

Abstract
Two-photon imaging using high-speed multi-channel detectors is a promising approach for optical recording of cellular membrane dynamics at multiple sites. A main bottleneck of this technique is the limited number of photons captured within a short exposure time (~1ms). Here, we implement temporal gating to improve the two-photon fluorescence yield from holographically projected multiple foci whilst maintaining a biologically safe incident average power. We observed up to 6x improvement in the signal-to-noise ratio (SNR) in Fluorescein and cultured hippocampal neurons showing evoked calcium transients. With improved SNR, we could pave the way to achieving multi-site optical recording of fluorogenic probes with response times in the order of ~1ms.
OCIS codes: (110.0110) Imaging systems, (170.2520) Fluorescence microscopy
1. Introduction
Optical recording of cellular activity via two-photon (2P) microscopy is now widely used in neuroscience. With the development of a genetically encoded calcium indicators [1,2] and bulk loaded calcium-sensitive dyes [3,4] that report changes in intracellular calcium, one can reconstruct the network connectivity by looking at the correlated activity of neurons. In addition, there are also genetically encoded voltage indicators [5] and voltage-sensitive dyes [6] that precisely report the fast changes in the membrane potential allowing one to study the propagation of action potentials along the dendritic tree of a neuron via single [7–9] and two-photon excitation [10]. Multi-site fluorescence imaging can be achieved via single-photon wide-field illumination using a conventional light source, patterned illumination using holographic projection [11,12] or a micro-LED array [13]. However, using light with a shorter wavelength has limited penetration depth making it less preferred for in vivo studies of mammalian brains. Two-photon excitation is highly localized within a tight focal volume and hence requires additional techniques to position the focus to record fluorescence changes from multiple sites along the sample.
Multi-site 2P imaging can be achieved by scanning the beam or by holographically splitting a single laser beam into multiple foci (i.e. one focus per site). In beam scanning, a diffraction-limited laser focus is scanned along a line or random path using: resonant mechanical scanners or acousto-optic deflectors (AODs) [14]. The fluorescence signal is recorded by a single-channel detector (e.g. photo multiplier tube), sequentially. With AODs, scanning 94 down to 34 sites can be achieved with sampling rates from 180 up to 500 Hz, respectively [15]. On the other hand, holographic projection technique uses a spatial light modulator (SLM) to shape the incident beam into different light patterns (e.g. multiple foci) on the sample plane and the fluorescence emitted from the multiple excitation sites can be recorded simultaneously using a multi-channel detector (e.g. sensitive complementary metal oxide semiconductor (sCMOS) camera or an electron multiplying charged coupled device (EMCCD) camera) [16–18]. Recently, multi-plane 2P imaging of an intact brain was reported using holographic projection with a tunable lens [19]. In addition, multi-focal patterns generated by an SLM can be sequentially switched using a digital micro mirror device (DMD) and the fluorescence is sequentially detected using a single-channel detector. With a high-speed DMD (operated at 16.6kHz, 60μs dwell-time per site), the fluorescence signal from 11 sites can be decoded via S-code matrix (11 sites plus 1 site for background correction) with a temporal resolution of 0.72 ms (12×60μs) [20]. While the combination of holographic projection and DMD results in the desired sampling conditions, independent switching of the multi-foci requires placing the DMD at the Fourier plane making it vulnerable to non-linear photo-damage especially when the laser power exceeds the damage thresholds of the micro mirrors. Note that the total excitation power required to generate 2P fluorescence after the objective lens is only a fraction of the power illuminating the DMD, which is dependent on the relatively low transmission efficiency of the objective lens at near infrared (NIR) wavelengths.
Holographic projection allows for simultaneous illumination of multiple foci onto the sample. However, by conservation of energy, the intensity of each focus decreases as the number of foci is increased [21]. Due to the non-linearity of 2P excitation, the fluorescence per site drops by ~N −2. Another factor to consider is the exposure of the sample to the laser beam. Unlike in beam scanning where the beam’s focus illuminates a point on the sample for a short dwell time (~100 ns), the multi-foci pattern in holographic projection constantly exposes the regions on the sample throughout the recording. Increasing the incident laser power can provide higher fluorescence intensity from each focus but it also increases the photon flux, which can cause photo-damage of the sample. Thus, there is a need to efficiently excite fluorescence (via 2P absorption) from each site while maintaining low photon flux to minimize photo-damage.
