

Abstract
The Drosophila melanogaster warts/lats tumour suppressor has two mammalian counterparts LATS1/Warts-1 and LATS2/Kpm. Here, we show that mammalian Lats orthologues exhibit distinct expression profiles according to germ cell layer origin. Lats2−/− embryos show overgrowth in restricted tissues of mesodermal lineage; however, lethality ultimately ensues on or before embryonic day 12.5 preceded by defective proliferation. Lats2−/− mouse embryonic fibroblasts (MEFs) acquire growth advantages and display a profound defect in contact inhibition of growth, yet exhibit defective cytokinesis. Lats2−/− embryos and MEFs display centrosome amplification and genomic instability. Lats2 localizes to centrosomes and overexpression of Lats2 suppresses centrosome overduplication induced in wild-type MEFs and reverses centrosome amplification inherent in Lats2−/− MEFs. These findings indicate an essential role of Lats2 in the integrity of processes that govern centrosome duplication, maintenance of mitotic fidelity and genomic stability.
Keywords: centrosome, embryonic lethality, genomic instability, Kpm, Lats2
Introduction
The equal distribution of duplicated chromosomes to daughter cells is dependent on proper assembly and function of bipolar spindles during mitosis. Checkpoint defects that permit mis-segregation of mitotic chromosomes have the potential of permitting the generation of progeny with improper chromosome numbers and gene dosage (Hartwell and Kastan, 1994). Conditions favouring the continued propagation of cells with unbalanced segregation facilitate neoplastic transformation through the exposure of recessive mutations by loss of heterozygosity. This chromosomal instability phenotype, or aneuploidy, is observed in a majority of tumour cells (Mitelman, 1971).
Several molecular mechanisms have been identified that potentiate chromosomal instability when defective (Hartwell and Kastan, 1994). The generation of aneuploid cells has been proposed to proceed in many cases through a tetraploid intermediate step (Galipeau et al, 1996; Borel et al, 2002; Meraldi et al, 2002) that can arise by the disruption of mitotic chromosomal segregation (Minn et al, 1996) or cytokinesis (Andreassen et al, 2001). Supernumerary centrosomes that facilitate the generation of multipolar spindles would allow the generation of aneuploid daughter cells through unequal chromosome segregation (Doxsey, 2001; Lange, 2002; Nigg, 2002). Supernumerary centrosomes present in tumour cells have been associated with the formation of multipolar spindles and genomic instability (Lingle et al, 1998, 2002; Brinkley, 2001).
The study of components important in developmental control of growth regulation in model organisms such as Drosophila has greatly facilitated our understanding of corresponding pathways that are relevant in cancer biology. The warts (Bryant et al, 1993; Justice et al, 1995) or lats (Xu et al, 1995) gene was originally identified in genetic screens designed to identify mutations that result in tissue overgrowth in Drosophila. Warts/Lats is a serine–threonine kinase that is structurally related to proteins that regulate mitotic exit or septation initiation in Saccharomyces cerevisiae and Schizosaccharomyces pombe, respectively (Nigg, 2001). Embryonic lethality and overproliferation in lats mutant flies were suppressed by reducing cdc2 or cyclin A gene dosage (Tao et al, 1999). Excessive cyclin A/cdk1 activity in the absence of lats was proposed to contribute to the excessive overgrowth that appears in warts/lats mutant cells. However, cell cycle deregulation as a sole mechanism of overgrowth has not been supported by other studies (Weinkove and Leevers, 2000). sav has been shown to cooperate with warts/lats in a novel pathway that simultaneously impacts control of cell death and proliferation (Tapon et al, 2002). Cells mutant for either warts/lats or sav were found to exhibit delays in cell cycle exit and diminished apoptosis, which were attributed to elevated levels of cyclin E and DIAP1 levels, respectively.
Mammalian orthologues of sav and warts/lats have been implicated in the regulation of cell cycle progression and tumour suppression. Mutations in a human homologue of sav (hWW45) were identified in three cancer cell lines (Tapon et al, 2002). Two mammalian orthologues of warts/lats have been identified in mammals: LATS1/Warts-1 (Nishiyama et al, 1999; Tao et al, 1999) and LATS2/Kpm (Hori et al, 2000; Yabuta et al, 2000). LATS1/Warts-1 protein is phosphorylated during the M phase of the cell cycle, colocalizes with centrosomes and the mitotic spindle, and associates with Cdk1 and Zyxin (Nishiyama et al, 1999; Tao et al, 1999; Hirota et al, 2000). Mice mutant for Lats1 developed pituitary hyperplasia and were susceptible to sarcomas and ovarian cancer (St John et al, 1999). However, hyperplasia and tumour development were observed in a restricted number of tissues in these mice, compared to the overgrowth phenotype observed in a wide range of tissues and developmental stages of warts/lats mutant flies. Hence, Lats1 might not encompass the entire mammalian equivalent of warts/lats activity.
