

Abstract
Experimental infection of inbred mouse strains with Candida albicans provides a good model system to identify host genetic determinants that regulate onset of, response to, and ultimate outcome of disseminated candidiasis. The A/J mouse strain is exquisitely sensitive to infection with C. albicans, while the C57BL/6J strain is relatively resistant, as measured by survival following intravenous injection of Candida blastospores. This differential susceptibility is caused by an A/J-specific loss-of-function mutation in the C5 component of the complement pathway. C5 plays several critical roles in host response to infection, including target lysis and phagocyte recruitment. Therefore, to determine which of its functions were required for host resistance to candidiasis, a detailed comparative analysis of pathophysiology and host response to acute C. albicans infection was conducted in A/J and C57BL/6J mice. C5-sufficient C57BL/6J mice were found to succumb late in infection due to severe kidney pathology, typified by fungal replication and robust neutrophil-based inflammatory response associated with extensive tissue damage. In contrast, A/J mice were moribund within 24 h postinfection but displayed little if any kidney damage despite an inability to mobilize granulocytes and a high fungal load in the kidney. Rather, C5 deficiency in A/J mice was associated with higher levels of circulating cytokines tumor necrosis factor alpha, interleukin-6, monocyte chemotactic protein 1 (MCP-1), MCP-5, and eotaxin in response to C. albicans. Transfer of the C5-defective allele from A/J onto a C57BL/6J genetic background in recombinant congenic strain BcA17 recapitulated the phenotypic aspects of the susceptibility of A/J mice to C. albicans, confirming the causative role of C5 deficiency in the dysregulated cytokine response.
Candida albicans is part of the normal gastrointestinal flora in most healthy individuals. Thus, the host and the pathogen have a commensal relationship which is dependent on a balance between effective host defense mechanisms and pathogenicity and virulence determinants of the pathogen that can overcome weakened host defenses. The innate and adaptive host defense mechanisms against Candida infection are only partly understood but can manifest as genetic determinants of susceptibility in human populations (32, 43) and in animal models of experimental infection (3, 36). The laboratory mouse is an acknowledged model for the study of disseminated candidiasis. Intravenous infection with C. albicans blastospores causes a pathology that resembles the human condition and is characterized by active fungal replication in the kidney, brain, and heart, with death usually resulting from kidney failure (1).
In inbred mouse strains, susceptibility to experimental candidiasis is under genetic control. However, the control mechanisms appear to be complex, as phenotypic indicators of susceptibility do not always cosegregate in inbred strains (3, 20, 27, 28). An analysis of the strain distribution pattern of resistance and susceptibility to infection with the fungal pathogens C. albicans and Cryptococcus neoformans indicates that susceptibility cosegregates with a known deficiency allele (Hc) for the C5 component of the complement pathway (1, 21, 29, 30, 33). The major effect of complement deficiency is further modulated by genetic background in inbred strains (3, 6, 26).
Ashman et al. (4) presented a genetic model based on the strain distribution pattern of inbred strains and of subsets of recombinant inbred strains in which two Candida resistance loci, Carg1 (2) and Carg2 (5), influence the outcome of intravenous infection with C. albicans. Carg1 was proposed to influence overall tissue damage and to some extent kidney colonization. On the other hand, Carg2 appears to affect disease progression in the kidney. A combination of a susceptibility allele at Carg2 together with C5 deficiency results in increased fungal load and high mortality. The mode of action of Carg1/Carg2, including possible interaction with the C5 pathway, remains unknown and awaits the mapping and cloning of the putative Carg1 and Carg2 loci.
The A/J strain is C5 deficient and has putative susceptibility alleles for Carg2 (1). It is acutely sensitive to systemic infection with C. albicans, as revealed by high kidney fungal load and high mortality (3). In genome-wide linkage studies in informative F2 crosses derived from A/J mice, we observed that susceptibility to C. albicans in this cross segregates as a single locus with C5 status, accounting for the total phenotypic variance of the F2 mice (39a).
The complement pathway plays several critical roles in the innate as well as adaptive mechanisms of defenses against many infections (14). These include the formation of a membrane attack complex that can cause direct proteolysis of the target microbe. In addition, complement components (e.g., C3 subfragments) can function effectively as opsonins that facilitate phagocytosis of microorganisms. Furthermore, the C3a and C5a proteolytic fragments display potent proinflammatory properties and trigger several aspects of the inflammatory response (11, 15, 41). Although the functions of C5a as a proinflammatory peptide are well established, its absence is not always characterized by decreased inflammation. There are reports of pathological levels of inflammation in C5-deficient models (7, 31). This then confers on C5a an important role not only in the initiation of inflammation, but also in the subsequent control of the response.
To determine which of the many activities of C5 are required for host resistance to candidiasis, we conducted a detailed comparative study of the pathophysiology and host responses to acute C. albicans infection in C5-deficient A/J and C5-sufficient C57BL/6 mice. We report that C5 deficiency in A/J mice results in a complex phenotype characterized by rapid fungal proliferation in target organs, failure to mobilize granulocytes, and development of a unique and rapidly fatal (within 24 h) allergic-type response. By contrast, C57BL/6J mice die of kidney failure associated with microbial replication and a tissue-damaging inflammatory response 2 to 3 weeks following infection. Our studies provide insight into the role of C5a in the regulation of host inflammatory responses.
MATERIALS AND METHODS
Mice.
Eight- to 12-week-old A/J and C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, Maine). The recombinant congenic strain BcA17 was purchased from Emerillon Therapeutics (Montréal, Québec, Canada). Mice were age and sex matched for all experiments. They were housed at the Biotechnology Research Institute. Housing and all experimental procedures were approved by the Biotechnology Research Institute Animal Care Committee, operating under the guidelines of the Canadian Council of Animal Care.
C. albicans infections.
C. albicans strain SC5314 was grown overnight in YPD medium (1% yeast extract, 1% Bacto Peptone [Difco Laboratories, Detroit, Mich.], and 2% dextrose [Sigma]) at 30°C and harvested by centrifugation. The blastospores were washed twice in phosphate-buffered saline (PBS) and resuspended in PBS at the required density. For experimental infections, A/J and C57BL/6J mice were injected via the tail vein with 200 μl of a suspension of 3 × 105 C. albicans blastospores in PBS. Mice were closely monitored for clinical signs such as lethargy, loss of appetite, hunched back, and ruffled fur. Mice exhibiting extreme lethargy were deemed moribund and were euthanized.