An approach to improve fluorescence yield in 2P excitation while keeping a low average power on the sample is by Triplet-State relaxation via pulse picking from a 80MHz femto-second laser [22]. Using a much lower pulse repetition rate (~0.5MHz) and with a high peak power yields brighter fluorescence. Moreover, the ~2µs off-state arising from the low repetition rate allows the fluorophores to relax back to ground state. Bunched pulse single-photon excitation has also been shown to achieve a similar fluorescence enhancement [23]. In addition, we have previously reported that 0.8-2.4 MHz gating frequencies show fluorescence enhancement from 2P raster-scan imaging of neurons while maintaining cell viability [24].
Here, we use temporal gating to improve multi-site 2P excitation [25] and enhance the fluorescence yield for 2P functional imaging using a multi-channel detector. This allows for simultaneous projection of parked excitation foci while keeping the average power per focus minimal. By temporally gating the incident laser at frequencies of 0.8 and 1.6 MHz, we observed up to 6-fold higher signal-to-noise ratio (SNR) from temporally gated beam using a CMOS camera with 60% quantum efficiency. This improvement in SNR was also observed with triggered calcium activity in neuronal culture. Incorporating temporal gating with holographic projection can potentially be applied for high-speed (~1kHz) 2P functional imaging of cell membrane dynamics where the detection is shot-noise limited.
2. Theory
Multiple foci in a single z-plane are generated using prism phase maps encoded on the SLM [26]. Previously, it has been shown [21] that the input power of a Gaussian beam, Pin, before phase modulation is distributed to N number of foci, and the power of each foci, P(r) is given by:
| (1) |
where δ(r) is a radially symmetric measure of the first-order diffraction efficiency and r is the radial coordinates of the multiple foci at the Fourier plane measured with respect to the zero-order or the undiffracted beam at the optical axis. The 2P fluorescence, F(r), generated from each focus at distance r is given by:
| (2) |
where α accounts for all the relevant quantities (e.g. dye concentration, 2P cross-section, numerical aperture of the focusing lens, etc) for generating two-photon fluorescence from a single foci [27].
With a temporally gated beam, we set an incident beam with an “on” state within a bunch width, τ, while the “off” state is given by fR−1 – τ, where fR is the gating frequency. From (2), the time average 2P fluorescence per foci, <F(r)>, within an exposure time, To, is given by
| (3) |
where k = TofR is the number of gating cycles within To assuming fR−1 < To. The fluorescence from temporally gated beam observes a linear relationship with the duty cycle, τfR .
Prior to holographically dividing the beam, the laser is temporally gated and from conservation of energy, the time-average laser power, <Pout>, (within fR−1) of the temporally gated beam is given by:
| (4) |
where <Pin> is the average power within the “on” state, τ, used for generating 2P fluorescence. For an ungated beam, τ = fR−1, setting <Pout> = <Pin>. On the other hand, for τ << fR−1 then <Pout> << <Pin>, which results in a low average power but with high <F(r)> set by <Pin>. Therefore, within the exposure time To, the average excitation laser before holographically dividing the beam is <Pout> and its relation with <F(r)> is derived by combining Eqs. (3) and (4) arriving at
| (5) |
Because of the non-linear relationship of the fluorescence with the input power <Pin> described in (2), we see an improvement in the fluorescence by a factor of 1/(τfR). In the experiments, we enhance <F(r)> by maintaining <Pout>/N and setting fR to 0.8 MHz and 1.6 MHz whilst keeping τ = 140ns. We also set To within the maximum allowable exposure (sampling) time of the camera.
We measure the improvement in the fluorescence yield by the SNR. The SNR(r) at each site is given by
| (6) |
where <F(r)> was measured from the camera’s gray level (8-bit) readings after background subtraction and σ = (σon2 + σoff2 )1/2 is the quadrature of the standard deviation of the fluorescence, σon, and the dark noise, σoff, assuming a normal distribution.
The calcium activity is visualized with the relative change in the fluorescence, which is calculated as
| (7) |
where <Fb>max is the baseline fluorescence just before the first triggered fluorescence activity, <Fb>max is the maximum of the pooled baseline fluorescence from all sites.
