2015), having mechanisms- such as different acetylation substrates- to distinguish between acetyl-CoA produced by each enzyme could be advantageous to cells. For example, such mechanisms could potentially cue cells to grow when ACLY serves as the acetyl-CoA source and to mediate adaptive responses when ACSS2 is the primary acetyl-CoA source. The roles of these enzymes in gene regulation appear to be complex, and in-depth analysis of the respective roles of ACLY and ACSS2 in genome-wide histone acetylation and acetylation of other protein substrates is needed to begin addressing these questions.
Recent work has shown that the PDC is present in the nucleus and is able to convert pyruvate to acetyl-CoA for use in histone acetylation (Sutendra et al., 2014), raising the question of how the findings of the current study can be aligned with the described role of nuclear PDC. We suggest two potential models that are consistent both with our data and a role for nuclear PDC in histone acetylation: In the first, ACLY is the primary acetyl-CoA producer for regulation of global levels of histone acetylation, while PDC (and potentially other nuclear acetyl-CoA sources such as CrAT) could participate in mediating histone acetylation at specific target genes but not globally. A recent report that PDC forms a complex with PKM2, p300, and the arylhydrocarbon receptor (AhR), to facilitate histone acetylation at AhR target genes, is consistent with such a possibility (Matsuda et al., 2015). In the second, the role of ACLY in glucose-dependent histone acetylation regulation could be context-dependent, with a larger role for PDC emerging in certain conditions or cell types. This possibility is supported by observations that PDC nuclear translocation is stimulated by conditions such as growth factor stimulation and mitochondrial stress (Sutendra et al., 2014). Further investigation will be needed to evaluate these models.
In sum, this study points to a crucial interplay between glucose and acetate metabolism to supply the nuclear-cytosolic acetyl-CoA pool for fatty acid synthesis and histone acetylation. At the same time it shows that, despite compensatory mechanisms, ACLY is required for optimal proliferation, and simply increasing nuclear-cytosolic acetyl-CoA production is insufficient to fully replace ACLY. This could point to the importance of ACLY’s other product oxaloacetate, a build-up of ACLY’s substrate citrate, deficiencies in anapleurosis and/or mitochondrial function upon loss of a major catapleurotic pathway, or a signaling mechanism that is specific to ACLY. Clearly, more work is needed both to understand the mechanisms through which ACLY facilitates cell proliferation and to further define the ways that cells partition and use acetyl-CoA produced by different enzymes. The findings of this study raise a number of important questions for future investigation, as discussed. They also clarify the importance of ACLY in glucose-dependent acetyl-CoA production outside of mitochondria and provide key insights into the mechanisms of metabolic flexibility used for production of nuclear-cytosolic acetyl-CoA. Understanding these compensatory mechanisms will be important to consider for therapeutic targeting of acetyl-CoA metabolic pathways.
EXPERIMENTAL PROCEDURES
Generation of Aclyf/f and AclyFAT−/− mice
A Knockout first targeting vector was obtained from the KnockOut Mouse Project (KOMP) that targets exon 9 of Acly (ID: 80097), predicted to result in a truncated protein subject to nonsense-mediated decay. The Knockout First allele is initially null, but can be converted to a conditional floxed allele upon Flp recombination (Skarnes et al., 2011). Recombinant 129/B6 hybrid ES cells were generated in Penn’s Gene Targeting Core and blastocysts injected at Penn’s Transgenic and Chimeric Mouse Core. Upon acquisition of the chimeric mice, animals were bred to obtain germline transmission. Aclyf/+ progenies were selected through sequential breeding with wild-type C57Bl/6J mice (purchased from Jackson labs) and mice expressing Flp recombinase (B6.Cg-Tg(ACTFLPe)9205Dym/J, Jackson laboratories). Finally, Aclyf/f mice were generated by interbreeding and selected by genotyping (see Supplement). Immortalized Aclyf/f MEFs were generated from these mice (see Supplement). To produce AclyFAT−/− mice, Aclyf/f mice were bred to Adiponectin-Cre transgenic mice (Stock No: 010803; B6;FVB-Tg(Adipoq-cre)1Evdr/J, Jackson labs). The University of Pennsylvania’s Institutional Animal Care and Use Committee (IACUC) approved all animal experiments.
