

Abstract
Phosphorylation of the p53 tumor suppressor at Ser20 (murine Ser23) has been proposed to be critical for disrupting p53 interaction with its negative regulator, MDM2, and allowing p53 stabilization. To determine the importance of Ser23 for the function of p53 in vivo, we generated a mouse in which the endogenous p53 locus was targeted to replace Ser23 with alanine. We show that, in mouse embryonic fibroblasts generated from Ser23 mutant mice, Ser23 mutation did not dramatically reduce IR-induced p53 protein stabilization or p53-dependent cell cycle arrest. However, in Ser23 mutant thymocytes and in the developing cerebellum, p53 stabilization following IR was decreased and resistance to apoptosis was observed. Homozygous Ser23 mutant animals had a reduced lifespan, but did not develop thymic lymphomas or sarcomas that are characteristic of p53−/− mice. Instead, Ser23 mutant animals died between 1 and 2 years with tumors that were most commonly of B-cell lineage. These data support an important role for Ser20/23 phosphorylation in p53 stabilization, apoptosis and tumor suppression.
Keywords: apoptosis, Chk2, lymphoma, p53, phosphorylation
Introduction
The p53 tumor suppressor becomes stabilized and activated in response to diverse cellular stresses such as DNA damage, hypoxia, ribonucleotide depletion and oncogene activation (reviewed in Levine, 1997; Giaccia and Kastan, 1998). Normally, p53 is present at low levels in the cell due to its rapid turnover; however, in response to activating signals, p53 becomes stabilized and enriched in the nucleus. p53 is regulated by post-translational modifications including phosphorylation and acetylation events that contribute both to the stabilization of p53 and to the conversion of p53 from a latent to an active transcription factor. Activation of p53 can cause either a cell cycle arrest or apoptosis, responses that are largely mediated by activation of p53-responsive target genes. The product of the p53 target gene MDM2 is involved in p53 ubiquitination and destruction via the proteasome, keeping p53 at low levels in unstressed cell (Haupt et al, 1997; Kubbutat et al, 1997). MDM2 also inhibits p53 by binding to the transactivation domain of p53 (Oliner et al, 1993).
Disruption of the negative regulation of p53 by MDM2 is thought to be critical to stabilizing p53, and phosphorylation of p53 has been thought to contribute to the disruption of p53/MDM2 interaction (Shieh et al, 1997; Chehab et al, 1999; Unger et al, 1999a). Human p53 is phosphorylated on multiple N-terminal residues following DNA damage, including serines 6, 9, 15, 20, 33, 37, 46 and threonine 18 (reviewed in Appella and Anderson, 2001). The physiological role of most of these phosphorylation events has not been clearly defined. It has been shown in vivo that the ataxia-telangectasia mutated (ATM) kinase is important for p53 stabilization, as in cells lacking functional ATM there was a delay in p53 stabilization and reduced Ser15 phosphorylation following ionizing radiation (Siliciano et al, 1997). Data from in vitro systems suggest that the phosphorylation of Ser15 of p53 by ATM may be direct (Banin et al, 1998; Canman et al, 1998). However, the functional consequences of Ser15 phosphorylation are controversial, with reports supporting an effect of Ser15 phosphorylation on the p53/MDM2 interaction (Shieh et al, 1997) and others concluding that Ser15 phosphorylation does not block this interaction (Dumaz and Meek, 1999). Phosphorylation of Ser15 has also been implicated in activating p53 for transcriptional transactivation (Dumaz and Meek, 1999). Effects of Ser15 phosphorylation independent of p53 stabilization may be critical in vivo; cells from mice with germline point mutation of Ser15 exhibited normal p53 stabilization, but thymocytes exhibited partially impaired p53-dependent apoptosis (Chao et al, 2003, Sluss et al 2004).
Ser20 of human p53 is also phosphorylated upon DNA damage (Shieh et al, 1999; Unger et al, 1999a), and this phosphorylation has been reported to be critical for p53 stabilization. Of the sites thought to be phosphorylated upon DNA damage, Ser20 and Thr18 (but not Ser15) lie directly in the segment of p53 that interacts with MDM2 (Kussie et al, 1996). Transfection of the p53 Ser20Ala phosphorylation site mutant into various human cell lines completely prevented induction of p53 protein levels in response to gamma or UV radiation (Chehab et al, 1999). Other experiments involving the transfection of exogenous p53 point mutants have also demonstrated defects in p53 stability as well as apoptosis when Ser20 was mutated (Unger et al, 1999a; 1999b). However, similar experiments have shown that multiple N-terminal phosphorylation sites, including Ser15 and Ser20, can be mutated to alanine without dramatic effect on p53 stability (Ashcroft et al, 1999; Blattner et al, 1999). Interpretation of the data from experiments using ectopically expressed p53 is confounded by possible nonphysiological regulation of p53 in this context. That is, the balance between p53 and MDM2, which is likely to be critical for proper p53 regulation, may not be properly achieved when p53 or MDM2 is ectopically expressed. Indeed, in one report in which p53 point mutants were co-transfected with MDM2, the importance of Ser20 mutation for p53 stability differed depending on the amount of p53 transfected (Dumaz et al, 2001). This illustrates the importance of examining p53 phosphorylation sites in vivo at endogenous levels to definitively determine the function and importance of the phosphorylation site. Importantly, while there is ample evidence for phosphorylation of human p53 on Ser20, phosphorylation of the equivalent residue on murine p53 (Ser23) has been less rigorously established due to the lesser quality of phospho-specific antibodies for the mouse.
Chk2 has emerged as a critical regulator of p53 downstream of ATM, and it is phosphorylated and activated by ATM in response to ionizing radiation (Matsuoka et al, 1998; Melchionna et al, 2000). A genetic connection between Chk2 and p53 was revealed by the finding of Chk2 mutations in patients with Li–Fraumeni syndrome (LFS), which predisposes patients to tumors in multiple tissues, including sarcomas, breast and brain tumors (Bell et al, 1999). LFS is normally associated with germline mutations in p53; however, the patients with Chk2 mutation had wild-type germline p53 (Bell et al, 1999). Studies of murine cells lacking Chk2 indicate that it is important for p53 function. Chk2−/− animals show strong impairment in apoptosis in the irradiated nervous system and thymus (Hirao et al, 2000; 2002; Takai et al, 2002). In one study, Chk2−/− thymocytes were completely defective for p53 stabilization following DNA damage (Hirao et al, 2000). However, more recently, germline Chk2−/− animals that showed a mild defect in p53 stabilization and strong defects in activation of p53 target genes were generated (Takai et al, 2002). Ser20 phosphorylation has been proposed to mediate the effects of Chk2 on p53 induction. Chk2 can phosphorylate Ser20 of p53 in vitro (Chehab et al, 2000; Hirao et al, 2000; Shieh et al, 2000). Transfection of a dominant-negative Chk2 inhibited Ser20 phosphorylation on p53 following DNA damage (Chehab et al, 2000). In vitro, Chk2 expression disrupted preformed complexes of p53 and MDM2, except when Ser20 was mutated to alanine (Chehab et al, 2000). However, Chk2 can phosphorylate multiple sites on p53 in vitro, including Ser15, Thr18 and Ser20 as well as undetermined sites in the C-terminus (Shieh et al, 2000); the sites phosphorylated by Chk2 on p53 in vivo have not been rigorously defined. Also, Chk2 may phosphorylate targets other than p53 itself that help to regulate p53 stability. Finally, recent data demonstrate that Chk2 function is not essential for Ser20 phosphorylation, as p53 can be efficiently phosphorylated on Ser20 in Chk2−/− cells (Takai et al, 2002; Jallepalli et al, 2003). In addition to Chk2, Chk1 (Shieh et al, 1999) and PLK3 (Xie et al, 2001) have been reported to phosphorylate Ser20 in vitro. The specific kinase responsible for Ser20 phosphorylation in vivo has yet to be determined.