3. Materials and methods
3.1 2P holographic microscope
We constructed a holographic 2P microscope for multi-site functional calcium imaging shown in Fig. 1. The system utilizes a femtosecond Ti:S laser (Coherent MIRA 900 pumped with a 12W Coherent Verdi G) for 2P excitation set at wavelength, λ = 800 nm. The laser is gated using an acousto-optic modulator (AOM, AA Opto ST110-A1-B4) driven by a radio frequency driver (AA Opto MODA110-B4-33) with τ = 140 ns pulse width and fR set to 0.8 and 1.6 MHz. Our choice of repetition rates were decided from prior work [24], which showed negligible phototoxic effect on the cell membrane. The laser beam is expanded 4x (L2/L1 = 200mm/50mm) and projected to a reflective phase-only SLM (Meadowlark Optics XY-512). A half-wave plate is placed before the beam expander to align the beam polarization with the preferred polarization angle of the SLM to achieve the maximum diffraction efficiency. The SLM is encoded with a phase-only hologram, which shapes the beam’s wavefront to produce multiple foci at the Fourier plane of lens, L3(f = 150 mm). Lens L4(f = 300 mm) and the objective lens (OL, Carl Zeiss 20x, NA = 1.0) form a 4f lens configuration to image the Fourier plane (of L3) to the sample plane of the OL. A dichroic mirror, DM1(Thorlabs FM202), reflects the NIR excitation laser towards the OL while transmitting the collected fluorescence towards L5 and the camera. A shortpass filter (FES700, Thorlabs) was placed before the camera to further filter the laser beam. The fluorescence signal was imaged using a 200 mm tube lens(L5) and on to a CMOS camera (Thorlabs DCC3240M) with quantum efficiency of ~60% at λ = 550 nm. We also incorporateda single-photon epifluorescence path consisting of: a blue light emitting diode (LED) operating a λ = 470 nm (Thorlabs M470L3), lens(L6, f = 125 mm), and a dichroic mirror (DM2, Thorlabs DMLP490R). Due to slight systematic optical misalignments in the optical train, we encoded aberration corrections (2nd order Astigmatism Z(2,4) = 8 and Spherical aberrations Z(0,4) = 6) into the SLM to achieve the maximum fluorescence signal.
Fig. 1.
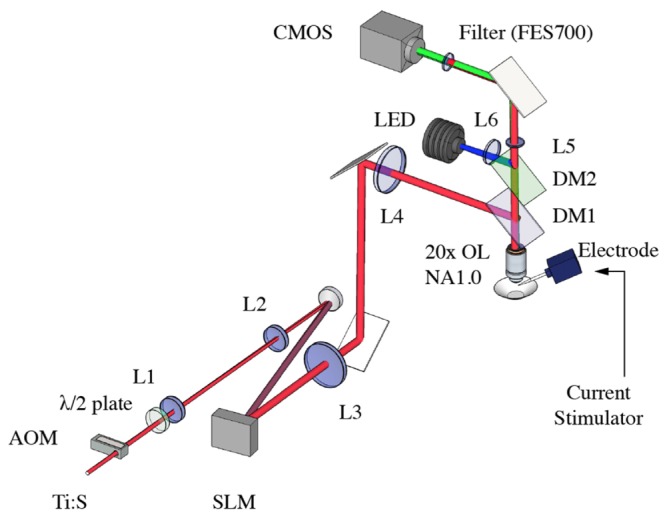
The holographic two-photon microscope for multisite fluorescence monitoring using a CMOS camera. Multi-site fluorescence imaging is achieved by holographically splitting the laser into multi-foci using a spatial light modulator (SLM). The laser is temporally gated with an acousto-optic modulator (AOM), to enhance the fluorescence yield from the multi-foci.
3.2 Calibration with Fluorescein dye
We prepared 10 mM of Fluorescein dye solution in a 100 μm-thick well mounted on a glass slide to demonstrate the relationship described in Eq. (5). The holographically projected multiple foci were positioned at the sample plane. The fluorescence was captured at 20 frames per second with 50 ms exposure time. The camera was set at 3.0x sensor gain, 2x2 binning, 120 black level offset and a region of interest (480x336 pixels) covering the area where the spots are projected. The fluorescence images as a function of time were analyzed using ImageJ (National Institute of Health). Within each image, we selected 5x5 pixels to measure the average grey levels of each focus as a function of time. Further analysis of was done in Matlab (Mathworks). The signal to noise per spot (SNR) was determined for ungated and temporally gated beam (0.8 and 1.6 MHz) from 2 to 6 mW average power per site.