For determination of fungal loads in target organs at predetermined times, organs were removed aseptically and homogenized in 5 ml of PBS. Then 50 μl of an appropriate dilution was plated on Sabouraud broth-agar plates containing 0.35 mg of chloramphenicol per liter. For experiments involving heat-killed C. albicans, a 5-ml overnight culture was divided into two tubes, which were centrifuged to pellet the blastospores; 2.5 ml of PBS was added to each pellet. One of the two blastospore suspensions was heated at 70°C for 1 h, while the other was stored at 4°C. At the end of the 1-h period, both samples were washed and resuspended in PBS at the desired density. For intraperitoneal injections, 108 heat-killed Candida organisms were injected in 1 ml of PBS. Sixteen hours later, the peritoneal cavity was rinsed with 5 ml of sterile PBS. The peritoneal exudate was clarified by centrifugation, and the peritoneal fluid was stored at −80°C until used for cytokine level determination.
Flow cytometry.
Spleens were removed from control and C. albicans-infected mice 24 h postinfection, and single-cell suspensions were prepared in PBS under aseptic conditions. Cells were collected by centrifugation (350 × g, 10 min), and erythrocytes were lysed with 0.175 M NH4Cl followed by one wash in PBS supplemented with 2% fetal bovine serum. Spleen cells at a density of 106 cells/ml were incubated with R-phycoerythrin-conjugated rat anti-mouse Ly-6G (Gr-1) and Ly-6C monoclonal antibody (clone RB6-8C5; BD Biosciences, Mississauga, Canada) according to the manufacturer's instructions. Excess antibody was removed by washing the cells once with PBS. Specific antibody binding was measured as total fluorescence of the cell population compared to the binding of a phycoerythrin-labeled isotypic negative control (Cedarlane Laboratories Limited, Hornby, Ontario, Canada). All flow cytometry was performed on an Epics XL flow cytofluorometer equipped with a 15-mW argon laser operated at 488 nm as an excitation source (Beckman Coulter, Fullerton, Calif.). The analysis was done on the live cell population based on propidium iodide dye exclusion and the forward scatter signal with a 488-nm dichroic long-pass filter and a 645-nm dichroic long-pass filter plus a 620-nm band pass filter to discriminate the propidium iodide fluorescence.
Evaluation of kidney function.
At predetermined time intervals, mice were exsanguinated by cardiac puncture under anesthesia, and blood was collected in microtubes with separation gel (Sarstedt, Montreal, Canada). Serum was isolated by centrifugation and stored at −20°C until used for blood urea nitrogen (BUN) determination. A commercially available kit that allows quantitative urease/Berthelot determination was used to measure BUN levels (Sigma).
Cytokine detection.
The levels of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) in serum were determined with two-site sandwich assays with commercially available enzyme-linked immunosorbent assay (ELISA) kits (BD Biosciences). Additional determination of 32 cytokines and chemokines was carried out on serum and peritoneal exudates with RayBio mouse cytokine array II membranes (Cedarlane). Incubation, washes, and exposure to film were done according to the instructions of the manufacturer. Briefly, the arrays were blocked at 4°C overnight before being incubated with 400 μl of mouse serum or peritoneal fluid at 20°C for 2 h. The membranes were washed and incubated with biotin-conjugated primary antibody and horseradish peroxidase-conjugated streptavidin according to the instructions of the manufacturer. A semiquantitative analysis of the comparative intensity of the spots was performed with the NIH Image 1.63 software.
Statistical analysis.
The statistical significance of the differences observed for tissue fungal load (Fig. 1B, 2B, and 7A), BUN levels (Fig. 2A), percentage of Gr-1+ cells (Fig. 3), or cytokine levels (Fig. 7B and C) between the various experimental and control samples was assessed with the Mann-Whitney test, which is a nonparametric two-sample ranking analysis.
FIG. 1.

Differential susceptibility of A/J and C57BL/6J mice to C. albicans infection. (A) Survival of A/J and C57BL/6J mice after intravenous infection with C. albicans. Twelve A/J and 25 C57BL/6J (B6) mice were injected intravenously with a C. albicans suspension and subsequently monitored for clinical signs as described in Materials and Methods. The figure shows the percent survival at various intervals postinfection. The figure presents data from one of three independent experiments. All three experiments gave comparable results. (B) Tissue distribution of C. albicans in A/J and C57BL/6J mice 24 h postinfection. Six mice each of the two strains were injected intravenously with 3 × 105 Candida blastospores, and tissue fungal load was determined 24 h postinfection as described in Materials and Methods. Results represent a compilation of two independent experiments. The standard error of the mean is indicated for each sample. Asterisks indicate statistically significant differences (P = 0.001).
FIG. 2.

Kidney pathophysiology in C. albicans-infected A/J and C57BL/6J mice. (A) Kidney function. BUN levels were determined in serum from uninfected and C. albicans-infected A/J and C57BL/6J mice (A/Jinf and B6inf., respectively) collected 24 h postinfection and in serum from C57BL/6J mice collected between days 7 and 30 postinfection (d7-30), when they were moribund (B6m). Isolation of serum and BUN determination were carried out as described in Materials and Methods. The standard error of the mean is indicated for each sample. Numbers in parentheses represent the number of mice analyzed in each experimental group. The results represent a compilation of two independent experiments. Asterisks indicate statistically significant differences (P = 0.001). (B) Kidney fungal load. Mice of the two strains were infected with 3 × 105 Candida blastospores, and kidney fungal load was determined 24 h postinfection (day 1) in both A/J and C57BL/6J mice. Between days 7 and 10 postinfection (days 7 to 10), moribund C57BL/6J (B6m) and healthy C57BL/6J mice were euthanized, and the fungal load was determined as described in Materials and Methods. The standard error of the mean is indicated for each sample. Numbers in parentheses represent the number of mice analyzed in each experimental group. Asterisks indicate statistically significant differences (P = 0.005).
FIG. 7.