Despite structural similarities and similar M-phase phosphorylation (Hori et al, 2000; Yabuta et al, 2000), the biological role of LATS2/kpm remains unknown. Conflicting results have been reported describing the effects of LATS2/kpm on cell cycle progression, in that LATS2/kpm overexpression has been described to result in a G2/M-phase arrest and apoptosis (Kamikubo et al, 2003) or inhibition of the G1–S transition (Li et al, 2003).
In order to clarify the biological function of LATS2/kpm, we have generated mice that carry a targeted mutation of the Lats2 locus. In this study, we show that lats/warts orthologues display distinct expression profiles according to germ cell layer origin. Furthermore, we show that disruption of Lats2 exerts contrasting roles in proliferation depending on the cellular context. Although Lats2−/− embryos exhibit an arrest in proliferation prior to embryonic lethality, Lats2−/− mouse embryonic fibroblasts (MEFs) acquire growth advantages that appear to be strikingly similar to those of mutant warts/lats cells in Drosophila. We also show that Lats2 is a centrosomal protein that negatively regulates centrosome duplication. Lats2−/− embryonic cells exhibit an increased frequency of cytokinesis defects, micronuclei accumulation, supernumerary centrosomes and aneuploidy, suggesting a role for Lats2 in maintenance of mitotic fidelity and genomic integrity.
Results
Expression of mammalian warts/lats homologues during embryogenesis
Murine Lats1 and Lats2 embryonic expression was examined by Northern analysis and in situ hybridization of whole-mount and sectioned embryo specimens. Lats1 and Lats2 expression appeared equivalent throughout development (embryonic days (E) 7–17) by Northern analysis (Figure 1A–C); however, in situ hybridization revealed a disparity in the spatial distribution of expression (Figure 1D–S). Prominent expression of Lats1 and Lats2 was found in tissues of ectodermal and mesodermal origin, respectively. At E8.5–9.5, Lats1 was highly expressed in the neural tube, head fold neuroepithelium and auditory vesicles (Figure 1D–F), with prominent expression in the mesencephalon at E10.5 (Figure 1G, L and M). Lats2 was prominent in lateral mesodermal plate, somites and cardiac outflow tract at E8.5 (Figure 1H and I), and in the general heart field (inflow and outflow tracts, ventricle and atrium) by E10.5 (Figure 1R and S). Moderate Lats2 expression was observed in the head mesenchyme, dorsal aorta area, developing gut, lungs and dermatome of the somites. Overlapping expressions of Lats1 and Lats2 were observed in branchial arches and limb buds, as well as the cardiogenic crescent at E8.5.
Figure 1.
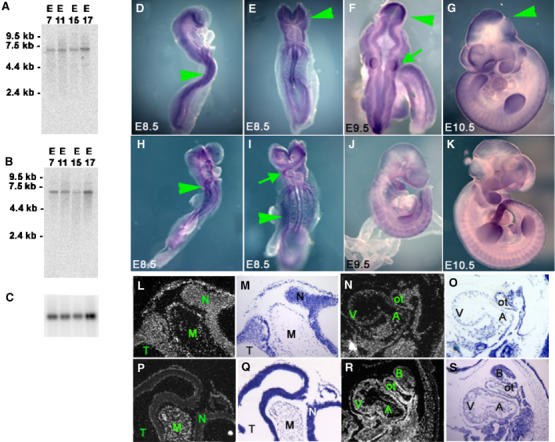
Northern analysis of Lats1 (A) and Lats2 (B) expression during embryonic development. Loading control for Northern blot shown with GADPH (C). Whole-mount in situ hybridization of Lats1 (D–G) and Lats2 (H–K) during embryonic developmental stages E8.5–10.5. Prominent Lats1 expression was detected in neuroepithelium (arrows, D, E), mesencephalon (arrows, F, G) and otic vesicles (lower arrow, F), whereas Lats2 expression was prominent in the lateral mesodermal plate (arrows, H, I) and the cardiac outflow tract (upper arrow, I). In situ hybridization (and corresponding counterstaining) of E10.5 embryonic histological sections with Lats1 (L–O) and Lats2 (P–S) showing prominent expression of Lats1 in neuroepithelium (L, M). Lats1 expression in the heart (N, O) was relatively lower compared to Lats2 expression (R, S). N, neuroepithelium; V, ventricle; A, atrium; B, first branchial arch; ot, outflow tract.
Targeted disruption of Lats2
To investigate the biological function of Lats2, a targeted disruption of Lats2 was generated in embryonic stem (ES) cells (Figure 2A–D). An insertional strategy was devised to replace serine 371 of Lats2 with an in-frame stop codon followed by an inverted neomycin cassette in order to disrupt translation of the carboxy-terminal kinase domain. Northern analysis revealed the expected loss of the wild-type 6-kb Lats2 mRNA in homozygous embryos, and the presence of a longer 8-kb Lats2 mRNA species produced from the targeted allele (Supplementary Figure 1). To ensure success of the targeting strategy, cDNAs (1.6 kb) derived from the start site of translation to the inserted neo sequence of the mutant allele were obtained by RT–PCR from Lats2+/− mouse mRNA. Two cDNA clones were sequenced entirely to verify that the mutant mRNA species contained sequence replacing serine 371 and early stop codons (Figure 2C). Western blot analysis using cellular extract from Lats2−/− MEFs and polyclonal antisera against Lats2 failed to detect the full-length Lats2 protein or any truncated peptide, indicating that the targeting strategy resulted in a null allele (Figure 2D).