Owing to many indications that Ser20 phosphorylation is important for p53 function, we sought to determine the effects of preventing phosphorylation at the murine equivalent of this residue (Ser23). We targeted the endogenous p53 locus and mutated Ser23 to Ala, a residue that cannot be phosphorylated. A similar approach was recently described by Wu et al (2002), who also targeted the murine p53 locus to generate Ser23 to Ala mutant ES cells. This group also derived mouse embryonic fibroblasts (MEFs) and thymocytes through blastocyst complementation approaches and concluded from their studies that Ser23 phosphorylation is not important for murine p53 function. Importantly, p53 function was studied under cell culture conditions and germline Ser23 mutant animals were not generated. Here we report the generation of germline Ser23 to Ala mutant mice. Our studies of these mice suggest an important role for Ser23 phosphorylation in p53 response to DNA damage in vivo and in tumor suppression.
Results
Generation of mice with p53 S23A mutation
To generate a mouse with the endogenous serine 23 mutated, we used homologous recombination to introduce a mutation into the p53 genomic locus. The targeting construct contained the Ser23 mutation in exon 2 and a puromycin gene flanked by loxP sites in intron 1 (Figure 1A and B). ES cells were targeted and homologous recombination was confirmed by Southern blotting (Figure 1C, lane 2). Treatment of targeted ES cells by transient transfection with a Cre-expression plasmid led to excision of the puromycin cassette, which was also confirmed by Southern blotting (Figure 1C, lane 3). ES cells from four independently targeted ES lines were injected into blastocysts to generate chimeras. Five chimeras were generated from two of the independently targeted ES lines. Four of these chimeras transmitted the mutant allele (termed S23A) to the germline and their progeny were used in the experiments described. S23A heterozygous mutant mice were intercrossed to obtain S23A homozygotes. These animals were born at Mendelian ratios and had no overt phenotypes at birth (similar to p53 null animals). RT–PCR for p53 was performed on MEFs derived from S23A animals and sequencing of the entire p53 open reading frame demonstrated the presence of the targeted mutation and confirmed that no additional mutations had occurred in the coding sequence of the gene (data not shown). Animals described here (and cells obtained from these) are of mixed (129/sv × C57BL/6) background.
Figure 1.
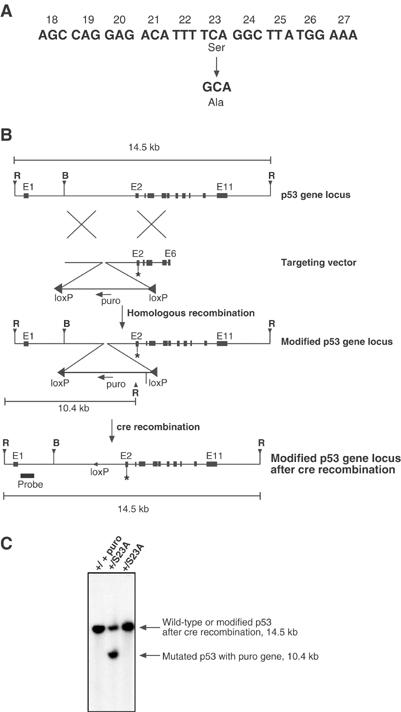
Targeting of endogenous p53 locus to introduce a Ser23 to Ala mutation. (A) The S23A mutation that is introduced by a single-nucleotide substitution is represented. (B) Schematic representation of targeting strategy. The targeting construct introduces the exon 2 point mutation and also introduces into intron 1 a puromycin cassette in the reverse orientation flanked by loxP (◂) sites, and containing an EcoR1 site. The modified p53 locus following homologous recombination is also shown. Following homologous recombination, Cre recombinase was introduced by transient transfection to delete the Puro cassette, leaving a single loxP site. The position of the probe external to the targeting cassette is shown, and the EcoR1-cut sites, internal and external to the cassette, are also indicated. Abbreviations used are: R, EcoR1; B, BamH1; puro, puromycin-resistance gene. (C) Genotyping of ES cell clones that survived puromycin selection. Southern blot analysis was performed on EcoR1-digested genomic DNA. The probe hybridizes to a 14.5 kb EcoR1 fragment from the unmodified locus (+/+). Correctly targeted heterozygous ES cells surviving puromycin selection also show the 10.4 kb EcoR1 fragment (+/S23Apuro). Following the addition of Cre to the targeted ES cell, the Puro cassette is excised (+/S23A).
S23A MEFs stabilize p53 and undergo cell cycle arrest in response to DNA damage
We generated S23A/S23A MEFs from embryonic day 13.5 (E13.5) embryos to determine if Ser23 phosphorylation was required for p53 stabilization. Surprisingly, we found that p53 protein levels could clearly be induced in S23A/S23A MEFs following 5 Gy gamma irradiation, although in some experiments at slightly decreased levels compared to wild-type controls (Figure 2A). Similar results were found with a dose of 10 Gy (not shown). Furthermore, p53 was fully functional in its ability to induce expression of p21 and MDM2 (Figure 2B). p53 null MEFs are resistant to a G1 arrest in response to gamma irradiation (Kastan et al, 1992), and this effect is in large part mediated by p21 (Brugarolas et al, 1995; Deng et al, 1995). In contrast, S23A/S23A MEFs arrested in G1 with no clear defect compared to wild-type cells when assayed 14 h (Figure 2C) or 24 h (data not shown) following gamma irradiation.
Figure 2.
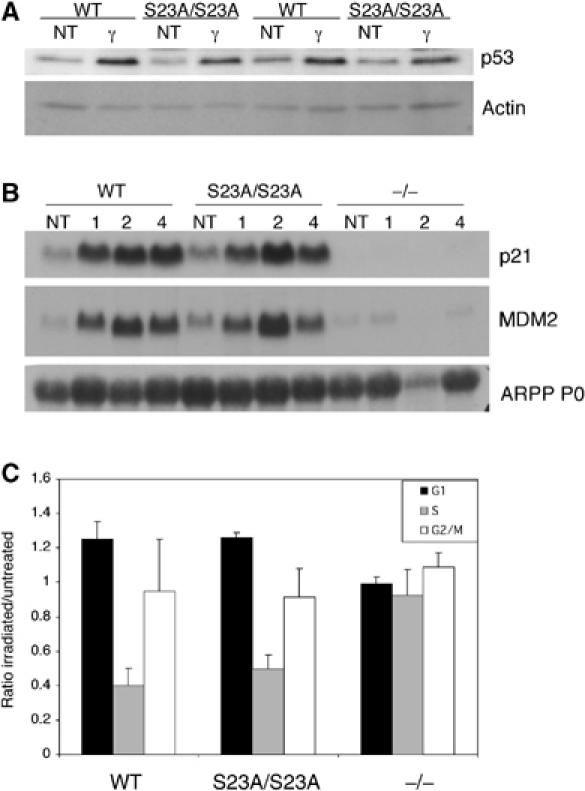
Ser23 phosphorylation is not required for p53-dependent stabilization or cell cycle arrest in MEFs treated with ionizing radiation. (A) Western blot analysis of p53 stabilization 1 h following treatment with 5 Gy IR. Two independent MEF lines are shown. (B) Northern blot analyses of the p53 target genes p21 and MDM2 in WT and S23A/S23A MEFs 1, 2 and 4 h following 5 Gy IR. (C) Cell cycle analysis of S23A/S23A and control MEFs following gamma irradiation. G1, S and G2/M cell populations were sorted by flow cytometry 14 h following treatment with 5 Gy IR. Numbers are relative to those obtained from unirradiated cells. Data are from three experiments and standard deviations are indicated (error bars).