3.3 Preparation of cultured hippocampal neurons
Preparation of cultured primary hippocampal neurons was performed in accordance to the protocol approved by the Australian National University Animal Ethics committee. Hippocampal tissue was dissected and dissociated from the brains of post-natal day 1-2 rats (Wistar). The tissue was then incubated in dissociating Papain solution at 37 °C in a water bath for 20 min. It was then triturated, incubated in DNAase solution at room temperature for 10 min, followed by resuspending in the plating medium (Dulbecco’s modified Earl’s medium supplemented with 10% fetal bovine serum, 1% L-glutamine, 1% penicillin-streptomycin and 1% B-27 supplement). The cells were then plated onto the pre-treated, poly-L-lysine coated glass coverslips and allowed to grow in an incubator at 37 °C and 5% CO2.
3.4 Calcium imaging on cultured hippocampal neurons
Temporal gating was applied to image calcium activity of primary hippocampal neuronal cultures (Days In Vitro, DIV26). On the day of imaging, the cultures were bathed in 10 μM Cal-520 (AAT Bioquest, CA) in culture medium for 15 mins. The cultures were then washed with culture external medium, consisting of (in mM): 125 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 25 HEPES and 10 D-Glucose, for 30 mins.
To determine the locations of the neurons, the sample was first imaged with the single-photon epifluoresence path of the microscope. Neurons were identified as elongated cells about 10 µm in diameter and exhibit higher levels of baseline fluorescence compared to their glial neighbours. Based from the wide-field image, eight foci were positioned on the somas and dendrites of the neurons. An extracellular bipolar electrode was placed near the recorded region to stimulate neuronal activity and correspondingly trigger calcium transients from the neurons. A train of current pulses (15 µA, 100 ms pulse at 0.5 Hz) was delivered by a custom-built current stimulator. The stimulator and acousto-optic modulator were triggered using a digital acquisition (DAQ) device (PCI-6363, National Instruments). The fluorescence from the holographically projected sites was captured at 20 Hz (50 ms exposure time) for 10 seconds with the CMOS camera. The relative change in fluorescence was calculated using Eq. (6) and it was then filtered with wavelet denoising (first order db1 wavelet, Matlab).
4. Results
We first compared the fluorescence yield of eight holographically generated foci of temporally gated and ungated beam incident in a chamber with Fluorescein dye for a preset average power per site. A time-series and histogram of the fluorescence at <Pout>/N = 2 to 6 mW for temporally gated and ungated beam are shown in Fig. 2(a). Note that <Pout>/N is an approximate measure of the average power per focus. Also note that the spread of <F(r)> measured for each <Pout>/N takes into account differences in excitation average power per focus due to δ(r) in (1). Figure 2(b) shows the relationship of the fluorescence with <Pout>/N in a log-log plot with base 2. The slopes (1.9 ± 0.1, 1.79 ± 0.08, 1.82 ± 0.05 for 0.8MHz, 1.6MHz, and ungated beam, respectively) are in good agreement with a two-photon excitation process (slope = 2.0). The fluorescence signals from 0.8MHz and 1.6MHz temporally gated beam were 6.7(P < 2E-16) and 3.7(P < 2E-16) fold higher compared to the ungated beam. Using chi-square goodness of fit test (at 5% significance level of rejection, Matlab), we verified that 93%(167/180) of the fluctuation in the fluorescence followed a normal distribution. This indicates that Gaussian statistics can be used to describe the fluctuations in the fluorescence. It was observed that the standard deviation showed a weak correlation (max slope 0.17 ± 0.02, ungated beam) with power, which may be due to the laser noise. Next, we determined the pooled ratio SNR of the temporally gated beam with respect to the ungated beam shown in Fig. 2(d). We observed 6.6 ± 0.1 and 3.9 ± 0.1 fold increase in the SNR for 0.8 and 1.6 MHz, respectively.
Fig. 2.