Figure 2.
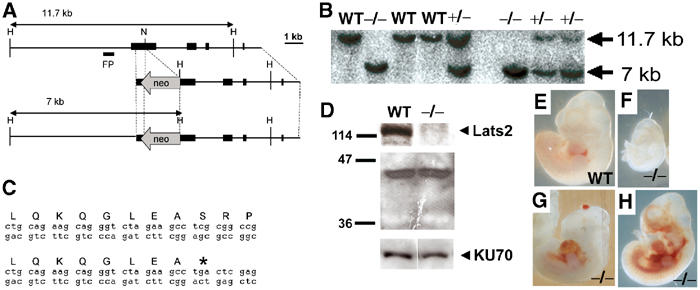
Lats2 targeted disruption and Lats2−/− embryo abnormalities. (A) Restriction maps depicting mouse Lats2 genomic fragment, targeting construct and predicted structure of targeted Lats2 allele. Exons are represented by filled rectangles. Restriction enzymes: H, HindIII; N, NotI. Digestion of genomic DNA with HindIII generates an 11.7-kb fragment from the wild-type allele and a 7-kb fragment from the mutant allele, both of which are detected using the 5′ flanking probe shown. (B) Southern analysis of genomic DNA extracted from E9.5 embryos obtained from heterozygote crosses. (C) Sequence of cDNAs corresponding to Lats2 mRNA derived from wild-type (upper) and mutant (lower) alleles. (D) Western analysis of protein extracts from wild-type and Lats2−/− MEFs. Polyclonal antibodies against Lats2 failed to detect full-length protein (upper panel) or a shorter Lats2 peptide (middle panel) in the mutant MEFs. Ku70 is shown as a loading control (lower panel). (E, F) Representative E10.5 embryos (photographed at the same magnification as at the time of dissection) depicting reduced size of Lats2−/− embryos. (G, H) Lats2−/− E11.5 embryos with distended pericardium with blood accumulation in the abdominal cavity.
Targeted disruption of Lats2 results in embryonic lethality
Analysis of live-born progeny obtained from 129/C57BL/6 F1 heterozygote crosses revealed that Lats2 is essential for the development of viable progeny (Table I). No viable Lats2−/− offspring were recovered at birth (67 wild type, 37%; 116 Lats2+/−, 63%; 0 Lats2−/−, 0%), and this lethality was independent of strain when outbred in C57BL/6 and ICR backgrounds. Heterozygous mice were phenotypically indistinguishable from wild-type littermates and a cohort of 40 monitored heterozygotes remained disease free following 1 year of observation.
Table 1.
Genotype of progeny collected from Lats2 heterozygote crosses and classification of Lat2−/−defects
| Gestational age (days postconception) | Wild type | +/− | −/− | Total typed | Resorptions | Total collected | |
|---|---|---|---|---|---|---|---|
| 8.5–8.75 | Total | 14 | 22 | 9 | 45 | 0 | 45 |
| Viablea | 14 | 22 | 9 | ||||
| Dead | 0 | 0 | 0 | ||||
| 9.0–9.5b | Total | 38 | 58 | 48 | 144 | 3 | 147 |
| Viable | 38 | 56 | 46 | ||||
| Dead | 0 | 2 | 2 | ||||
| 10.5c | Total | 27 | 43 | 19 | 89 | 2 | 91 |
| Viable | 27 | 41 | 16 | ||||
| Dead | 0 | 2 | 3 | ||||
| 11.5d | Total | 13 | 30 | 18 | 61 | 2 | 63 |
| Viable | 12 | 30 | 8 | ||||
| Dead | 1 | 0 | 10 | ||||
| 12.5e | Total | 10 | 29 | 13 | 52 | 4 | 56 |
| Viable | 10 | 27 | 1 | ||||
| Dead | 0 | 2 | 12 | ||||
| Weanling | |||||||
| 129/J × C57BL/6 (F1 × F1) | 30 | 56 | 0 | 86 | |||
| C57BL/6 outbred (F2 × F2) | 6 | 11 | 0 | 17 | |||
| C57BL/6 outbred (F3 × F3) | 6 | 8 | 0 | 14 | |||
| ICR outbred (F3 × F3) | 25 | 41 | 0 | 66 | |||
| Total | 67 | 116 | 0 | 183 | |||
| aAt dissection, embryos were determined to be alive based on the presence of a heartbeat. | |||||||
| bAt E9.5, 25/40 Lats2−/− embryos were either smaller or developmentally delayed compared to wild-type or heterozygous siblings. | |||||||
| cAt E10.5, Lats2−/− embryos were typically smaller (14/19), delayed in development (2/19) and presented with distended blood-filled pericardia (4/19) and/or haemorrhaging (5/19) in the body/head region. | |||||||
| dAt E11.5, Lats2−/− embryos were typically smaller (14/18), along with head and/or body cavity haemorrhaging (5/18), a distended and/or blood-filled pericardia (8/18) and irregular kinked neural tubes (5/18). | |||||||
| eAt E12.5, 12/13 Lats2−/− e410mbryos examined were dead, smaller than littermates, especially in the head (hindbrain/neck), and appeared developmentally arrested at E10.5–11.0. | |||||||
Embryonic lethality of Lats2−/− mice was observed to occur during E10.5–12.5 (Table I). Progeny from Lats2+/− intercrosses developed normally until E8.5; however, Lats2−/− embryos exhibited pleiotrophic developmental defects at E9.5 and thereafter (Figure 2E–H). Lats2−/− embryos were typically smaller or developmentally delayed (Figure 2E and F) and occasionally were found to contain distended, blood-filled pericardia, haemorrhaging in the body or head region (Figure 2G and H), and irregular kinked neural tubes (Table I).