Defective apoptosis in S23A mutant thymocytes
While MEFs respond to gamma irradiation by undergoing p53-dependent cell cycle arrest and not apoptosis, thymocytes undergo apoptosis in response to the same stimulus. p53 null thymocytes are completely resistant to gamma irradiation-induced apoptosis under the experimental conditions assayed (Clarke et al, 1993; Lowe et al, 1993). Thymocyte apoptosis assays were performed on S23A thymocytes to determine if phosphorylation of Ser23 is essential for p53 induction and function in this system. Isolated thymocytes were plated, treated with IR and apoptosis was assessed 6, 12 and 24 h later. S23A/S23A thymocytes were not completely resistant to IR-induced apoptosis, in contrast to p53 null cells. However, they showed an intermediate level of resistance at the 12 and 24 h time points (Figure 3A). This partial resistance was also seen at each dose of radiation tested in a dose–response experiment (Figure 3B). It has been previously shown that the induction of apoptosis in thymocytes is sensitive to the levels of p53 induced, as p53 heterozygotes were partially resistant to apoptosis in this assay (Clarke et al, 1993; Lowe et al, 1993). S23A/+ thymocytes showed an intermediate phenotype between +/+ and S23A/S23A thymocytes (Figure 3A and B), supporting the notion that apoptosis in this assay is sensitive to the dose of p53. In contrast to the resistance following gamma irradiation, no resistance was shown to the p53-independent apoptosis induced by dexamethasone (Figure 3A and B). To determine if the defect in IR-induced apoptosis correlated with decreased p53 protein induction, we isolated thymocytes, irradiated them under cell culture conditions and performed a time course examining p53 stabilization in this tissue. A modest but clear defect in p53 protein levels induced following irradiation was observed (Figure 3C). Examination of p53 protein levels in the thymus following whole-body irradiation in vivo revealed an even stronger difference between control and S23A heterozygous or homozygous mutant cells (Figure 3D). Basal levels of p53 in uninduced thymocytes was low; however, it was apparent that there was decreased basal levels of p53 protein with the Ser23 mutation (Figure 3D). Importantly, mRNA levels of p53 in the thymus were not changed with Ser23 mutation (not shown). The decreased protein level of p53 induced in the thymus following IR, either in vitro or in vivo, suggests that the apoptotic defect is due to decreased p53 stability when Ser23 is mutated. To investigate this further, we examined p53 protein stability following DNA damage. The protein synthesis inhibitor cyclohexamide was added to isolated thymocytes 2 h following treatment with 5 Gy gamma radiation, and p53 protein levels were then followed over time. As shown in Figure 3E, the half-life of p53 in S23A/S23A cells was significantly reduced compared to wild-type p53. S23A/+ cells exhibited an intermediate phenotype. Thus, S23A mutation reduces p53 stability following DNA damage in thymocytes.
Figure 3.
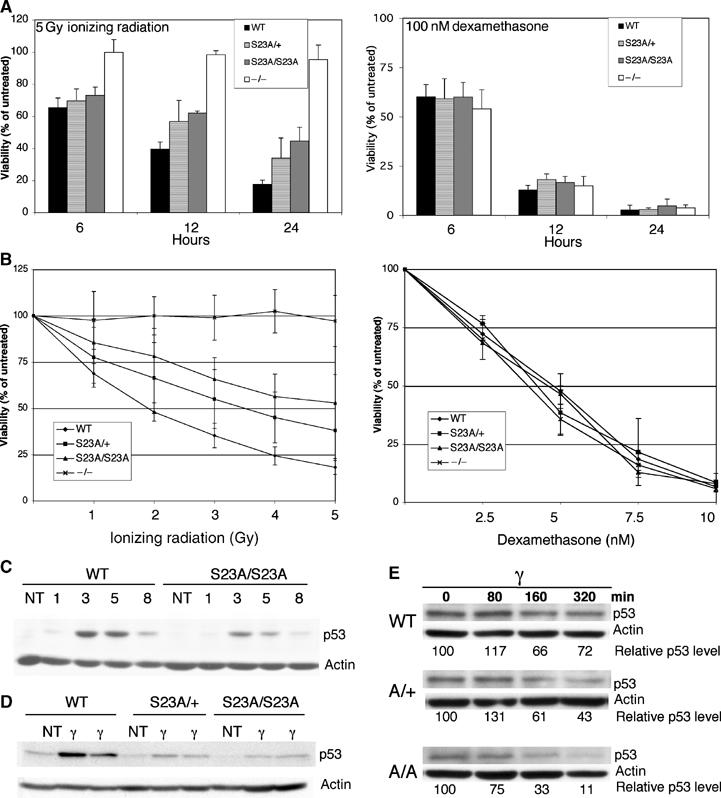
Decreased apoptosis and p53 stabilization in S23A mutant thymocytes following DNA damage. (A) Flow-cytometric analysis of apoptosis in isolated thymocytes irradiated with 5 Gy or treated with 100 nM dexamethasone. Cells were plated at 1 × 106 cells/ml prior to treatments, and apoptosis was assessed at various time points using two-color cytometry following staining of cells with PI and annexin-FITC. Viable cells were both annexin and PI negative. All values were expressed relative to the viability in untreated samples, which was 65–70% at 24 h and did not differ between the genotypes examined. For WT, S23A/+ and S23A/S23A data, littermates were used in each experiment. (B) Thymocyte viability 24 h following varying doses of ionizing radiation, or dexamethasone. For all apoptosis experiments, values represent the averages of five experiments, with error bars indicating the standard deviation. (C) p53 protein levels were determined by immunoblot in isolated thymocytes irradiated in vitro with 5 Gy and then cultured for 1, 3, 5, or 8 h. (D) p53 protein levels were determined by immunoblot in thymus extracts from WT, S23A/+ or S23A/S23A mice that had been irradiated with 5 Gy 5 h before thymus collection. One untreated and two irradiated samples of each genotype are shown. (E) p53 stability in WT, S23A/+ or S23A/S23A isolated irradiated thymocytes. Cyclohexamide was added to inhibit protein synthesis 2 h following 5 Gy irradiation. Band intensities were normalized to actin levels and levels relative to the T=0 point for each genotype are shown. This figure is representative of four experiments.