(a) Sample time-series across <Pout>/N ~2 to 6mW for ungated and temporally gated beam and sample negative images of the fluorescence from the holographic sites at <P>/N = 4mW, (b) The plot of fluorescence of each site with power, (c) The fluctuations of the fluorescence with power (d) The pooled SNR of each site for gated beam (fR = 0.8 and 1.6 MHz) relative to the SNR of the ungated beam (U).
We then applied temporal gating for imaging calcium activity of cultured hippocampal neurons loaded with Cal-520 AM dye. Triggered activity was evoked by applying a train of current pulses using an extracellular bipolar electrode near two neurons of interest (see Fig. 3(a)). Eight foci were positioned at the somas and dendrites of two neurons. Site one was placed outside the two neurons and its fluorescence was used to subtract the correlated noise with the other sites. The raw traces in Fig. 3(b) shows that the temporally gated beam yielded higher fluorescence than the ungated. In addition, triggered calcium activity was observed. A sample trace of the triggered calcium activity with 0.8 MHz temporally gated beam is shown in Fig. 3(c). Except for site 4, the six other sites showed fast fluorescent transients (~500ms time constant) that were well timed with the current stimulus (see Fig. 3(c)). The pooled SNR (48 samples from <Pout>/N ~3, 4, and 6 mW) increased by 3.9 ± 0.4 (P = 3E-12) and 3.7 ± 0.3 (P = 5E-12) for 0.8 and 1.6 MHz temporally gated beam, respectively (see Fig. 3(d)).
Fig. 3.

(a) Epifluorescence image of the neuronal cultures loaded with Cal-520 dye with the positions of the eight holographically projected two-photon sites. (b) A sample time-series plot of the fluorescence of each site for ungated and temporally gated beam at 4mW average power (c) The relative change in the fluorescence under 0.8 MHz and 4 mW temporally gated beam (d) The pooled SNR ratio of the temporally gated beam relative to the ungated beam. The scale bar is 50µm.
5. Discussion
We increased the fluorescence yield while maintaining a low average power on the sample from holographically generated multiple foci by modulating the 80 MHz femto-second pulses with an AOM driven at 0.8 and 1.6 MHz gating frequencies. The AOM dumps the beam at the off-states thereby throwing away most of the laser power. This puts an upper limit to the laser power and consequently the number of foci that can be generated. Alternatively, one can use an expensive regenerative amplifier (RegA) which is capable of delivering 200 kHz pulse repetition rate without significant loss of average power [28]. However, a RegA operates at a pre-set wavelength adding complexity when tuning the excitation to different wavelengths. Nevertheless, temporal gating with an AOM and a femtosecond pulse laser (e.g Ti:S) with an average output power of >2W presents a flexible alternative to achieve enhancement in fluorescence.
The increase in fluorescence with temporal gating translates to improvements in penetration depth, SNR, and reduction in photo-damage. Increasing the 2P fluorescence yield from the multiple foci leads to a higher average SNR. Since information is derived from the changes in the fluorescence of these foci, it is important that each focus reliably excites the fluorogenic probe within the focal volume. Temporal gating allows the excitation of parked multiple foci without inducing noticeable cell damage. Our results showed up to 6.6 fold increase in the fluorescence yield with temporally gated beam while observing triggered calcium transients from the neurons. The boost in SNR with temporal gating is consistent with that observed in triplet-state relaxation illumination [22,23].
Although we have not looked at penetration depth through a scattering medium in this particular work, previous reports were able to image deeper with lower repetition rates by a factor of 2.0x (40μm/20μm, fR = 1.2 MHz) in brain slices [24] and ~1.4x (860μm/600μm, fR = 200 kHz) in living intact brain [28]. However, one potential limitation of parallel detection of the 2P multi-site fluorescence is the cross talk from scattered fluorescence from each focus especially when imaging through deep opaque tissue. To avoid the cross talk, the minimum distances between the various foci needs to be calibrated, as it will be dependent on the degree of light scattering. Our future work will to look into the application of our technique through a scattering medium (or tissue) by calibrating the minimum distances between the various foci as a function of depth and sample scattering properties. This could limit the application of our technique to brain tissues of younger rodents as their scattering properties increases with age.