C. albicans infection of BcA17 mice. A/J, C57BL/6J, and BcA17 mice were injected with 3 × 105 C. albicans organisms intravenously. Twenty-four hours later, the C. albicans-infected and uninfected A/J, C57BL/6J, and BcA17 mice were euthanized by exsanguination, and the kidneys were harvested. Kidney fungal load (A) and serum cytokine levels (B and C) were determined as described in Materials and Methods. The standard error of the mean is indicated for each sample. Numbers in parentheses indicate the number of mice analyzed in each experimental group. Asterisks indicate statistically significant differences (P = 0.005).
FIG. 3.

Flow cytometric analysis of Gr-1+ cells in the spleens of A/J and C57BL/6J mice during C. albicans infection. Spleens were removed from control and C. albicans-infected A/J and C57BL/6J (A/Jinf. And B6inf., respectively) mice 24 h postinfection. Spleen cell preparations were stained with R-phycoerythrin-conjugated rat anti-mouse Gr-1 antibody. The percentage of cells positive for R-phycoerythrin fluorescence was determined by flow cytometry. The standard error of the mean is indicated for each sample. Numbers in parentheses indicate the number of mice analyzed per experimental group. Asterisks indicate statistically significant differences (P = 0.005).
RESULTS
Phenotypic expression of differential susceptibility to C. albicans infection in A/J and C57BL/6J mice.
Twelve A/J (C5 deficient) and 25 C57BL/6J (C5 sufficient) mice were infected with 3 × 105 Candida blastospores intravenously, and survival was recorded over time. Mice were considered moribund when extremely lethargic and were therefore euthanized as per the guidelines of the Canadian Council of Animal Care. Results from a representative experiment are shown in Fig. 1A.
A/J mice demonstrated acute sensitivity, with 100% of animals succumbing within 24 h following infection. By contrast, C57BL/6J mice are much more resistant, do not start dying until day 7, and succumb to infection over a protracted period from day 7 to day 30, with 32% of the infected mice showing no clinical signs of disease 30 days postinfection. The extent of replication of C. albicans in the heart, brain, lung, liver, kidney, and spleen was also determined in A/J and C57BL/6J mice. For this, six animals of each strain were infected intravenously with 3 × 105 Candida blastospores, and 24 h later, animals were sacrificed and CFU were determined on organ homogenates (Fig. 1B). A/J mice showed higher fungal loads in the heart, lung, liver, and kidney compared to C57BL/6J mice, and this was significantly higher than the fungal load in the corresponding tissues in C57BL/6J mice by a factor varying between 10-fold (liver) and 500-fold (heart) (statistical significance, P = 0.001). Kidneys from A/J mice showed the highest fungal load. Active replication was also noted in the brain and spleen, although it was similar in A/J and C57BL/6J mice (Fig. 1B). These results show that susceptibility of A/J mice is associated with increased fungal replication in target organs very early during infection.
Kidney pathophysiology in A/J and C57BL/6J mice.
Since A/J mice display a high fungal load in the kidney (compared to C57BL/6J mice), kidney pathophysiology was examined further in A/J and C57BL/6J mice. Kidney function was assessed by measuring BUN (Fig. 2A). Surprisingly, BUN levels were similarly low in both control and moribund A/J mice, suggesting that kidney function was not compromised in C. albicans-infected A/J mice at the time of death (24 h postinfection). In sharp contrast, BUN levels in most moribund C57BL/6J mice (n = 20) were remarkably elevated in comparison to the uninfected controls (P = 0.001) and moribund A/J mice (P = 0.001), suggesting kidney dysfunction in infected C57BL/6J mice.
Impaired kidney function in moribund C57BL/6J mice was associated with high fungal loads, which in some cases (n = 7) were at least 10 times greater than those recorded for moribund A/J mice (B6m, P = 0.005; Fig. 2B). In addition, determination of kidney fungal load at days 7 to 10 in several C57BL/6J mice displaying no external signs of disease (B6, Fig. 2B) showed values similar to those seen in moribund A/J mice (log CFU, ≈5.5 to 6.0; P ≈ 0.005). These results indicate that moribund C57BL/6J mice have higher fungal loads than moribund A/J mice. Moreover, at similar fungal loads, several C57BL/6J mice were symptom-free, while A/J mice were succumbing to infection.
Histological examination of kidney sections from moribund A/J mice showed C. albicans colonies detectable as periodic acid-Schiff stain-positive thread-like structures. Such structures were absent from kidney sections of infected C57BL/6J mice examined at 24 h (data not shown). In addition, normal tubular and cellular architecture in the cortex and medulla was evident in kidney sections from both A/J and C57BL/6J mice. By contrast, kidney sections from moribund C57BL/6J mice (identified at days 7 to 30) showed gross abnormalities, including destruction of tissue organization and massive cellular infiltration (data not shown). Together, these results strongly suggest that the abrupt death of A/J mice appears to have a different pathological etiology from that of C57BL/6J mice, which clearly succumb to C. albicans infection due to kidney failure.
Complement-dependent activities in C. albicans-infected A/J and C57BL/6J mice.
To gain further insight into the mechanism of C5-associated premature death of A/J mice, complement-dependent granulocyte recruitment was evaluated. We used flow cytometry to examine cellular mobilization of neutrophils in the spleens of C. albicans-infected A/J and C57BL/6J mice (Fig. 3). Nucleated spleen cells were obtained prior to and 24 h after infection, incubated with phycoerythrin-conjugated anti-Gr-1 antibody, and analyzed by flow cytometry. Although there appears to be a small difference in the number of Gr-1+ cells in the uninfected mice from the two strains, the difference was not statistically significant. Twenty-four hours postinfection, there was a doubling of the number of Gr-1+ cells in the spleens of C57BL/6J mice (P = 0.005), while in the spleens of A/J mice, the number of Gr-1+ cells remained unchanged. Therefore, despite the presence of equivalent fungal loads in the spleens of A/J and C57BL/6J mice at 24 h (Fig. 1B), A/J mice cannot recruit neutrophils at that site, a finding suggestive of an altered inflammatory response.
Cytokine response in A/J and C57BL/6J mice.