We also examined apoptosis in the splenic white pulp, which contains both B- and T-lymphocytes and is a site of p53-dependent apoptosis following irradiation in vivo (Komarova et al, 2000). TUNEL assays showed that, 6 h following irradiation with 5 Gy in vivo, there were high levels of apoptosis in both the control and S23A/S23A splenic white pulp without dramatic differences between these genotypes (Figure 4A). By 18-h post-irradiation, overall levels of apoptosis were lower in both the wild-type and S23A/A spleens, facilitating quantification. There was a 3.6-fold decrease in white-pulp apoptosis in S23A/S23A animals versus wild-type controls (P<0.001; Student's t-test) (Figure 4A and B). We performed FACS analyses using the B-cell marker B220 to quantify depletion of splenic B-lymphocytes 24 h following irradiation in vivo. As shown in Figure 4C, while the proportion of B220-positive splenocytes in untreated wild-type and S23A/S23A animals was similar, by 24 h following irradiation the S23A/S23A animals were significantly resistant to loss of the B-cell population (P=0.025; Student's t-test). Thus, Ser23 mutation leads to partial resistance of splenic B-cells to irradiation-induced apoptosis.
Figure 4.
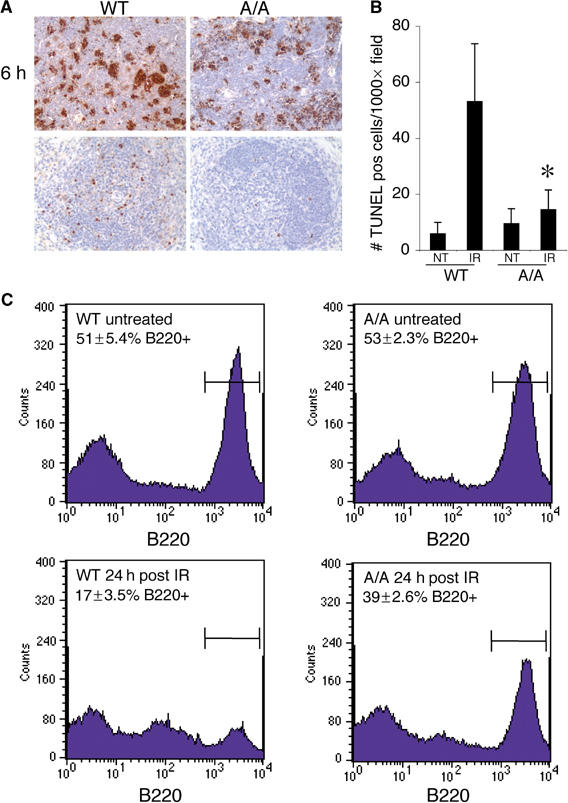
Decreased apoptosis in splenic B cells of S23A/S23A mice exposed to gamma radiation in vivo. (A) TUNEL staining to assess apoptosis in the splenic white pulp 6 h, and 18 h following gamma irradiation in S23A/S23A or control animals. (B) Quantification of TUNEL-positive cells in the splenic white pulp at 18-h time point post-irradiation or in untreated wild-type or S23A/S23A animals. (C) FACs analysis of B220 positivity in wild-type and S23A/S23A splenocytes 24 h post 5 Gy irradiation or from untreated controls in vivo. Percentage of B220-positive cells±s.d. from three independent experiments is indicated.
S23A developing cerebellum is resistant to DNA damage-induced apoptosis
We next examined the effects of S23A mutation on p53 function in another cell population that undergoes p53-dependent apoptosis in vivo. We focused our analysis on gamma radiation-induced apoptosis in the external granule layer in the developing cerebellum, a neuronal population of cells that undergo apoptosis in both a p53- and ATM-dependent manner (Herzog et al, 1998). Mice (5-days-old; P5) were irradiated and we assessed apoptosis 18 h later by TUNEL analysis. As shown in Figure 5A, significantly decreased CNS apoptosis in S23A/S23A animals compared to wild type was observed. Quantification of TUNEL-positive nuclei revealed that there was 3.7-fold more apoptosis in the wild type compared to S23A/S23A cerebellum (Figure 5B). As with thymocytes, the phenotype seen with the point mutant did not recapitulate the degree of resistance observed in p53 null mice (Figure 5A and B). To determine if the defect in apoptosis is due to a defect in p53 stability, we irradiated P5 S23A/S23A, wild-type or p53 null mice and collected the cerebellum 3 h later to examine p53 levels. p53 protein levels were strikingly decreased in the S23A/S23A samples (Figure 5C).
Figure 5.
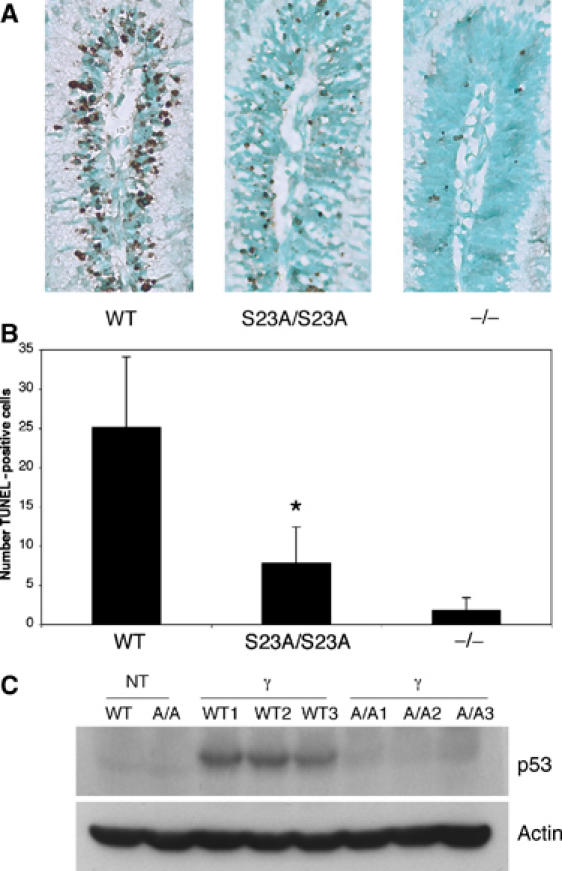
Decreased p53 levels and apoptosis in the developing cerebellum of S23A/S23A mice exposed to gamma radiation. (A) TUNEL staining to assess apoptosis in the external granule layer of P5 developing cerebellum collected 18 h following 4 Gy of gamma radiation. (B) Quantification of apoptosis in external granule layer cells. TUNEL-positive cells were quantified in equivalent areas. Data are from eight S23A/S23A animals, eight WT animals and four p53 null animals. S23A/S23A and WT mice used in this study were littermates. *P=0.001 comparing WT to S23A/S23A. (C) Western blot shows significantly decreased p53 protein levels in S23A/S23A irradiated cerebellum. Postnatal day 5 (P5) mice were treated with 4 Gy IR and their cerebellum was collected 3 h post-irradiation. Three wild-type and three S23A/S23A irradiated samples are shown along with untreated controls.