High fluorescence yield and SNR are crucial for imaging fast changes in fluorescence (~1ms) such as signals, which correspond to action potentials wherein the number of photons from fluorescence from the event (ΔF/F) is low. When imaging with fast fluorogenic probes (e.g voltage-sensitive indicators), an EMCCD camera can be used and can be set near camera saturation by applying gain or reducing the frame rate to overcome imaging noise. An increase in fluorescence from the excitation offers high SNR while maintaining high frame rates (>1 kHz).
As a proof-of-principle demonstration, we used a standard CMOS camera (max QE 60%, 3x Gain) to capture enhancement of the fluorescence with temporally gated excitation. Further improvement can be made at the detection by using a more sensitive camera (e.g sCMOS or EMCCD). In Fig. 4, we extrapolate how this enhancement in fluorescence performs with an EMCCD camera, to allow for high-speed detection of fast fluorogenic probes. Consider the relation of the photo-electrons with time via photoconversion N(t)~QE·G·photons(t) where G is the electronic gain from the camera. With temporal gating, the increase in the number of photo-electrons is due to the higher number of photons generated from a higher fluorescence yield. Since there are more photo-electrons with 0.8 MHz temporally gated beam with respect to the ungated beam, the exposure time of the camera can be reduced to collect the same total number of photo-electrons, N0.8MHz/Nungated = 1, (see point B in Fig. 4). In this case, the resulting exposure time of the CMOS camera with the temporally gated beam is 7.58 ms allowing 130 Hz frame rate. Furthermore, if the camera is replaced with an EMCCD camera (max QE 96%, 50x EM Gain), the relative number of photo-electrons generated increases by 26.7-fold ([QE·G]EMCCD/[QE·G]CMOS) allowing a much shorter exposure time of 0.28 ms corresponding to 3.6 kHz frame rate (see point A in Fig. 4) for collecting the same number of photo-electrons as with the ungated beam. By substituting the CMOS camera with a high (~96% at 550nm and 50x EM Gain) quantum efficiency Electron-Multiplied CCD (EMCCD), the frame rate can be increased to kHz range allowing one to capture fast changes in the fluorescence such as action potentials in neurons with the use of fast fluorogenic probes.
Fig. 4.
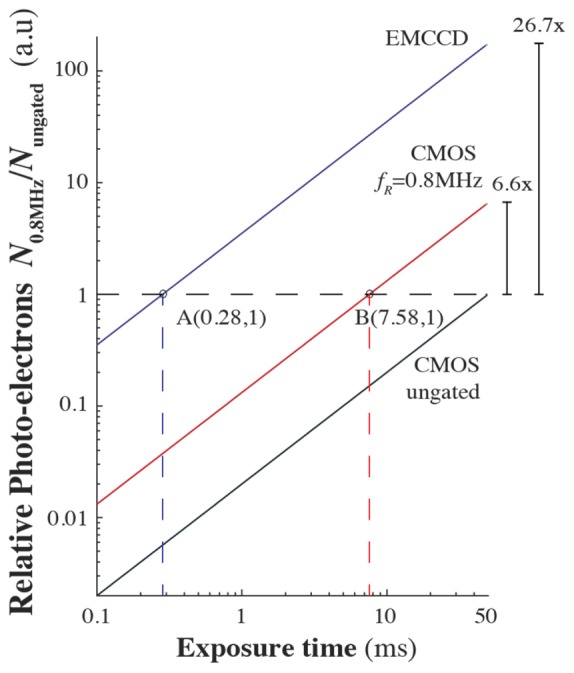
Diagram of the relative number of photoelectrons with exposure time for 0.8 MHz (red line) temporally gated and ungated beam (black line) captured by the CMOS camera, and for 0.8 MHz temporally gated beam using an EMCCD(blue line). The exposure time can be reduced down to 7.58 ms (red broken line) with 0.8 MHz temporally gated beam for collecting the same number of photo-electrons (N0.8MHz/Nungated = 1, horizontal broken line) as that of the ungated beam at 50ms exposure. Using an EMCCD with temporal gating, the exposure time can be further reduced to ~0.28 ms (blue broken line).