Histological analyses revealed no specific defect in the development of yolk sac, yolk sac blood islands, blood cells, embryonic vascularization, chorionic-allantois fusion, placental vascularization or differentiation (Supplementary Figure 1). Immunohistochemical staining with anti-PECAM did not reveal any defects in blood vessel formation or appearance (Supplementary Figure 1). Lats2−/− embryos expressed normal levels of placental PPARγ (Supplementary Figure 2), a placental differentiation marker that can lead to myocardial thinning due to incomplete placental vascularization if deficient (Barak et al, 1999).
Lats2−/− embryos display contrasting defects in cardiac growth
Lats2−/− E9.5 embryos had correctly looped hearts; however, 25% (n=12) contained irregular supernumerary cells invading the interior of the common atrial chamber. The supernumerary cells were primarily located in the myocardium, and extended into the open chamber, with the endocardial layer apparently disrupted (Figure 3A and B). Similar outgrowths were also observed in 33% (n=12) of Lats2−/− E10.5 embryos examined (Figure 3C and D). In contrast, a marked ventricular hypoplasia with defective trabeculae and myocardia was observed in 36% (n=14) of E10.5 Lats2−/− embryos (Figure 3E and F). Cardiac development appeared normal with respect to expression of cardiac α-actin (Figure 3G–J), Bmp4 (Figure 3K–N) and Nkx2.5 (Supplementary Figure 2). The atrial and ventricular defects, pericardial distension and blood pooling suggest that cardiac insufficiency contributed to embryonic lethality in a subset of Lats2−/− embryos.
Figure 3.
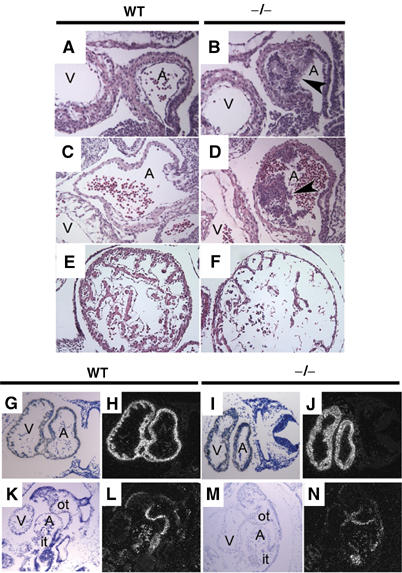
Haematoxylin and eosin-stained sagittal sections of heart (A, atrium; V, ventricle) from wild-type (A, C) and mutant (B, D) embryos at E9.5 (A, B) and E10.5 (C, D) with atrial hyperplasia (arrows). (E, F) Ventricular hypoplasia in Lats2−/− embryos. Representative haematoxylin and eosin-stained transverse sections of ventricles from E10.5 wild-type (E) and Lats2−/− (F) embryos depicting trabeculation defect and thinner myocardium. Expression of cardiac actin at E9.5 (G–J) and Bmp4 at E10.5 (K–N) in wild-type (G, H, K, L) and Lats2−/− embryos (I, J, M, N) by in situ hybridization (A, atrium; V, ventricle; it, inflow tract; ot, outflow tract).
Lats2−/− embryos exhibit an arrest in proliferation
Proliferation and apoptosis in embryos were examined using BrdU incorporation and Tdt-mediated dUTP nick end labelling (TUNEL) analysis, respectively. BrdU incorporation was equivalent in E8.5 embryos (Figure 4). However by E9.5, decreased BrdU incorporation was evident in 75% (n=12) of Lats2−/− embryos and by E10.5, all live Lats2−/− embryos examined (n=12) displayed a proliferation defect with respect to decreased (75%) or absent (25%) BrdU incorporation when compared to wild-type and Lats2+/− siblings (n=14). A decreased level of cyclin E was observed in Lats2−/− embryos showing decreased proliferation, while other cyclin levels examined did not differ from the wild-type littermates (Supplementary Figure 3). Despite the proliferation arrest, apoptotic profiles as evaluated by TUNEL analysis and enumeration of DAPI-stained picnotic nuclei were equivalent (Supplementary Figure 4). The proliferation arrest was found to be restricted to the embryonic conceptus, as extraembryonic tissues showed robust BrdU incorporation in Lats2−/− embryos (Supplementary Figure 5). As the decreased BrdU incorporation preceded the appearance of other morphological and histological defects, including the previously described atrial hyperplasia, the proliferation arrest observed is likely responsible for hypoplasia, morphological defects and ultimate lethality in Lats2−/− embryos.