In vivo de novo lipogenesis
13-week-old male Aclyf/f (n=6) and AclyFAT−/− (n=7) mice (C57Bl/6 backcrossed) were I.P. injected with 0.035mls/g of body weight of 0.9% NaCl deuterated water (D2O) (Sigma). For 3 subsequent weeks, mice were provided water bottles containing 8% D2O. At the end of 3 weeks, mice were fasted for 6 hours, sacrificed, and plasma, liver, VWAT, and SWAT collected and snap frozen. Plasma from 4 additional mice (2 Aclyf/f and 2 AclyFAT−/−) not given D2O was used as controls. See Supplement for details of analysis.
Cell culture and proliferation assays
MEFs (generation described in Supplement) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% Cosmic Calf Serum (CS) (Hyclone SH30087.03, Lot. AXA30096). LN229 cells were cultured in Roswell Park Memorial Institute 1640 Medium (RPMI-1640) (Gibco) supplemented with 10% CS (Hyclone SH30087.03, Lot. AXA30096) and 2 mM L-glutamine. For experiments using dialyzed FBS, cells were cultured in glucose-free DMEM + 10% dFBS (Gibco 26400044), with indicated concentrations of glucose and sodium acetate added. For proliferation assays, cells were plated in triplicate at the indicated density and allowed to adhere overnight. Culture medium was changed the following day and cells were allowed to proliferate until indicated days following plating. Cells were collected and counted on a hemocytometer. Cell lines used for viral production included Phoenix E and HEK293T cells, which were purchased from ATCC. Cells were cultured in DMEM + 10% CS and used at low passage. All cell lines were routinely monitored and confirmed to be free of mycoplasma.
Acyl-CoA quantification and isotopologue analysis
Acyl-CoA species were extracted in 1 ml 10% (w/v) trichloroacetic acid (Sigma-Aldrich, St. Louis, MO cat. #T6399). Isotopologue enrichment analysis to quantify the incorporation of 10 mM [U-13C]glucose and 100 μM [U-13C]acetate into acyl-CoA thioesters was performed by LC-MS/HRMS. For quantitation, internal standards containing [13C315N1]-labelled acyl-CoAs generated in pan6-deficient yeast culture (Snyder et al., 2015) were added to each sample in equal amounts. Samples were analyzed by an Ultimate 3000 autosampler coupled to a Thermo Q Exactive Plus instrument in positive ESI mode using the settings described previously (Frey et al., 2016). A more detailed method can be found in the Supplemental Material.
Statistics
Student’s two-tailed t tests (two sample equal variance, two-tailed distribution) were used for all analyses directly comparing two data sets, unless otherwise indicated. Significance was defined as: *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Please also see Supplemental Experimental Procedures.
Supplementary Material
HIGHLIGHTS.
ACSS2 is upregulated upon genetic deletion of Acly in vitro and in vivo.
Acetate sustains viability in Acly-deficient MEFs, but proliferation is impaired.
Low levels of acetate can supply abundant acetyl-CoA in the absence of ACLY.
Acetate partially rescues lipogenesis and histone acetylation in ACLY deficiency.
Acknowledgments
This work was supported by R01CA174761, a Pew Biomedical Scholar Award, an American Diabetes Association Junior Faculty Award (7-12-JF-59), and the Linda Pechenik Montague Investigator Award to KEW. SZ is supported by predoctoral training grant 5T32CA115299. AMT is supported by a Penn-PORT IRACDA Postdoctoral Fellowship K12 GM081259. JVL is supported by predoctoral fellowship F31 CA189744. IAB is supported by NIH grant P30ES013508. KEW and IAB also acknowledge support from the Abramson Cancer Center Basic Science Center of Excellence in Cancer Metabolism. AJA is supported by NIH grant GM102503. NWS is supported by K22ES26235 and R21HD087866. CMM acknowledges R01CA188652. We thank Tobias Raabe from the Penn Gene Targeting Core and Jean Richa from the Transgenic and Chimeric Mouse Core for generation of the ES cells and chimeric mice that were used to produce the Aclyf/f mice. We thank Tony Mancuso from the AFCRI quantitative metabolomics core for guidance on GC-MS metabolite analysis. We thank James Alwine, Chi Dang, and Wellen lab members for helpful discussions during the preparation of this manuscript.