The altered recruitment of inflammatory cells observed in the spleens of infected A/J mice prompted us to measure the levels of circulating cytokines associated with host inflammatory response, namely TNF-α, IL-12, and IL-10. Cytokine levels were measured by ELISA in serum from A/J and C57BL/6J mice obtained either prior to or at different times following C. albicans infection (Fig. 4). TNF-α was below the detection level in both mouse strains prior to infection. By contrast, TNF-α levels in moribund A/J mouse serum (24 h, n = 3) were significantly higher than in moribund C57BL/6J mouse serum (days 7 to 20, n = 4). Elevated TNF-α did not appear to be a nonspecific consequence of the moribund status of A/J mice, as similarly elevated levels were detected 19 h postinfection (Fig. 4; n = 4), when A/J mice do not show severe clinical signs. While IL-12 and IL-10 were not detectable in the serum from either strain either before or after infection (data not shown), high TNF-α levels suggest an exaggerated inflammatory response in A/J mice. In contrast, a more modest elevation of TNF-α levels was noted in C57BL/6J mice. However, these levels remained constant throughout the infection, both at early time points (19 and 24 h) and in moribund animals (B6m, days 7 to 20).
FIG. 4.

TNF-α levels in the circulation of C. albicans-infected A/J and C57BL/6J mice. Mice of the two strains were infected with 3 × 105 Candida blastospores and exsanguinated at the indicated times. TNF-α levels were determined in serum from control and C. albicans-infected mice as described in Materials and Methods. The standard error of the mean is indicated for each sample. Numbers in parentheses represent the number of mice analyzed in each experimental group.
A more exhaustive survey of 32 cytokines and chemokines was carried out in the serum of control and C. albicans-infected A/J and C57BL/6J mice with a commercial blot array (see Materials and Methods) (Fig. 5). In control serum, only soluble TNF receptor I (s TNFRI) and tissue inhibitor of metalloproteinases 1 (TIMP-1) were detected in both strains. Levels of both cytokines were similar in the two strains (A/J versus C57BL/6J = 1.4-fold for sTNFRI, and A/J versus C57BL/6J = 0.5-fold for TIMP-I) and increased to similar extents in A/J and C57BL/6J mice 24 h postinfection (A/J versus C57BL/6J = 1.3-fold for sTNFRI, and A/J versus C57BL/6J = 0.9-fold for TIMP-I). Infection induced a number of other cytokines in plasma, the levels of which were consistently higher in A/J than in C57BL/6J mice, most notably, monocyte chemotactic protein 1 (MCP-1), MCP-5, macrophage inflammatory protein 2 (MIP-2), eotaxin, and IL-6, which were all elevated (3.9-, 2.3-, 2.2-, 2.0-, and 1.8-fold, respectively) in A/J compared to C57BL/6J mouse serum. The differences in levels of granulocyte colony-stimulating factor and KC, also known as small inducible cytokine subfamily B member 1, were more modest (1.4- and 1.5-fold, respectively). This observation suggests an unusual inflammatory response in A/J mice that resembles an allergic response.
FIG. 5.

Cytokine profile in the circulation of C. albicans-infected A/J and C57BL/6J mice. Four mice each of the two strains were injected intravenously with 3 × 105 Candida blastospores and exsanguinated 24 h postinfection. Serum was isolated and pooled for each of the four experimental groups, control and C. albicans-infected A/J and C57BL/6J mice. Four hundred microliters of serum was used to determine the levels of cytokines and chemokines with the RayBio mouse cytokine array II membranes as described in Materials and Methods. The figure presents one of three experiments, all of which gave similar results. GCFS, granulocyte colony-stimulating factor.
To distinguish between intrinsic qualitative differences in the cytokine response of A/J and C57BL/6J mice as opposed to quantitatively different responses elicited by different fungal loads present at the time of analysis (Fig. 1B), cytokine response was also examined in the peritoneal cavity following injection of heat-killed C. albicans (Fig. 6). The cytokine profiles observed under these conditions were strikingly different in the two strains (Fig. 6). The peritoneal exudate of A/J mice contained significantly higher levels of MCP-5 (32-fold), MCP-1 (25-fold), and IL-6 (24-fold) compared to those in the peritoneal exudates of C57BL/6J mice, where these cytokines were present at the limit of detection. Similarly, IL-12 and granulocyte colony-stimulating factor were clearly detectable only in A/J peritoneal fluid, making it difficult to evaluate relative levels by densitometry measurements. The difference in levels of vascular endothelial growth factor (VEGF; 16-fold) and RANTES (7.5-fold) was less striking. 6Ckine (a chemokine with six cysteines) and TIMP-1 were present at comparable levels (1.3- and 1.1-fold, respectively) in exudates from both strains, whereas MIP-2 and cutaneous T-cell-attracting chemokine (CTACK) were slightly (2.5- and 2.3-fold, respectively) higher in A/J than C57BL/6J mice. Together, these experiments show that mice of the A/J and C57BL/6J strains mount very different cytokine responses following exposure to virulent or heat-killed C. albicans.
FIG. 6.