Tumorigenesis in S23A/S23A animals
We examined whether mutation of Ser23 would contribute to tumor formation by aging cohorts of S23A/S23A and wild-type animals. p53−/− animals die between 4 and 6 months, usually due to thymic lymphoblastic T-cell lymphomas and sarcomas (Donehower et al, 1992; 1995; Jacks et al, 1994), S23A/S23A mice lived to a much longer median survival time of 444 days (Figure 6). Necropsies and histopathological analyses were performed on moribund mice. Surprisingly, none of these animals developed thymic T-cell lymphoblastic lymphoma. In contrast, a wide spectrum of B-cell lineage tumors were found in S23A/S23A animals, classified based on histological criteria as follicular, diffuse large-cell and splenic marginal zone lymphomas, as well as plasmacytomas (Figure 7A–E, Table I). The most common of these tumors was follicular lymphoma. Mice with this category of tumor typically had predominantly nodal disease (often involving the mesenteric lymph node), with less involvement of the spleen. Extranodal tumor involvement in tissues such as kidney and liver was often found. Most lymph nodes affected by these tumors had a vaguely nodular architecture. In some cases, the nodularity was highlighted by immunostaining for plasma cells that are located at the periphery of the nodules (Figure 7A). The infiltrate was composed predominantly of small, irregular lymphocytes (centrocytes) with a variable, but typically small, fraction of larger nucleolated lymphocytes (centroblasts) (Figure 7B). Occasional tumors had higher mitotic rates and increased fractions of large cells. In some cases, plasma cells were abundant both within and outside the infiltrate of lymphoma cells. In the spleen, lymphoma cells largely replaced the white pulp. Follicular lymphomas were positive for the B-cell marker B220 (not shown) and exhibited clonal or oligoclonal IgJH rearrangements confirming their B-cell origin (Figure 7G). We suggest that the plasmacytic cells, which were often atypical in morphology, are part of the clonal tumor. However, we cannot rule out the possibility that the plasmacytic component is a separate lesion as other animals without lymphoma also exhibited plasmacytic hyperplasia. In one animal, plasmacellular proliferation and morphology was overtly cytologically malignant, with extensive invasion of extranodal sites (Figure 7E). This tumor was classified as a malignant plasmacytoma (Table I). Spleens involved by marginal zone lymphoma had a largely preserved architecture, with the periarteriolar lymphoid area cuffed by small-to-intermediate-sized lymphocytes with oblong to slightly folded nuclei and a moderate amount of pale cytoplasm. Numerous plasma cells were located at the periphery of the infiltrate, often extending into splenic the red pulp (Figure 7C and D). Two animals had histiocytic sarcomas, which stained positively for the macrophage marker F480 (not shown) and did not exhibit clonal IgJH rearrangement (Figure 7F and G). We note that both of these cases of macrophage tumors occurred in a background of splenic red pulp hyperplasia, including the myeloid, erythroid and megakaryocytic lineages, suggesting that this hyperplasia may be a pre-malignant lesion. Marked red pulp hyperplasia without histiocytic tumors was also observed in four other S23A/S23A animals that did not have lymphomas. Other nonhematopoietic tumors were also observed in S23A/S23A animals (Table I). During the course of the study, five wild-type mice died, one due to a hemangioblastoma and the other with a hepatoma. Our tumorigenesis study indicates that S23A/S23A animals are prone to tumor development in a cell-type-specific manner.
Figure 6.
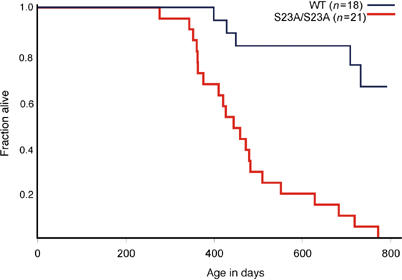
Survival of S23A mutant mice. Kaplan–Meier analysis of tumor-free survival in wild-type and S23A/S23A mice. Animals were killed when moribund, or if tumor burden was apparent.
Figure 7.
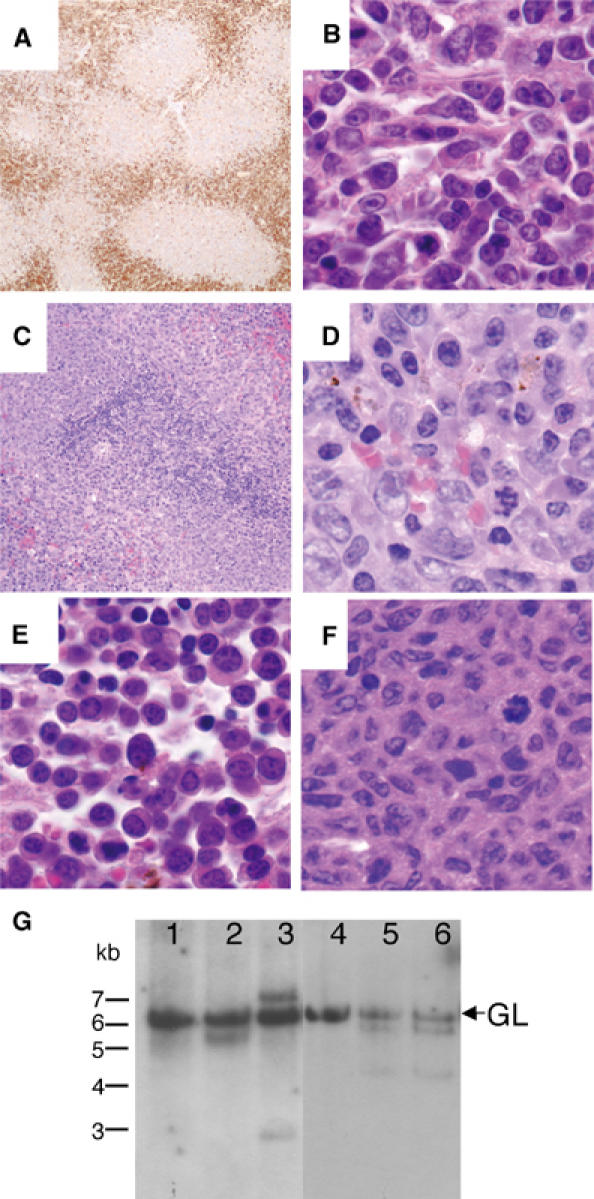
B-cell tumors in S23A mutant mice. (A) Plasma cell immunostaining (anti-Ig kappa light chain) of a follicular lymphoma at 40 × , showing nodular tumor architecture. (B) Follicular lymphoma (H+E 1000 ×). (C) Splenic marginal zone lymphoma (MZL) showing expanded marginal zone with pale eosinophilic cells (H+E 200 ×). (D) High-power view of splenic MZL showing the appearance of cells with plasmacytic morphology (H+E 1000 ×). (E) Malignant plasmacytoma (H+E 1000 ×). (F) Histiocytic sarcoma (H+E 1000 ×). (G) Southern blot analysis of immunoglobulin heavy chain recombination. DNA from S23A/S23A tumors (lanes 2–6) or wild-type lymph nodes (lane 1) was digested with EcoR1 and hybridized with a JH region probe. The 6.2 kb germline band is indicated (GL). (1) Normal lymph node from wild-type mouse; (2) follicular lymphoma; (3) follicular lymphoma; (4) histiocytic sarcoma; (5, 6) follicular lymphoma in different lymph nodes from same S23A/S23A animal. B-cell tumors in lanes 2, 3, 5 and 6 show one or two novel bands indicating tumors are monoclonal with one or both JH alleles rearranged, or biclonal.