Figure 4.
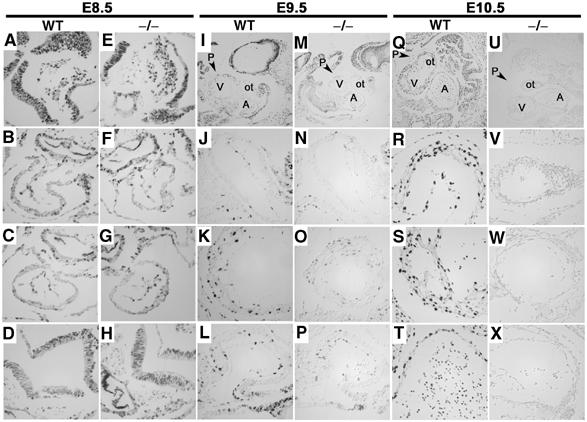
Stage-specific proliferation defect in Lats2−/− embryos. E8.5 transverse, E9.5 sagittal and E10.5 sagittal embryo sections stained for BrdU. All images for a given embryonic stage were captured at the same exposure settings and × 40 magnification, unless otherwise stated. (A, E) Left side of head fold. (B, F) Heart outflow tract. (C, G) Heart ventricle. (D, H) Neural tube. Top panels for E9.5 and E10.5 depict heart area at × 10 magnification (V, ventricle; A, atrium; ot, outflow tract; P, pericardium) with representative outflow tract (J, N, R, V) heart ventricle (K, O, S, W) and atrium (L, P, T, X) below.
Lats2−/− MEFs display perturbations of growth control
The observed arrest in proliferation during development might be attributed to a cell-intrinsic block in proliferation or the result of perturbed developmental cues. Despite the observed developmental arrest of Lats2−/− in situ, we were able to derive and passage MEFs from Lats2−/− E10.5 embryos. A total of three Lats2−/− E10.5 MEF cultures were derived and compared to wild-type littermate control cultures. Early passage Lats2−/− MEFs showed no impairment in proliferation as assessed by [3H]thymidine incorporation or cell cycle analysis when compared to wild-type littermate cultures of equivalent passage (Supplementary Figure 6). Initial growth rates were comparable to sibling MEF cultures in a standard 3T3 protocol of passage (Figure 5A), but at later passages Lats2−/− MEFs grew more rapidly whereas control MEFs entered the expected lag phase of growth. Lats2−/− MEFs displayed a greater propensity to achieve a higher saturation density than control MEFs of equivalent passage (Figure 5B). The plating efficiency, as assessed by colony formation of low-passage Lats2−/− MEFs, was found to be markedly increased compared to control MEFs of equivalent passage seeded at the same density (Figure 5C). Furthermore, Lats2−/− MEFs displayed a partial loss of contact inhibition of growth, as confluent cultures continued to divide and either detach from the culture dish or form foci (Figure 5D–F). Foci were readily stained with crystal violet (Figure 5D) and were composed of rounded and highly refractile cells (Figure 5E and F).
Figure 5.
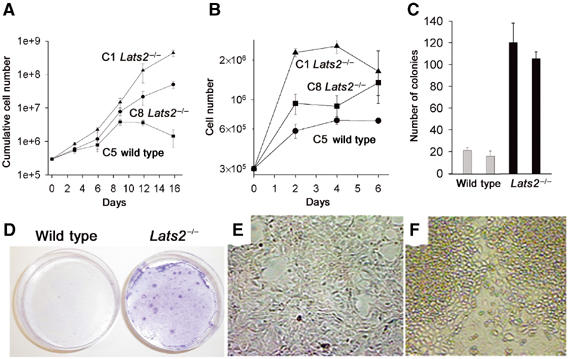
Growth kinetics in Lats2−/− MEFs. (A) Growth curve representative of passage 2 MEFs derived from wild-type (▪) and two independently derived Lats2−/− MEF lines (▴ and •). (B) Representative saturation density of growth for passage 2 MEFs derived from wild-type (•) and two independently derived Lats2−/− MEF lines (▴ and ▪). (C) Plating efficiency at low seeding density (colony formation/3000 cells plated) for two wild-type MEF lines (grey bar, n=3) and two Lats2−/− MEF lines (black bar, n=3). (D) Representative determination of contact inhibition of growth. A total of 5 × 104 cells of either wild-type or Lats2−/− MEFs were plated in 6 cm dishes. Cells were maintained in culture without passage for 2 weeks and stained with crystal violet. (E, F) Morphology of wild-type MEFs (E) and foci observed in Lats2−/− MEFs (F).