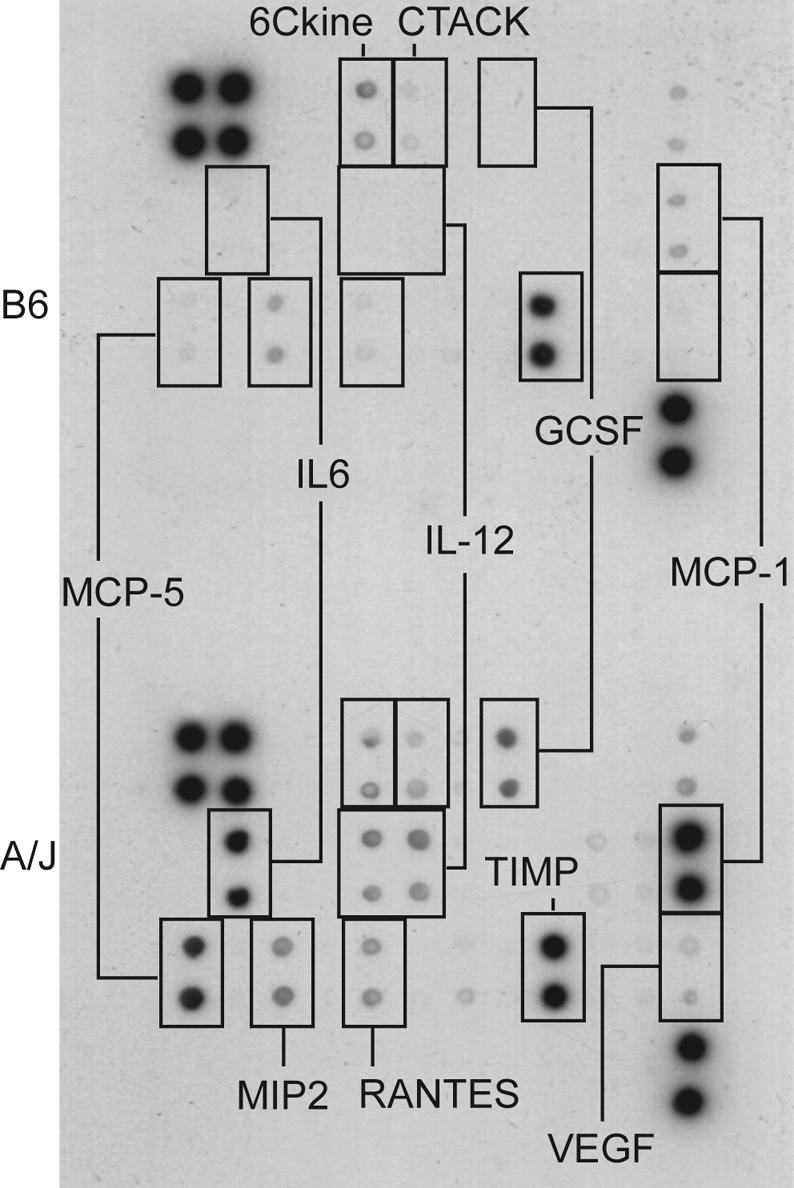
Cytokine profile in the peritoneal exudate of A/J and C57BL/6J mice injected intraperitoneally with heat-killed C. albicans blastospores. Four mice each of the two strains were injected intraperitoneally with 108 heat-killed Candida blastospores in 1 ml of PBS. Sixteen hours postinjection, a peritoneal lavage was performed with 5 ml of PBS. Four hundred microliters of peritoneal fluid was used to determine the levels of cytokines and chemokines with the RayBio mouse cytokine array II membranes as described in Materials and Methods. The figure presents results from one of two experiments, both of which gave similar results.
Studies in recombinant congenic strain BcA17.
A/J and C57BL/6J mice show major interstrain differences in the pathophysiology of C. albicans infection and in host response to C. albicans. This could be a direct cause of C5 deficiency in A/J mice or could be determined by additional genetic factors acting alone or in combination with the C5 deficit. These hypotheses were distinguished by phenotyping a recombinant congenic strain, BcA17 (Fig. 7 and 8), referred to as BcA70 by Fortin et al. (12), which belongs to the AcB/BcA set of 37 recombinant congenic strains recently derived by systematic inbreeding from a double backcross (N3) between A/J and C57BL/6J parents (12). In this breeding scheme, each strain contains a small amount (12.5%) of DNA from one parent fixed as a set of discrete congenic segments on the background (87.5%) of the other parent. BcA17 mice have the genetic background of C57BL/6J mice but contain a small chromosome 2 congenic fragment derived from A/J mice, including a mutant allele at the C5 locus (12).
FIG. 8.

Circulating cytokines in BcA17 mice. Six A/J, C57BL/6J, and BcA17 mice each were injected with 3 × 105 C. albicans intravenously (A/J inf., C57BL/6J inf, and BcA17 inf., respectively). Twenty-four hours later, the C. albicans-infected and six uninfected BcA17 mice were euthanized by exsanguination. Serum cytokine profiles were determined with the RayBio mouse cytokine array II membranes as described in Materials and Methods. The figure presents one of two experiments, both of which gave similar results.
BcA17 mice were found to be susceptible to C. albicans infection and demonstrated fungal loads at 24 h postinfection that were similar to those found in A/J mice but clearly different from those measured in C57BL/6J mice (P = 0.005) (Fig. 7A). BcA17 also showed the typical A/J-like response to C. albicans with respect to elevated plasma levels of TNF-α and IL-6 cytokines (Fig. 7B and C), significantly elevated in comparison to levels in C57BL/6J mice (P = 0.005). Additional comparisons of cytokine profiles in the plasma of C. albicans-infected C57BL/6J, A/J, and BcA17 mice with array blots clearly showed the diagnostic A/J-like profile, consisting of elevated IL-6, eotaxin, granulocyte colony-stimulating factor, MIP-2, and KC (Fig. 8). Together, these results demonstrate that the host response of A/J mice to C. albicans can be recapitulated in BcA17 mice, confirming that C5 deficiency is indeed responsible for the unique pathophysiology and host response to C. albicans observed in this strain.
DISCUSSION
As opposed to the C57BL/6J mouse strain, the A/J mouse strain carries a loss-of-function mutation in the structural gene for the C5 component of the complement pathway (29, 30). Studies by our group (this article) and others on inbred strains (3) and of informative segregating F2 mice have shown that C5 deficiency imparts susceptibility to fungal infections in the A/J strain (39a), which translates into extreme susceptibility, resulting in uniform death of the infected mice within 24 h. By contrast, C5-sufficient C57BL/6J mice are considerably more resistant, with only a fraction of infected animals succumbing over a 3- to 4-week period.
To ensure that it was in fact C5 deficiency and not other strain-dependent differences that were responsible for the phenotypes observed, we employed a BcA recombinant congenic strain that allowed us to evaluate the consequence of C5 deficiency in a C57BL/6J genetic background. Our results show clearly that all the phenotypes studied (high kidney fungal load and cytokine profile, including high TNF-α) are linked to C5 deficiency.
In the present study, the pathogenesis and parameters of the host response to C. albicans infection have been characterized in acutely infected A/J and C57BL/6J mice. Overall, we observed completely different pathophysiologies of infection and host response to C. albicans in the two mouse strains. In C57BL/6J mice, we observed fungal replication over a 2- to 3-week period in a number of organs, especially the kidney. Replication results in high fungal loads, robust inflammatory response in situ (including recruitment of Gr-1+ cells), and major tissue damage at the time of death. Morbidity is an apparent result of kidney failure. By contrast, A/J mice die very quickly after infection, and death does not seem to be linked to loss of kidney function. Rather, A/J mice show a major “dysfunctional” inflammatory response revealed by altered cytokine profiles in response to live or heat-killed C. albicans.