6. Conclusion
We have used temporal gating to enhance the 2P fluorescence yield from holographically generated multi-foci. With our technique, we were able to achieve higher fluorescence intensity whilst maintaining a low average power per site. The high SNR observed with the calcium activity in cultures shows that temporal gating can be applied without damaging the cells. By substituting the CMOS camera with a high (~96% at 550nm and 50x EM Gain) quantum efficiency EMCCD camera, the frame rate can be increased to kHz range allowing one to capture fast changes in the fluorescence such as action potentials in neurons with the use for fast fluorogenic probes. Temporal gating together with multi-site holographic projection could provide high-speed multi-site recording of fluorescence fluctuations corresponding to neuronal activity.
Funding
This work was supported by the Australian Research Council Discovery Project (contract number DP140101555).
References and links
- 1.Chen T. W., Wardill T. J., Sun Y., Pulver S. R., Renninger S. L., Baohan A., Schreiter E. R., Kerr R. A., Orger M. B., Jayaraman V., Looger L. L., Svoboda K., Kim D. S., “Ultrasensitive fluorescent proteins for imaging neuronal activity,” Nature 499(7458), 295–300 (2013). 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakai J., Ohkura M., Imoto K., “A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein,” Nat. Biotechnol. 19(2), 137–141 (2001). 10.1038/84397 [DOI] [PubMed] [Google Scholar]
- 3.Ohki K., Chung S., Ch’ng Y. H., Kara P., Reid R. C., “Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex,” Nature 433(7026), 597–603 (2005). 10.1038/nature03274 [DOI] [PubMed] [Google Scholar]
- 4.Tada M., Takeuchi A., Hashizume M., Kitamura K., Kano M., “A highly sensitive fluorescent indicator dye for calcium imaging of neural activity in vitro and in vivo,” Eur. J. Neurosci. 39(11), 1720–1728 (2014). 10.1111/ejn.12476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong Y., Huang C., Li J. Z., Grewe B. F., Zhang Y., Eismann S., Schnitzer M. J., “High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor,” Science 350(6266), 1361–1366 (2015). 10.1126/science.aab0810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan P., Acker C. D., Zhou W. L., Lee P., Bollensdorff C., Negrean A., Lotti J., Sacconi L., Antic S. D., Kohl P., Mansvelder H. D., Pavone F. S., Loew L. M., “Palette of fluorinated voltage-sensitive hemicyanine dyes,” Proc. Natl. Acad. Sci. U.S.A. 109(50), 20443–20448 (2012). 10.1073/pnas.1214850109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popovic M., Vogt K., Holthoff K., Konnerth A., Salzberg B. M., Grinvald A., Antic S. D., Canepari M., Zecevic D., “Imaging Submillisecond Membrane Potential Changes from Individual Regions of Single Axons, Dendrites and Spines,” Adv. Exp. Med. Biol. 859, 57–101 (2015). 10.1007/978-3-319-17641-3_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuart G. J., Palmer L. M., “Imaging membrane potential in dendrites and axons of single neurons,” Pflugers Arch. 453(3), 403–410 (2006). 10.1007/s00424-006-0149-3 [DOI] [PubMed] [Google Scholar]
- 9.Zhou W. L., Yan P., Wuskell J. P., Loew L. M., Antic S. D., “Dynamics of action potential backpropagation in basal dendrites of prefrontal cortical pyramidal neurons,” Eur. J. Neurosci. 27(4), 923–936 (2008). 10.1111/j.1460-9568.2008.06075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acker C. D., Yan P., Loew L. M., “Single-voxel recording of voltage transients in dendritic spines,” Biophys. J. 101(2), L11–L13 (2011). 10.1016/j.bpj.2011.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foust A. J., Zampini V., Tanese D., Papagiakoumou E., Emiliani V., “Computer-generated holography enhances voltage dye fluorescence discrimination in adjacent neuronal structures,” Neurophotonics 2(2), 021007 (2015). 10.1117/1.NPh.2.2.021007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutz C., Otis T. S., DeSars V., Charpak S., DiGregorio D. A., Emiliani V., “Holographic photolysis of caged neurotransmitters,” Nat. Methods 5(9), 821–827 (2008). 10.1038/nmeth.1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poher V., Grossman N., Kennedy G. T., Nikolic K., Zhang H. X., Gong Z., Drakakis E. M., Gu E., Dawson M. D., French P. M. W., Degenaar P., Neil M. A. A., “Micro-LED arrays: a tool for two-dimensional neuron stimulation,” J. Phys. D Appl. Phys. 41(9), 094014 (2008). 10.1088/0022-3727/41/9/094014 [DOI] [Google Scholar]