Table 1.
Histological classification of spontaneous tumors in S23A/S23A mice
| Tumor type | Number of cases (%) |
|---|---|
| B-cell lineage tumors | 13 (62) |
| Follicular lymphoma | 7 (33) |
| Splenic marginal zone lymphoma | 2 (9.5) |
| Diffuse large B-cell lymphoma | 2 (9.5) |
| Plasmacytoma—well differentiated | 1 (4.8) |
| Malignant plasmacytoma—moderately differentiated | 1 (4.8) |
| Other tumors | |
| Histiocytic sarcoma | 2 (9.5) |
| Hemangiosarcoma | 1 (4.8) |
| Lung adenoma | 1 (4.8) |
| Lung adenocarcinoma | 1 (4.8) |
| Hepatoma | 1 (4.8) |
| Hair matrix tumor | 1 (4.8) |
Discussion
We demonstrate here that mutating Ser23 to alanine affects both p53 stabilization and function. Mutant thymocytes were partially defective in p53-dependent apoptosis in response to ionizing radiation, and strong resistance was observed in the developing cerebellum. These data show in vivo the importance of this residue for p53 function and have helped define the pathway leading to IR-induced apoptosis in these tissues. Our finding that S23A/S23A animals are tumor prone suggests that Ser23 phosphorylation is important for the tumor suppressor function of p53. This is the first report of a p53 phosphorylation site mouse mutant that is prone to spontaneous tumorigenesis.
A previous report of p53 S23A mutant function in targeted murine cells concluded that Ser23 phosphorylation is not important for p53 stabilization and function in various cell types, including ES cells, MEFs and thymocytes (Wu et al, 2002). While we have not found strong effects of this mutation on p53 function in MEFs, we observe significant defects in p53 stabilization and apoptosis in irradiated thymocytes. The study by Wu et al (2002) was different in that germline S23A/S23A animals were not generated, and the thymocytes they studied were generated through a RAG2-deficient blastocyst complementation approach. In addition, this group studied S23A/− cells in comparison to p53+/− controls, while we compared S23A/S23A and S23A/+ cells to wild-type controls. As p53+/− cells exhibit significant resistance to apoptosis compared to wild-type cells (Clarke et al, 1993; Lowe et al, 1993), it is possible that differences in thymocyte apoptosis and p53 stabilization were not found due to the smaller window these authors may have had for detecting a subtle difference. Also, we found the strongest effects of this mutation following irradiation in vivo. This was especially true in the irradiated developing cerebellum, which this group was unable to study, and in tumorigenesis.
Ser23 mutation did not have major effects on p53 stabilization under some conditions. For example, in S23A/S23A MEFs, IR-induced p53 stabilization was not dramatically reduced and p53-dependent cell cycle arrest was intact. This is in contrast to the strong defect in levels of p53 protein induced in the thymus and cerebellum irradiated in vivo. Tissue culture stress, including high oxygen concentrations, may allow p53 to be sensitized to IR-induced stabilization even in the absence of Ser23 phosphorylation. Indeed, in the thymus irradiated in vivo, a stronger defect in induction was observed compared to thymocytes irradiated in vitro (Figure 3C and D).
The defect in apoptosis in the irradiated Ser23 mutant thymus and CNS clearly correlates with defective p53 induction. The Chk2 kinase has been implicated as a Ser23 kinase in response to ionizing radiation and Chk2−/− mice are defective for p53-dependent apoptosis in the irradiated thymus and CNS (Hirao et al, 2000; 2002; Takai et al, 2002). Much stronger resistance to thymocyte apoptosis was found in Chk2−/− cells compared to our findings in S23A/S23A thymocytes, indicating that in these cells Chk2 has effects on p53 that are not mediated by Ser23 phosphorylation. Data from targeted Chk2−/− cells suggested that Chk2−/− thymocytes are defective for p53 stabilization following IR; however, the extent of this effect differed from mild (Takai et al, 2002) to a complete lack of induction (Hirao et al, 2000). Whether the defective apoptosis in Chk2−/− animals is mediated through effects on p53 stabilization is not clear, as defects in transactivation of p53 target genes in Chk2−/− cells have been reported in one study (Takai et al, 2002). In vitro, Chk2 has been reported to phosphorylate multiple sites on p53, including Ser15, Thr18 and Ser37 (Shieh et al, 2000). Chk2 can also phosphorylate undetermined C-terminal sites of p53 mutants lacking the N-terminus, which could mediate effects of Chk2 independent of p53 stabilization (Shieh et al, 2000). It is also possible that Chk2 could affect p53 indirectly. It is apparent that p53 can be efficiently phosphorylated on Ser20/23 in Chk2−/− cells (Takai et al, 2002; Jallepalli et al, 2003), and it remains to be determined how much of the effects of S23 mutation are due to an inability of Chk2 to phosphorylate p53 on this residue. It will be important to determine if older Chk2−/− animals show a tumor spectrum similar to S23A/S23A mice.
It was striking that S23A/S23A mice developed B-cell tumors but not T-cell lymphomas or frequent sarcomas. B-cell tumors have been observed in p53−/− mice (Donehower et al, 1995; Ward et al, 1999); however, fatal thymic lymphoblastic T-cell lymphomas and sarcomas may have limited the development and progression of B-cell tumors in p53−/− animals. It is possible that there is a different threshold for the amount of p53 signaling needed to suppress tumorigenesis in different tissues. For example, thymocytes may be sensitized to p53-dependent apoptosis such that even low levels of p53 are sufficient to induce apoptosis. Indeed, in thymocytes, while there was resistance to apoptosis, a time course showed that S23A/S23A thymocytes do eventually die in response to radiation while, at those doses, p53−/− cells were completely resistant (Figure 3A and B). In response to tumor-initiating events, cells in other tissues may need a higher degree of p53 signaling for apoptosis and tumor suppression. Alternatively, the upstream pathways to p53 activation may differ in different cell types in response to different stresses such that Ser20/23 phosphorylation is critical in some but not other situations. The specific role of Ser23 in tumor suppression in B cells could be due to impaired apoptosis in Ser23 mutant B cells, as splenic B cells showed partial resistance to irradiation-induced apoptosis. p53 function has also been implicated in B-cell proliferation and maturation (Shaulsky et al, 1991; Aloni-Grinstein et al, 1995; Shick et al, 1997). A close examination of the role of the p53/Ser23 pathway in the B-cell differentiation program, and study of differentiation marker expression in the B-cell lineage tumors of Ser23 mutant mice is warranted. As we observed a long latency to tumorigenesis in these animals, it is likely that cells needed time to acquire genetic changes to develop into B-cell tumors. It will also be important to identify the genetic changes that lead to the B-cell lymphomas in this model, and to investigate how mutation of the Ser23/p53 pathway cooperates with such events. Moreover, based on these results, the role of p53 pathway mutations in human B-cell lymphoma bears further examination.