Since membrane attack complex-mediated lysis of pathogens is an important complement activity known to be abolished by C5 deficiency, the fungistatic and fungicidal activity of plasma from A/J and C57BL/6J mice was compared. Plasma was found to have potent fungistatic activity, but no major differences were observed between A/J and C57BL/6J mice (data not shown). These findings are consistent with previously published reports showing that, although pathogenic fungi can activate the complement pathway (24), the direct killing of Candida organisms by complement in vitro has remained difficult to demonstrate (28). In addition, we were unable to detect major differences in the clearance of C. albicans from blood, with all organisms cleared from A/J and C57BL/6J blood 1 h following intravenous injection (data not shown), in agreement with previous reports (44).
Another well-recognized role of C5a is as a chemoattractant for inflammatory cells, in particular for neutrophils (9, 15). Thus, C5a deficiency could be manifested as reduced recruitment of neutrophils (16). In the present study, we noted that Gr-1+ cell mobilization in the C5-deficient A/J strain was reduced compared to that in the C57BL/6J strain (Fig. 3). This observation is consistent with the paucity of detectable cellular infiltration in kidney sections of moribund A/J mice despite the presence of Candida hyphae (data not shown). Higher levels of the circulating chemokine MIP-2, a functional homologue of human IL-8 and an important factor in neutrophil recruitment, does not appear to compensate for C5a deficiency in A/J mice. Reduced recruitment of inflammatory cells to the site of infection most likely contributes to the higher fungal load detected in A/J mouse tissues, including the kidney, at the time of death. However, our results show that higher fungal loads in the kidney at 24 h are unlikely to be the cause of death in A/J mice based on the relatively intact kidney architecture noted in A/J mice at that time, relatively intact kidney function (as measured by BUN), and the fact that infected C57BL/6J mice with few external signs of disease show fungal loads greater than those seen in moribund A/J mice.
One intriguing possibility is that complement deficiency may in fact lead to an exaggerated or inappropriate inflammatory response in A/J mice, possibly through a loss of negative feedback regulation of this pathway by C5a. Recent studies of C5a receptor-deficient mouse mutants suggested that C5a and its receptor may be capable of both promoting and reducing the extent of acute inflammation (7, 31). It is of note that C5a and formyl peptide receptors are the only receptors known to mediate heterologous desensitization (8, 13), an important mechanism for the control of inflammation. Therefore, once an inflammatory response has been triggered by C5a or another agent, C5a must play an important role in the subsequent limitation of the response. In our studies of C. albicans infection, we noted higher levels of TNF-α in serum from moribund A/J mice compared to moribund C57BL/6J mice. This result argued against reduced inflammation in A/J mice but was consistent with a “hyperinflammatory” response.
This prompted us to carry out a more comprehensive analysis of the cytokine profile induced by C. albicans infection in the two strains. It was clear from this analysis that the A/J mouse does not suffer from a dampened inflammatory response. Within 24 h of infection, the A/J mouse has higher levels of several cytokines in the circulation. TNF-α was notable by its absence from this list. Since we had already established differences in TNF-α levels in the serum of Candida-infected A/J and C57BL/6J mice by ELISA, this result could have served to validate the use of the cytokine arrays. However, we were unable to detect TNF-α in the serum from either mouse strain with the cytokine array. The explanation probably lies in the concentration in our samples, which was at the detection limit of the array (100 pg/ml). However the cytokine arrays revealed a remarkable difference in the levels of circulating IL-6 between Candida-infected A/J and C57BL/6J mice, and this difference was reproducible by an independent ELISA (IL-6 level in C. albicans-infected A/J and C57BL/6J mice measured 24 h postinfection, 1,593 ± 140 and 428 ± 42.6 pg/ml, respectively).
Striking among the other differences between the two cytokine profiles was the presence of high levels of the MCP subfamily members MCP-1, MCP-5, and eotaxin in A/J mouse circulation. MCP-1, MCP-5, and IL-6 levels were also notably higher in the peritoneal fluid from A/J mice compared to that of C57BL/6J mice, although eotaxin was undetectable in both samples of peritoneal fluid. MCP-1 and MCP-5 have functional homology and are both known for chemotactic recruitment of monocytes/macrophages to the site of inflammation (38). MCP-1 has been shown to activate mast cells and to induce the differentiation of Th0 cells to Th2 (reviewed in reference 19). In Th2-dominant disease models, lack of MCP-1 results in alleviation of clinical symptoms (17, 18, 42).
MCP-5 and eotaxin are eosinophil-mobilizing chemokines implicated in allergic inflammation. In addition to eosinophils, the receptor for eotaxin is present on other cell types, basophils (40), and Th2 T lymphocytes (25, 37).
We also detected low levels of other cytokines such as IL-12, VEGF, and RANTES in the peritoneal fluid of A/J mice but not C57BL/6J mice. Since they are barely detectable in our assay, it is difficult to know whether these represent major differences with the C57BL/6J mice and what the physiological or pathological consequences may be.
The cytokine profile in the circulation of Candida-infected A/J mice has distinct similarities to that observed during sepsis (10). However, there are notable differences in the absolute amounts and in the rapidity of induction of key cytokines such as TNF-α and IL-6 (23, 45). Similarly, high levels of IL-10, an anti-inflammatory cytokine that participates in the immune dysfunction characteristic of sepsis (22, 39), were not observed in Candida-infected A/J mice. Although the observed differences do not rule out the possibility that moribund A/J mice succumb to septicemia, a thorough investigation of the pathological consequences of the observed cytokine profile is necessary to address this issue conclusively. This is the focus of ongoing studies.
A preponderance of a Th2 response has been linked to susceptibility to C. albicans infection (34, 35). However, in our model the Th1/Th2 paradigm cannot fully explain the cytokine profile. For instance, IL-12 is known to direct a Th1 response but is detectable in A/J peritoneal fluid, rather than in C57BL/6J peritoneal fluid. Similarly, IL-4 has a Th2 bias but is present at similar levels in the two strains of mice.