Lats2−/− MEFs display cell division abnormalities
Warts/lats gene family members have homology to gene products that are required for mitotic exit in S. cerevisiae and septation in S. pombe (Nigg, 2001). As these processes may share common features with cytokinesis in mammalian cells, we investigated the possibility that Lats2 might impact processes that promote separation during cell division. Microscopic examination of wild-type, Lats2+/− and Lats2−/− MEFs following indirect immunofluorescence of α-tubulin or α-actin revealed that Lats2−/− MEFs exhibited an increased propensity (% of cells±s.d.) to retain cytoplasmic bridges with neighbouring cells (35.9±2.8%) compared to wild-type MEFs of equivalent passage (8.4±3.6%) (Figure 6A–H). During cytokinesis, bundled microtubules that comprise the central spindle form a slender tube called the midbody, which connects the two daughter cells. Successful completion of cytokinesis is dependent on the cleavage of the midbody, a process known as abcission (Glotzer, 2001). The increased retention of cytoplasmic bridges suggests that either Lats2−/− MEFs are defective in processes that ensure the correct completion of cytokinesis, or that cytokinesis is impaired by the presence of incorrectly segregated chromatin. In support of the latter possibility, chromatin spanning the midbody of daughter cells could be visualized following immunofluorescent detection of phospho-histone H3 (Figure 6E–G).
Figure 6.
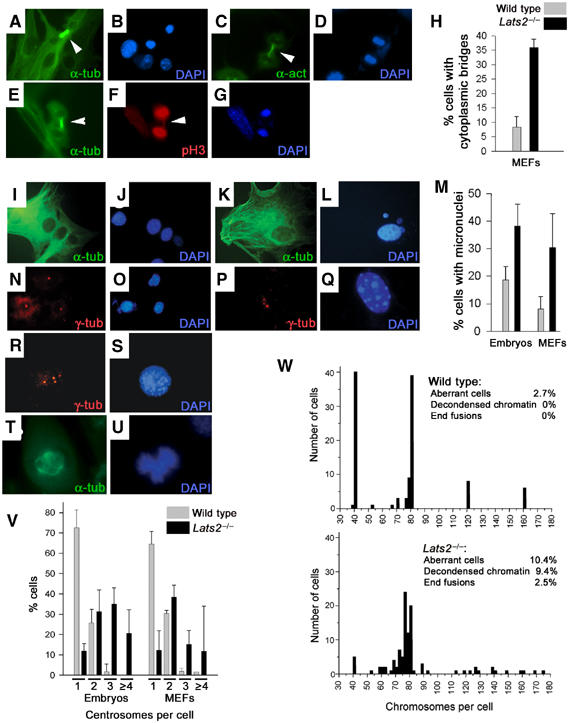
Cytokinesis abnormalities, micronuclei, aneuploidy and centrosome amplification in Lats2−/− embryos and MEFs. (A, B) Lats2−/− MEFs stained with FITC-conjugated anti-α-tubulin to visualize tubulin cytoskeleton (A) with microtubule bundles (arrowhead) and counterstained with DAPI (B) to visualize DNA. (C, D) Lats2−/− MEFs stained with anti-α-actin (C) and counterstained with DAPI (D) to visualize midbody (arrowhead) and DNA, respectively. (E–G) Bridged chromatin within the midbody (E, arrowhead) of Lats2−/− MEFs could be detected following staining for phospho-histone H3 (F, arrowhead) (DAPI counterstain shown in G). (H) Graph depicting the percentage of cells exhibiting cytoplasmic bridges from three independently derived Lats2−/− MEF lines (black bar) compared to three independent wild-type or heterozygous lines of equivalent passage (grey bar). Representative photomicrographs of wild-type MEFs (I) with normal nuclear morphology (J) and a Lats2−/− MEF (K) containing micronuclei (L) are depicted. (M) Graph depicting the percentage of cells exhibiting micronuclei from three wild-type (left grey bar) and Lats2−/− (left black bar) sibling embryos and three independently derived Lats2−/− MEF lines (right black bar) compared to three independent wild-type or heterozygous lines of equivalent passage (right grey bar). MEFs (N–S) were immunostained with anti-γ-tubulin (red) and counterstained with DAPI (blue) to visualize centrosomes and DNA, respectively. Three wild-type MEFs are depicted (N, O) with single centrosomes. Lats2−/− MEFs containing three centrosomes (P, Q) and four centrosomes (R, S) are depicted. (T, U) Abnormal spindle pole formation and chromosomal segregation defects during mitosis in Lats2−/− fibroblasts. Fibroblasts were immunostained with anti-α-tubulin (green) to visualize mitotic spindles and counterstained with DAPI (blue) to visualize chromatin. (V) Graph depicting the percentage of cells exhibiting 1, 2, 3 or ⩾4 centrosomes from three wild-type (grey bar) and Lats2−/− (black bar) sibling embryos and three independently derived Lats2−/− MEF lines (black bar) compared to three independent wild-type or heterozygous lines of equivalent passage (grey bar). (W) Pooled results of karyotypic analyses of wild-type MEF lines (upper panel) compared to Lats2−/− MEF lines (lower panel) with frequencies of specific chromosomal aberrations observed.