Footnotes
AUTHOR CONTIBUTIONS
Project was conceptualized and designed by SZ and KEW. SZ performed the majority of cell culture experiments and GC-MS metabolite analysis. AMT generated AclyFAT−/− mice and performed the majority of animal experiments. RAH, YMK and AJA conducted mass spectrometry analysis of histone acetylation. ST, AJF, IAB, and NWS conducted acyl-CoA measurements by LC-MS. JVL, SLC, and JMV conducted experiments. AC generated Aclyf/f MEFs and assisted with mouse experiments. AS and AMW conducted NMR acetate measurements. MW, NM, and CMM analyzed tissue rates of DNL from D2O-labeled mice. SZ and AMT prepared figures. KEW guided study, analyzed data, and wrote manuscript. All authors read and provided feedback on manuscript and figures.
Authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ballantyne CM, Davidson MH, Macdougall DE, Bays HE, Dicarlo LA, Rosenberg NL, Margulies J, Newton RS. Efficacy and safety of a novel dual modulator of adenosine triphosphate-citrate lyase and adenosine monophosphate-activated protein kinase in patients with hypercholesterolemia: results of a multicenter, randomized, double-blind, placebo-controlled, parallel-group trial. J Am Coll Cardiol. 2013;62:1154–1162. doi: 10.1016/j.jacc.2013.05.050. [DOI] [PubMed] [Google Scholar]
- Balmer ML, Ma EH, Bantug GR, Grahlert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, et al. Memory CD8(+) T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity. 2016;44:1312–1324. doi: 10.1016/j.immuni.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Kosinski C, Gavino B, Horton JD, Skarnes WC, Young SG. ATP-citrate lyase deficiency in the mouse. J Biol Chem. 2004;279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrer A, Wellen KE. Metabolism and epigenetics: a link cancer cells exploit. Current opinion in biotechnology. 2015;34:23–29. doi: 10.1016/j.copbio.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Xu M, Nagati JS, Hogg RT, Das A, Gerard RD, Garcia JA. The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment. PLoS One. 2015;10:e0116515. doi: 10.1371/journal.pone.0116515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, Wang T, Birsoy K, Sabatini DM. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell. 2016;166:1324–1337. e1311. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 2015;3:10. doi: 10.1186/s40170-015-0135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. Acetate dependence of tumors. Cell. 2014;159:1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016;5 doi: 10.7554/eLife.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, Snyder NW. LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and C-isotopic labeling of acyl-coenzyme A thioesters. Anal Bioanal Chem. 2016 doi: 10.1007/s00216-016-9448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Yang HB, Jiang SX, Yan GQ, Wang D, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun. 2016;7:11960. doi: 10.1038/ncomms11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MJ, Rosenberg NL, Macdougall DE, Hanselman JC, Margulies JR, Strange P, Milad MA, McBride SJ, Newton RS. Efficacy and safety of ETC-1002, a novel investigational low-density lipoprotein-cholesterol-lowering therapy for the treatment of patients with hypercholesterolemia and type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2014;34:676–683. doi: 10.1161/ATVBAHA.113.302677. [DOI] [PubMed] [Google Scholar]
- Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, Sukhatme VP. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol. 2012;227:1709–1720. doi: 10.1002/jcp.22895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanai JI, Doro N, Seth P, Sukhatme VP. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013;4:e696. doi: 10.1038/cddis.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Invest. 2006;116:1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S, Kahn BB. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann DB, Herz R, Frohlich J. Role of gastrointestinal tract and liver in acetate metabolism in rat and man. Eur J Clin Invest. 1985;15:221–226. doi: 10.1111/j.1365-2362.1985.tb00172.x. [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014;2:23. doi: 10.1186/2049-3002-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Jang H, Lee SM, Lee JE, Choi J, Kim TW, Cho EJ, Youn HD. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J. 2015;282:361–371. doi: 10.1111/febs.13139. [DOI] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist F, Tygstrup N, Winkler K, Mellemgaard K, Munck-Petersen S. Ethanol metabolism and production of free acetate in the human liver. J Clin Invest. 1962;41:955–961. doi: 10.1172/JCI104574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong A, Hannah VC, Brown MS, Goldstein JL. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2000;275:26458–26466. doi: 10.1074/jbc.M004160200. [DOI] [PubMed] [Google Scholar]
- Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics. 2009;4:399–403. doi: 10.4161/epi.4.6.9767. [DOI] [PubMed] [Google Scholar]
- Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Adachi J, Ihara M, Tanuma N, Shima H, Kakizuka A, Ikura M, Ikura T, Matsuda T. Nuclear pyruvate kinase M2 complex serves as a transcriptional coactivator of arylhydrocarbon receptor. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB, et al. Histone acetylation regulates intracellular pH. Mol Cell. 2013;49:310–321. doi: 10.1016/j.molcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- Pearce NJ, Yates JW, Berkhout TA, Jackson B, Tew D, Boyd H, Camilleri P, Sweeney P, Gribble AD, Shaw A, et al. The role of ATP citrate-lyase in the metabolic regulation of plasma lipids. Hypolipidaemic effects of SB-204990, a lactone prodrug of the potent ATP citrate-lyase inhibitor SB-201076. Biochem J. 1998;334(Pt 1):113–119. doi: 10.1042/bj3340113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217. doi: 10.1038/nature18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015;21:805–821. doi: 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Scheppach W, Pomare EW, Elia M, Cummings JH. The contribution of the large intestine to blood acetate in man. Clin Sci (Lond) 1991;80:177–182. doi: 10.1042/cs0800177. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016 doi: 10.1038/nrc.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Carriveau WJ, Li J, Campbell SL, Kopinski PK, Lim HW, Daurio N, Trefely S, Won KJ, Wallace DC, et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget. 2016 doi: 10.18632/oncotarget.9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skutches CL, Holroyde CP, Myers RN, Paul P, Reichard GA. Plasma acetate turnover and oxidation. J Clin Invest. 1979;64:708–713. doi: 10.1172/JCI109513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder NW, Tombline G, Worth AJ, Parry RC, Silvers JA, Gillespie KP, Basu SS, Millen J, Goldfarb DS, Blair IA. Production of stable isotope-labeled acyl-coenzyme A thioesters by yeast stable isotope labeling by essential nutrients in cell culture. Anal Biochem. 2015;474:59–65. doi: 10.1016/j.ab.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell. 2014;158:84–97. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Tollinger CD, Vreman HJ, Weiner MW. Measurement of acetate in human blood by gas chromatography: effects of sample preparation, feeding, and various diseases. Clin Chem. 1979;25:1787–1790. [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Nagati JS, Xie J, Li J, Walters H, Moon YA, Gerard RD, Huang CL, Comerford SA, Hammer RE, et al. An acetate switch regulates stress erythropoiesis. Nat Med. 2014;20:1018–1026. doi: 10.1038/nm.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii Y, Waki A, Furukawa T, Kiyono Y, Mori T, Yoshii H, Kudo T, Okazawa H, Welch MJ, Fujibayashi Y. Tumor uptake of radiolabeled acetate reflects the expression of cytosolic acetyl-CoA synthetase: implications for the mechanism of acetate PET. Nucl Med Biol. 2009;36:771–777. doi: 10.1016/j.nucmedbio.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Yun M, Bang SH, Kim JW, Park JY, Kim KS, Lee JD. The importance of acetyl coenzyme A synthetase for 11C-acetate uptake and cell survival in hepatocellular carcinoma. J Nucl Med. 2009;50:1222–1228. doi: 10.2967/jnumed.109.062703. [DOI] [PubMed] [Google Scholar]
- Zaidi N, Royaux I, Swinnen JV, Smans K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther. 2012;11:1925–1935. doi: 10.1158/1535-7163.MCT-12-0095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.