- 14.Bullen A., Patel S. S., Saggau P., “High-speed, random-access fluorescence microscopy: I. High-resolution optical recording with voltage-sensitive dyes and ion indicators,” Biophys. J. 73(1), 477–491 (1997). 10.1016/S0006-3495(97)78086-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grewe B. F., Langer D., Kasper H., Kampa B. M., Helmchen F., “High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision,” Nat. Methods 7(5), 399–405 (2010). 10.1038/nmeth.1453 [DOI] [PubMed] [Google Scholar]
- 16.Quirin S., Jackson J., Peterka D. S., Yuste R., “Simultaneous imaging of neural activity in three dimensions,” Front. Neural Circuits 8, 29 (2014). 10.3389/fncir.2014.00029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S. J., Allen W. E., Kauvar I., Andalman A. S., Young N. P., Kim C. K., Marshel J. H., Wetzstein G., Deisseroth K., “Extended field-of-view and increased-signal 3D holographic illumination with time-division multiplexing,” Opt. Express 23(25), 32573–32581 (2015). 10.1364/OE.23.032573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dal Maschio M., Difato F., Beltramo R., Blau A., Benfenati F., Fellin T., “Simultaneous two-photon imaging and photo-stimulation with structured light illumination,” Opt. Express 18(18), 18720–18731 (2010). 10.1364/OE.18.018720 [DOI] [PubMed] [Google Scholar]
- 19.Yang W., Miller J. E. K., Carrillo-Reid L., Pnevmatikakis E., Paninski L., Yuste R., Peterka D. S., “Simultaneous Multi-plane Imaging of Neural Circuits,” Neuron 89(2), 269–284 (2016). 10.1016/j.neuron.2015.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ducros M., Goulam Houssen Y., Bradley J., de Sars V., Charpak S., “Encoded multisite two-photon microscopy,” Proc. Natl. Acad. Sci. U.S.A. 110(32), 13138–13143 (2013). 10.1073/pnas.1307818110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daria V. R., Stricker C., Bowman R., Redman S., Bachor H. A., “Arbitrary multisite two-photon excitation in four dimensions,” Appl. Phys. Lett. 95(9), 093701 (2009). 10.1063/1.3216581 [DOI] [Google Scholar]
- 22.Donnert G., Eggeling C., Hell S. W., “Major signal increase in fluorescence microscopy through dark-state relaxation,” Nat. Methods 4(1), 81–86 (2007). 10.1038/nmeth986 [DOI] [PubMed] [Google Scholar]
- 23.Donnert G., Eggeling C., Hell S. W., “Triplet-relaxation microscopy with bunched pulsed excitation,” Photochem. Photobiol. Sci. 8(4), 481–485 (2009). 10.1039/b903357m [DOI] [PubMed] [Google Scholar]
- 24.Gautam V., Drury J., Choy J. M. C., Stricker C., Bachor H.-A., Daria V. R., “Improved two-photon imaging of living neurons in brain tissue through temporal gating,” Biomed. Opt. Express 6(10), 4027–4036 (2015). 10.1364/BOE.6.004027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Go M. A., Stricker C., Redman S., Bachor H. A., Daria V. R., “Simultaneous multi-site two-photon photostimulation in three dimensions,” J. Biophotonics 5(10), 745–753 (2012). 10.1002/jbio.201100101 [DOI] [PubMed] [Google Scholar]
- 26.Liesener J., Reicherter M., Haist T., Tiziani H. J., “Multi-functional optical tweezers using computer-generated holograms,” Opt. Commun. 185(1-3), 77–82 (2000). 10.1016/S0030-4018(00)00990-1 [DOI] [Google Scholar]
- 27.Xu C., Webb W. W., “Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm,” J. Opt. Soc. Am. B 13(3), 481–491 (1996). 10.1364/JOSAB.13.000481 [DOI] [Google Scholar]
- 28.Theer P., Hasan M. T., Denk W., “Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier,” Opt. Lett. 28(12), 1022–1024 (2003). 10.1364/OL.28.001022 [DOI] [PubMed] [Google Scholar]