In addition, it is important that IL-6, MCP-1, MCP-5, eotaxin, and MIP-2 are also increased in C5-sufficient C57BL/6J circulation within the first 24 h after the infectious challenge, albeit to lower levels than in C5-deficient A/J mice. These observations are easier to understand in the context of the anti-inflammatory rather than the proinflammatory activities of C5a. Therefore, our studies suggest that in the absence of C5, the inflammatory response lacks sufficient control mechanisms to be protective. Instead, it allows the unabated amplification of an initial response that may represent a default pathway. It is not clear which cell types are implicated in this allergic-type inflammatory response. Examination of the cytokine profile suggests that cells of the monocytic lineage (secreting MCP-1 and MCP-5) and eosinophils (recruited by eotaxin) must contribute to the elevated cytokine/chemokine levels. To identify the changes that are most crucial for morbidity in the Candida-infected A/J mice, it would be of interest to compare their cytokine response to that in AKR mice, a C5-deficient strain that does not succumb to C. albicans infection as readily as does the A/J mouse (3).
In conclusion, therefore, this study provides an exhaustive description of the pathogenesis of candidiasis in two inbred strains of mice differing in their C5 status. This allows an appreciation of the less-studied role of C5a as an anti-inflammatory factor.
Acknowledgments
We thank Edwin Chang for help in statistical analysis and Suzie Bergeron, Ann-Marie Boyer, and Nadine Jabbour for technical assistance.
This work was supported by the Genomics and Health Initiative of the National Research Council of Canada.
Editor: T. R. Kozel
Footnotes
This is National Research Council publication number 37728.
REFERENCES
- 1.Ashman, R. B. 1998. Candida albicans: pathogenesis, immunity and host defense. Res. Immunol. 149:281-288. [DOI] [PubMed] [Google Scholar]
- 2.Ashman, R. B. 1998. A gene (Carg1) that regulates tissue resistance to Candida albicans maps to chromosome 14 of the mouse. Microb. Pathog. 25:333-335. [DOI] [PubMed] [Google Scholar]
- 3.Ashman, R. B., E. M. Bolitho, and J. M. Papadimitriou. 1993. Patterns of resistance to Candida albicans in inbred mouse strains. Immunol. Cell Biol. 71:221-225. [DOI] [PubMed] [Google Scholar]
- 4.Ashman, R. B., A. Fulurija, and J. M. Papadimitriou. 1997. Evidence that two independent host genes influence the severity of tissue damage and susceptibility to acute pyelonephritis in murine candidiasis. Microb. Pathog. 22:187-192. [DOI] [PubMed] [Google Scholar]
- 5.Ashman, R. B., A. Fulurija, and J. M. Papadimitriou. 1998. A second Candida albicans resistance gene (Carg2) regulates tissue damage, but not fungal clearance, in sub-lethal murine systemic infection. Microb. Pathog. 25:349-352. [DOI] [PubMed] [Google Scholar]
- 6.Ashman, R. B., J. M. Papadimitriou, A. Fulurija, K. E. Drysdale, C. S. Farah, O. Naidoo, and T. Gotjamanos. 2003. Role of complement C5 and T lymphocytes in pathogenesis of disseminated and mucosal candidiasis in susceptible DBA/2 mice. Microb. Pathog. 34:103-113. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia, M., A. K. Saluja, V. P. Singh, J.-L. Frossard, H.-S. Lee, L. Bhagat, C. Gerard, and M. L. Steer. 2001. Complement factor C5a exerts an anti-inflammatory effect in acute pancreatitis and associated lung injury. Am. J. Physiol. Gastrointest. Liver Physiol. 280:G974-G978. [DOI] [PubMed] [Google Scholar]
- 8.Campbell, J. J., E. F. Foxman, and E. C. Butcher. 1997. Chemoattractant receptor crosstalk as a regulatory mechanism in leukocyte adhesion and migration. Eur. J. Immunol. 27:2571-2578. [DOI] [PubMed] [Google Scholar]
- 9.Chenoweth, D. E., and T. E. Hugli. 1978. Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc. Natl. Acad. Sci. USA 75:3943-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen, J. 2002. The immunopathogenesis of sepsis. Nature 420:885-891. [DOI] [PubMed] [Google Scholar]
- 11.Czermack, B. J., V. Sarma, N. M. Bless, H. Schmal, H. P. Friedl, and P. A. Ward. 1999. In vitro and in vivo dependency of chemokine generation on C5a and TNF-α. J. Immunol. 162:2321. [PubMed] [Google Scholar]
- 12.Fortin, A., E. Diez, D. Rochefort, L. Laroche, D. Malo, G. A. Rouleau, P. Gros, and E. Skamene. 2001. Recombinant congenic strains derived from A/J and C57BL/6J: a tool for genetic dissection of complex traits. Genomics 74:21-35. [DOI] [PubMed] [Google Scholar]
- 13.Foxman, E. F., J. J. Campbell, and E. C. Butcher. 1997. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J. Cell Biol. 139:1349-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank, M. M., and L. F. Fries. 1991. the role of complement in inflammation and phagocytosis. Immunol. Today 12:322-326. [DOI] [PubMed] [Google Scholar]
- 15.Gerard, C., and N. P. Gerard. 1994. C5a anaphylatoxin and its seven transmembrane-segment receptor. Annu. Rev. Immunol. 12:775. [DOI] [PubMed] [Google Scholar]
- 16.Gervais, F., M. Stevenson, and E. Skamene. 1998. Genetic control of resistance to Listeria monocytogenes: regulation of leukocyte inflammatory responses by the Hc locus. J. Immunol. 132:2078-2083. [PubMed] [Google Scholar]
- 17.Gonzalo, J.-A., C. M. Lloyd, D. Wen, J. P. Albar, T. N. C. Wells, A. Proudfoot, C. Martinez-A., M. Dorf, T. Bjerke, A. J. Coyle, and J.-C. Gutierrez-Ramos. 1998. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J. Exp. Med. 188:157-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu, L., S. C. Tseng, R. M. Horner, C. Tam, M. Loda, and B. J. Rollins. 2000. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature 404:407-411. [DOI] [PubMed] [Google Scholar]
- 19.Gu, L., S. C. Tseng, and B. J. Rollins. 1999. Monocyte chemoattractant protein-1. Chem. Immunol. 72:7-29. [DOI] [PubMed] [Google Scholar]