While p53 undergoes numerous phosphorylation events, the physiological role of these modifications has been difficult to assess. We have shown that p53 Ser23 phosphorylation is critical for p53 stabilization in response to ionizing radiation in vivo, and for tumor suppression. In contrast, Ser18 phosphorylation in mice is not required for tumor suppression or, in thymocytes, for p53 stabilization, but may have important roles in apoptosis and/or p53 target gene induction (Chao et al, 2003, Sluss et al, 2004). Bruins et al (2004) find that Ser389, a site phosphorylated after UV irradiation, is important for UV-induced p53 function, and germline Ser389 mutants showed increased sensitivity to UV-induced skin tumors. Collectively, these data demonstrate the importance of generating germline phosphorylation site mutants to dissect the roles of specific p53 phosphorylation events in vivo.
These and other data suggest a model in which, in response to ionizing radiation, p53 is stabilized via phosphorylation of Ser20/23. This may be mediated by Chk2, consistent with defective p53 stabilization in Chk2−/− cells, or other Ser23 kinases may be critical. Identification of the pathway upstream of Ser23 phosphorylation may lead to the discovery of genes that could be mutated in tumors in which p53 itself is intact. Given the importance of the p53 pathway in tumor development and the response of cells to chemotherapy and other DNA-damaging agents, it is critical to fully elucidate this pathway.
Materials and methods
Construction of targeting vector and generation of S23A mutant mice
Fragments of mouse genomic p53 sequence extending from intron 1 through exon 6 were cloned into the vector pBSKII. Ser23 is encoded in exon 2 and the single base pair mutation leading to an Ala substitution was introduced by site-directed mutagenesis using the QuikChange kit (Stratagene). A puromycin resistance gene driven by the PGK promoter and flanked by loxP sites (pBS.DAT-LoxStop) was introduced into an Xho1 site in intron 1. p53 exons and intron/exon boundaries in the targeting vector were sequenced to confirm that no additional, unexpected mutations were introduced during the cloning. The targeting vector was linearized with Not1 and electroporated into J1 ES cells (derived from strain 129/sv) before being selected for resistance to puromycin. Homologous recombination was confirmed by Southern blot with EcoR1-digested genomic DNA. The Southern probe was external to the targeting vector and was derived from a Bsm1 digest of a fragment of genomic DNA from intron1. GFP-Cre was transiently transfected into ES cells to excise the puro cassette leaving a single loxP site in intron1. Excision of the puro cassette was confirmed by Southern blot. Heterozygous ES cells were injected into C57BL/6 blastocysts that were then implanted into pseudopregnant CD1 females essentially as described (Bradley, 1987). The p53 cDNA sequence from S23A/S23A mice was determined by RT–PCR on RNA isolated from three independent MEF lines generated from E13.5 embryos using standard techniques. In each of the lines, the sequence was the same as wild type, except for the single T–G base substitution targeted to exon 2. All animals described are on a mixed 129/Sv × C57BL/6 background. PCR genotyping was based on the presence of a single loxP site remaining in intron 1 of the correctly targeted locus. The following primers flanking the remaining loxP site were used: dt020200.1 5′agcctgcctagcttcctcagg3′ and dt011200.3 5′cttggagacatagccacactg3′. These primers amplify a 540 bp mutant band in the presence of a single loxP site, and a 420 bp wild-type band.
Western blot analysis
For MEF or thymocyte experiments, cells were plated in DMEM supplemented with 10% FCS, L-glutamine (2 mM) and antibiotics. Cells were irradiated using a GammaCell40 with 137Cs source. Cells were lysed in lysis buffer (100 mM NaCl, 100 mM Tris (pH 8.0), 1% NP-40, complete protease inhibitor cocktail (Roche), sodium fluoride (10 mM), sodium vanadate (1 mM)). For in vivo thymus Western blots, whole mice were irradiated with a dose of 5 Gy, and 5 h later the thymus was collected for protein extraction. For half-life experiments, thymocytes were suspended at 10 million cells/ml. Cyclohexamide (40 μg/ml) was added 2 h post-irradiation and samples collected for Western analysis at various time points afterwards. For cerebellum Western blots, newborn mice 5 days after birth were irradiated with 4 Gy and, 3 h later, the cerebellum was dissected and frozen on dry ice before being lysed in lysis buffer. Following SDS–PAGE, proteins were transferred to PVDF membranes (Immobilon P, Millipore). Membranes were blocked in 5% nonfat dry milk in TBST. For p53 Western blots on MEFs, p53 Ab-3 (Oncogene Research Products) was used at a 1/1000 dilution. For p53 Western blots on thymus tissue and cerebellum tissue, the sheep polyclonal antibody Ab-7 (Oncogene Research Products) was used at a 1/2500 dilution, or the rabbit polyclonal antibody FL393 (Santa Cruz) at 1/1000 dilution. Blots were stripped and reprobed for actin (Santa Cruz) at a 1/1000 dilution. Enhanced chemiluminescence was used for signal detection (ECL Plus, Amersham) and blots were exposed to film (X-OMAT). For thymocyte half-life experiments, band intensities were quantified by scanning densitometry using NIH Image software.
Northern blot analysis
Exponentially growing cells were plated at a density of 1 × 106 cells/10 cm dish, and treated 24 h later with 5 Gy IR. At various time points, cells were lysed directly on the plate with TRIZOL reagent (Gibco BRL) and RNA was isolated following the manufacturer's protocol. RNA was denatured and run on a 1% agarose in 1 × MOPS with 6% formaldehyde and transferred to a Hybond N membrane (Amersham). The membrane was crosslinked using a UV Stratalinker (Stratagene) and hybridized with a 32P-labeled probe. Probes were made to MDM2, p21 and, p53 and ARPP P0 cDNAs using the Prime-It II random primer labeling kit (Stratagene). Blots were hybridized in ExpressHyb (Stratagene) solution at 65°C.
Cell cycle analysis
Cells were plated at 8 × 105 cells/10 cM plate and irradiated with 5 Gy IR 24 h later. Analysis of samples was performed 14 or 24 h after IR. Samples were processed with a FACScan (Becton Dickinson). Data analysis was carried out using ModFit LT software (Becton Dickinson).
Thymocyte apoptosis assay
Fresh thymocytes were isolated from 5–8-week-old mice and plated at a density of 1 × 106 cells/ml, and then incubated at 37°C. For time-course experiments, cells were treated at T=0 with either 100 nM dexamethasone or with 5 Gy of IR. For dose–response curves, thymocytes were also plated a 1 × 106 cells/ml, treated at T=0 and collected for apoptosis analysis 24 h later. Apoptosis was assessed using annexin-FITC (Becton Dickinson) and propidium iodide (PI; 50 μg/ml) staining of cells followed by two-color cytometry using a FACScan (Becton Dickinson) and CellQuest software (Becton Dickinson). Cells scored as viable were those that were negative in staining for both annexin and PI. All data were normalized to the viable fraction in the untreated cells at the given time point.