Lats2−/− embryos and MEFs display centrosome amplification, multipolar mitotic spindles and genomic instability
Although wild-type MEFs display low levels of genomic instability, especially a propensity to accumulate polyploid cells when passaged (Livingstone et al, 1992), we observed a relatively higher frequency (% of cells±s.e.m.) of Lats2−/− MEFs with micronuclei (Figure 6I–M) (30.4±7.2%, n=3) compared to wild-type MEF cultures (8.1±2.7%, n=3). Micronuclei are small extra nuclei within the cytoplasm that represent lagging or damaged chromosomal fragments caused by aberrant mitoses that are excluded from the main daughter nuclei at telophase (Therman and Susman, 1993). Lats2−/− cells derived from freshly harvested E10.5 embryos also showed a relatively higher frequency of cells with micronuclei (38.2±8.0%, n=3) compared to wild-type sibling embryos (18.6±4.9%, n=4) (Figure 6M). We also evaluated centrosome status in Lats2−/− and wild-type freshly harvested embryonic cells and cultured MEFs following γ-tubulin immunostaining. Cells with multiple copies of centrosomes (>2 centrosomes/cell) were rarely observed in wild-type cells, but were frequently observed in Lats2−/− embryonic cells (not shown) and early passage MEFs (Figure 6N–S). The frequency of Lats2−/− embryonic cells with >2 centrosomes was 56±14% (n=3, >200 cells scored per embryo) compared to 1.8±3.7% of wild-type cells (n=4, >200 cells scored per embryo) (Figure 6V). Similarly, the frequency of Lats2−/− passage 5 MEFs with >2 centrosomes was 34% (n=314) and 48% (n=319) in two independent lines compared to 5% (n=316) and 3% (n=307) in two independent wild-type lines of equivalent passage (Figure 6V). Functional supernumerary centrosomes might be expected to facilitate multipolar spindle formation during mitosis, leading to suboptimal chromosome segregation and aneuploidy induction. A subset of Lats2−/− MEF mitotic events in asynchronous culture (Figure 6T and U) exhibited multipolar mitotic spindles that were not observed in wild-type mitotic cells.
To more rigorously assess alterations in genome integrity, we conducted karyotypic analyses on Lats2−/− and wild-type littermate control MEF cultures of equivalent passage. As expected, a substantial number of wild-type MEFs showed loss of diploidy with approximately 56% of wild-type MEFs exhibiting tetraploidy or polyploidy (Figure 6W). In contrast, only 7% of Lats2−/− MEFs at the same passage were diploid. Lats2−/− MEFs displayed a nearly four-fold increase in the number of aberrant cells containing one or more structural chromosome rearrangements (chromosome/chromatid breaks, double minutes and centromeric fusions) and defects in chromatin condensation at mitosis. The association of Lats2 deficiency with genomic instability was not restricted to MEFs, as embryonic cells arrested in vivo showed a similar increase in the propensity to accumulate altered chromosome numbers (Supplementary Figure 7).
Lats2 is a centrosomal protein that negatively regulates centrosome duplication
As Lats2-deficient cells exhibited an increased propensity to contain supernumerary centrosomes, we sought to directly assess the relationship of this protein with centrosomes. Due to the lack of a specific antibody against Lats2, we compared the cellular localization of GFP-tagged Lats2 to GFP alone in wild-type MEFs following 24 h after transfection with the respective expression vectors. Cells transfected with GFP vector alone demonstrated no specific localization of GFP fluorescence (Figure 7A–C). In contrast, cells transfected with a GFP-Lats2 expression construct demonstrated GFP fluorescence almost exclusively restricted to one or two punctate dots per cell that were found to colocalize with centrosomes stained with anti-γ-tubulin (Figure 7D–K). In order to ascertain whether the kinase activity of Lats2 was required for centrosome localization, we transfected cells with a GFP-Lats2 expression construct mutated to ablate the kinase activity as previously described (GFP-Lats2KD, see Materials and methods). Cells transfected with GFP-Lats2KD also contained punctate staining that colocalized with anti-γ-tubulin, indicating that kinase activity was not required for centrosome localization (Figure 7L–N).
Figure 7.