- 20.Hector, R. F., J. F. Domer, and E. W. Carrow. 1982. Immune responses to Candida albicans in genetically distinct mice. Infect. Immun. 38:1020-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herzenberg, L. A., D. K. Tachibana, L. A. Herzenberg, and L. T. Rosenberg. 1963. A gene locus concerned with hemolytic complement in Mus musculus. Genetics 48:711-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hildebrand, F., H.-C. Pape, P. Hoevel, C. Krettek, and M. van Griensven. 2003. The importance of systemic cytokines in the pathogenesis of polymicrobial sepsis and dehydroepiandrosterone treatment in a rodent model. Shock 20:338-346. [DOI] [PubMed] [Google Scholar]
- 23.Jaimes, E. A., D. Del Castillo, M. S. Rutherford, and L. Raij. 2001. Countervailing influence of tumor necrosis factor-α and nitric oxide in endotoxemia. J. Am. Soc. Nephrol. 12:1204-1210. [DOI] [PubMed] [Google Scholar]
- 24.Kozel, T. R. 1996. Activation of the complement system by pathogenic fungi. Clin. Microbiol. Rev. 9:34-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lloyd, C. M., T. Delany, T. Nguyen, J. Tian, A. C. Martinez, A. J. Coyle, and J.-C. Gutierrez-Ramos. 2000. CC chemokine receptor CCR3/eotaxin is followed by CCR4/monocyte-derived chemokine in mediating pulmonary T helper lymphocyte type 2 recruitment after serial antigen challenge in vivo. J. Exp. Med. 191:265-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyon, F. L., R. F. Hector, and J. E. Domer. 1986. Innate and acquired immune responses against Candida albicans in congenic B10.D2 mice with deficiency of the C5 complement component. J. Med. Vet. Mycol. 24:359-367. [DOI] [PubMed] [Google Scholar]
- 27.Marquis, G., S. Montplaisir, M. Pelletier, P. Auger, and W. S. Lapp. 1988. Genetics of resistance to infection with Candida albicans in mice. Br. J. Exp. Pathol. 69:651-660. [PMC free article] [PubMed] [Google Scholar]
- 28.Morelli, R., and L. T. Rosenberg. 1971. Role of complement during experimental Candida infection in mice. Infect. Immun. 3:521-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson, U. R., and H. J. Muller-Eberhard. 1967. Deficiency of the fifth component of complement in mice with an inherited complement defect. J. Exp. Med. 125:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ooi, Y. M., and H. R. Colten. 1979. Deficiency of the fifth component of complement in mice with an inherited complement defect. Nature 282:207-208.492335 [Google Scholar]
- 31.Osaka, H. 1999. Expression of C5a receptor in mouse brain: role in signal transduction and neurodegeneration. Neuroscience 88:1073-1082. [DOI] [PubMed] [Google Scholar]
- 32.Pfaller, M. A. 2000. Bloodstream infections due to Candida species: SENTRY antimicrobial surveillance program in North America and Latin America. Antimicrob. Agents Chemother. 44:747-751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhodes, J. C., L. S. Wicker, and W. J. Urba. 1980. Genetic control of susceptibility to Cryptococcus neoformans in mice. Infect. Immun. 29:494-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romani, L. 1999. Immunity to Candida albicans: Th1, Th2 cells and beyond. Curr. Opin. Microbiol. 2:363-367. [DOI] [PubMed] [Google Scholar]
- 35.Romani, L. 2004. Immunity to fungal infections. Nat. Rev. Immunol. 4:1-13. [DOI] [PubMed] [Google Scholar]
- 36.Romani, L. 2002. Immunology of invasive candidiasis, p. 223-241. In R. A. Calderone (ed.), Candida and candidiasis. ASM Press, Washington, D.C.
- 37.Sallusto, F., C. R. Mackay, and A. Lanzavecchia. 1997. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science 277:2005-2007. [DOI] [PubMed] [Google Scholar]
- 38.Sarafi, M. N., E. A. Garcia-Zepeda, J. A. MacLean, I. F. Charo, and A. D. Luster. 1997. Murine monocyte chemoattractant protein (MCP-5): a novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med. 185:99-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sherry, R. M., J. L. Cue, J. K. Goddard, J. B. Paramore, and J. T. di Piro. 1996. Interleukin-10 is associated with the development of sepsis in trauma patients. J. Trauma 40:613-620. [DOI] [PubMed] [Google Scholar]
- 39a.Tuite, A., A. Mullick, and P. Gros. Genetic analysis of innate immunity in resistance to C. albicans. Genes Immun., in press. [DOI] [PubMed]
- 40.Ugguccioni, M., C. R. Mackay, B. Ochensberger, P. Loetscher, S. Rhis, G. J. LaRosa, P. Rao, P. D. Ponath, M. Baggiolini, and C. A. Dahinden. 1997. High expression of the chemokine receptor CCR3 in human blood basophils. Role in activation by eotaxin, MCP-4 and other chemokines. J. Clin. Investig. 100:1137-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward, P. A. 2004. The dark side of C5a in sepsis. Nat. Rev. Immunol. 4:133-142. [DOI] [PubMed] [Google Scholar]
- 42.Warmington, K. S., L. Boring, J. H. Ruth, J. Sonstein, C. M. Hogaboam, J. L. Curtis, S. L. Kunkel, I. R. Charo, and S. W. Chensue. 1999. Effect of C-C chemokine receptor 2 (CCR2) knockout on type-2 (schistosomal antigen-elicited) pulmonary granuloma formation: analysis of cellular recruitment and cytokine responses. Am. J. Pathol. 154:1407-1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wenzel, R. P. 1995. Nosocomial candidiasis: risk factors and attributable mortality. Clin. Infect. Dis. 20:1531-1534. [DOI] [PubMed] [Google Scholar]
- 44.Willcox, M. D. P., B. C. Webb, A. Thakur, and D. W. S. Harty. 1998. Interactions between Candida species and platelets. J. Med. Microbiol. 47:103-110. [DOI] [PubMed] [Google Scholar]
- 45.Yan, J.-J., J.-S. Jung, J. E. Lee, J. Lee, S.-O. Huh, H.-S. Kim, K. C. Jung, J.-Y. Cho, J. S. Nam, H. W. Suh, Y.-H. Kim, and D.-K. Song. 2004. Therapeutic effects of lysophosphatidylcholine in experimental sepsis. Nat. Med. 10:161-167. [DOI] [PubMed] [Google Scholar]