In vivo analysis of apoptosis
For CNS apoptosis studies, S23A/S23A or wild-type littermates were irradiated 5 days after birth with 4 Gy. Mice were killed 18 h later, at which time when their brains were removed and fixed in 10% formalin (3.7% formaldehyde solution in PBS) for 24 h. For analysis of apoptosis in the adult spleen, 6–8-week-old animals were irradiated with 5 Gy and killed 24 h later. Spleens were fixed in 10% formalin for 24 h. Tissues were processed and embedded in paraffin blocks. Sagittal sections were cut at 4 μm. Tdt dUTP-biotin Nick End Labeling (TUNEL) assays were performed to assess apoptosis. For quantification of CNS apoptosis, TUNEL-positive nuclei in equivalent areas of the cerebellar external granule layer of S23A/S23A and control animals were counted. For quantification of B-cell depletion following DNA damage, animals were irradiated with 5 Gy 24 h before collection of spleens and isolation of splenocytes. Splenocytes were stained with anti-B220 coupled to flourescein-isothiocyanate (Pharmingen) using standard protocols and subject to fluorescence-activated cell sorting analysis.
Tumor analysis
S23A/S23A and wild-type controls were aged. Mice were killed when moribund, or if visible tumor burden was apparent. Inner organs were fixed in 10% neutral buffered formalin. Bones were fixed in Bouins fixative. Tissue was embedded in paraffin and 4 μm sections were cut and stained with hematoxylin and eosin for pathological analysis. For a subset of tumors, immunohistochemical staining was performed with the antibodies B220 (Becton Dickinson), Ig kappa (Southern Biotechnology), and F480 (Serotec) on formalin-fixed, paraffin-embedded tissue. B220 and Ig kappa staining were performed with a citrate buffer unmasking step. F480 staining was performed with a trypsin unmasking step, and for F480 staining, all incubations and washes were carried out in 0.05% Triton X-100.
Southern blotting
Tumor tissue was digested with proteinase K overnight, and DNA was isolated following phenol chloroform extraction. Following EcoR1 digest, DNA was loaded on a 0.8% agarose gel and transferred to a nylon membrane N+ (Amersham). Membrane was hybridized to a 32P-labeled probe of a 1.9 kb BamH1–EcoR1 JH genomic fragment including mouse JH3 and JH4 sequences for detection of gene rearrangements.
Acknowledgments
We are grateful to Denise Crowley and Mike Brown for excellent histological assistance. We thank Dave Tuveson (University of Pennsylvania) for the pBS.DAT-LoxStop cassette and Brian Sauer (Stowers Institute) for the GFP-Cre expressing plasmid. TJ is an Investigator of the Howard Hughes Medical Institute.
References
- Aloni-Grinstein R, Schwartz D, Rotter V (1995) Accumulation of wild-type p53 protein upon gamma-irradiation induces a G2 arrest-dependent immunoglobulin kappa light chain gene expression. EMBO J 14: 1392–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E, Anderson CW (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 268: 2764–2772 [DOI] [PubMed] [Google Scholar]
- Ashcroft M, Kubbutat MH, Vousden KH (1999) Regulation of p53 function and stability by phosphorylation. Mol Cell Biol 19: 1751–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677 [DOI] [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA (1999) Heterozygous germ line hCHK2 mutations in Li–Fraumeni syndrome. Science 286: 2528–2531 [DOI] [PubMed] [Google Scholar]
- Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P (1999) DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene 18: 1723–1732 [DOI] [PubMed] [Google Scholar]
- Bradley A (1987) Production and analysis of Chimeric mice. In Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, Robertson EJ (ed), pp 113–152. Oxford, UK: IRL Press [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377: 552–557 [DOI] [PubMed] [Google Scholar]
- Bruins W, Zwart E, Attardi LD, Iwakuma T, Hoogervorst EM, Beems RB, Miranda B, Van Oostrom CThM, Van den Berg J, Van den Aardweg GJ, Lozano G, Van Steeg H, Jacks T, De Vries A (2004) Increased sensitivity to UV radiation in mice with a p53 point mutation at Ser389. Mol Cell Biol, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281: 1677–1679 [DOI] [PubMed] [Google Scholar]
- Chao C, Hergenhahn M, Kaeser MD, Wu Z, Saito S, Iggo R, Hollstein M, Appella E, Xu Y (2003) Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J Biol Chem 278: 41028–41033 [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev 14: 278–288 [PMC free article] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA 96: 13777–13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362: 849–852 [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82: 675–684 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA Jr, Park SH, Thompson T, Ford RJ, Bradley A (1995) Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinogen 14: 16–22 [DOI] [PubMed] [Google Scholar]
- Dumaz N, Meek DW (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J 18: 7002–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N, Milne DM, Jardine LJ, Meek DW (2001) Critical roles for the serine 20, but not the serine 15, phosphorylation site and for the polyproline domain in regulating p53 turnover. Biochem J 359: 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 12: 2973–2983 [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Herzog H, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ (1998) Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280: 1089–1091 [DOI] [PubMed] [Google Scholar]
- Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T, De Stanchina E, Bristow RG, Suda T, Lowe SW, Jeggo PA, Elledge SJ, Mak TW (2002) Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol 22: 6521–6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287: 1824–1827 [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7 [DOI] [PubMed] [Google Scholar]
- Jallepalli PV, Lengauer C, Vogelstein B, Bunz F (2003) The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 278: 20475–20479 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71: 587–597 [DOI] [PubMed] [Google Scholar]
- Komarova EA, Christov K, Faerman AI, Gudkov AV (2000) Different impact of p53 and p21 on the radiation response of mouse tissues. Oncogene 19: 3791–3798 [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274: 948–953 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362: 847–849 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Melchionna R, Chen XB, Blasina A, McGowan CH (2000) Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol 2: 762–765 [DOI] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B (1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362: 857–860 [DOI] [PubMed] [Google Scholar]
- Shaulsky G, Goldfinger N, Peled A, Rotter V (1991) Involvement of wild-type p53 in pre-B-cell differentiation in vitro. Proc Natl Acad Sci USA 88: 8982–8986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shick L, Carman JH, Choi JK, Somasundaram K, Burrell M, Hill DE, Zeng YX, Wang Y, Wiman KG, Salhany K, Kadesch TR, Monroe JG, Donehower LA, el-Deiry WS (1997) Decreased immunoglobulin deposition in tumors and increased immature B cells in p53-null mice. Cell Growth Differ 8: 121–123 [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14: 289–300 [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334 [DOI] [PubMed] [Google Scholar]
- Shieh SY, Taya Y, Prives C (1999) DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J 18: 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB (1997) DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev 11: 3471–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Armata H, Gallant J, Jones SN (2004) Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol Cell Biol 24: 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N (2002) Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J 21: 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T, Juven-Gershon T, Moallem E, Berger M, Vogt Sionov R, Lozano G, Oren M, Haupt Y (1999a) Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J 18: 1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T, Sionov RV, Moallem E, Yee CL, Howley PM, Oren M, Haupt Y (1999b) Mutations in serines 15 and 20 of human p53 impair its apoptotic activity. Oncogene 18: 3205–3212 [DOI] [PubMed] [Google Scholar]
- Ward JM, Tadesse-Heath L, Perkins SN, Chattopadhyay SK, Hursting SD, Morse HC III (1999) Splenic marginal zone B-cell and thymic T-cell lymphomas in p53-deficient mice. Lab Invest 79: 3–14 [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y (2002) Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol 22: 2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Wang Q, Wu H, Cogswell J, Lu L, Jhanwar-Uniyal M, Dai W (2001) Reactive oxygen species-induced phosphorylation of p53 on serine 20 is mediated in part by polo-like kinase-3. J Biol Chem 276: 36194–36199 [DOI] [PubMed] [Google Scholar]