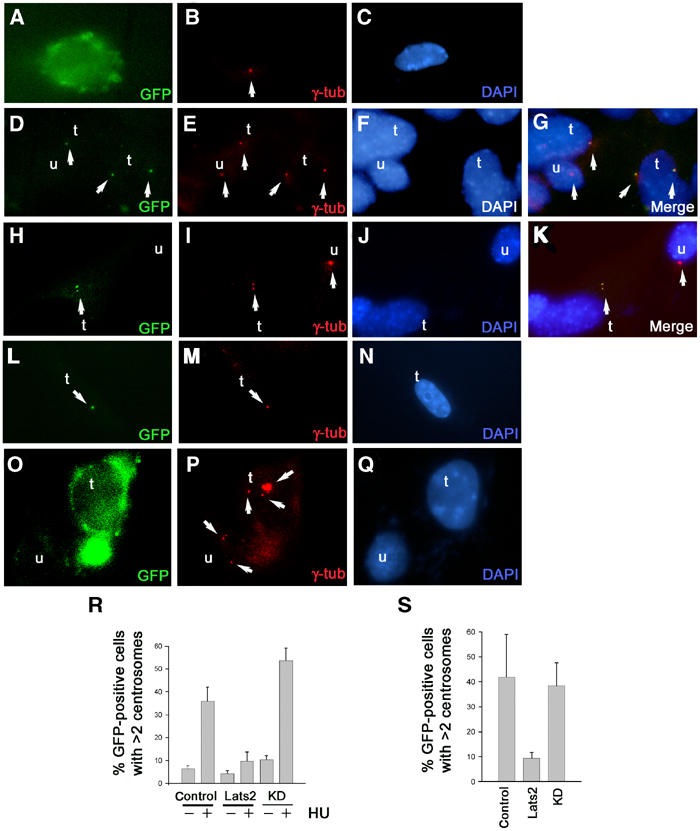
Lats2 is a centrosomal protein that suppresses centrosome overduplication. (A–K) Subcellular localization of GFP-Lats2 at centrosomes by indirect immunofluorescence. Wild-type MEFs transfected with either GFP (A–C), GFP-Lats2 (D–K) or GFP-Lats2KD (L–N) expression constructs were analysed 24 h later for GFP fluorescence (A, D, H, L) and γ-tubulin (red) immunofluorescence (B, E, I, M), and DNA was stained with DAPI (C, F, J, N). Merged images G and K confirming colocalization of GFP-Lats2 and γ-tubulin are derived from (D–F) and (H–J), respectively. Transfected cells (GFP-positive) and untransfected cells in (D–N) are denoted as t and u respectively; arrows indicate centrosomes (B, E, I, M, P), GFP-Lats2 fluorescence (D, H), GFP-Lats2KD fluorescence (L) and merged signals (G, K). (O–Q) Centrosome overduplication in the presence of HU is suppressed by GFP-Lats2. Representative GFP vector-transfected cell (t) and untransfected (u) cell (O) demonstrating supernumerary centrosomes (arrows) as visualized by γ-tubulin immunostaining (P). DNA is visualized by DAPI stain (Q). (R) Graph depicting the percentage of GFP-positive cells exhibiting >2 centrosomes from wild-type MEFs transfected with GFP vector, GFP-Lats2 (Lats2) or GFP-LatsKD (KD) in the absence or presence of 2 mM HU. (S) Centrosome overduplication in Lats2-deficient MEFs is suppressed by GFP-Lats2. Graph depicting the percentage of GFP-positive cells exhibiting >2 centrosomes from Lats2−/− MEFs transfected with GFP (control), GFP-Lats2 (Lats2) or GFP-Lats2KD (KD).
Given that GFP-Lats2 localizes to centrosomes, we sought to determine whether Lats2 directly impacts centrosome replication. Centrosome duplication initiates at the G1/S transition in mammalian cells, and is completed during S phase (Doxsey, 2001; Hinchcliffe and Sluder, 2001; Lange, 2002; Nigg, 2002). Under experimental conditions that impose a prolonged S-phase arrest in mammalian somatic cells, multiple rounds of centrosome duplication are facilitated in the absence of DNA replication or cytokinesis (Balczon et al, 1995). Using a previously described centrosome duplication assay (Meraldi et al, 1999), we investigated whether overexpression of GFP-Lats2 would suppress centrosome reduplication in wild-type MEFs following treatment with hydroxyurea (HU), an agent that facilitates S-phase arrest. HU treatment of cells transfected with GFP (Figure 7O–Q) resulted in a higher frequency of cells exhibiting three or more centrosomes per cell (35.8±6.3%, mean±s.d. of three experiments) compared to transfected cells without HU treatment (6.4±1.4%) as expected (Figure 7R). In contrast, HU-treated cells transfected with GFP-Lats2 showed a suppression of centrosome reduplication (9.8±4.0%) (Figure 7R). A requirement for the kinase activity of Lats2 in the suppression of centrosome reduplication was observed, in that HU-treated cells transfected with GFP-Lats2KD exhibited a higher frequency of cells exhibiting three or more centrosomes per cell (53.7±5.5%) compared to transfected cells without HU treatment (10.4±1.8%) (Figure 7R).
We then sought to determine whether expression of GFP-Lats2 would be sufficient to suppress the centrosome overduplication phenotype inherent in Lats2−/− MEFs. Lats2-deficient MEFs transfected with GFP-Lats2 were found to exhibit a lower frequency of cells with three or more centrosomes (9.4±2.4% of GFP-positive cells, mean±s.d. of three experiments) compared to Lats2-deficient MEFs transfected with GFP alone (41.8±17.1%). In contrast, expression of GFP-Lats2KD in Lats2-deficient MEFs did not suppress the frequency of cells with supernumerary centrosomes (38.4±9.1%), implicating Lats2 kinase activity in mediating the suppression of the centrosome overduplication phenotype (Figure 7S). These results support a role for Lats2 in maintenance of normal centrosome copy number through negative regulation of centrosome overduplication.
Discussion
Distinct requirements for mammalian warts/lats orthologues in embryonic development
The spatial disparity in embryonic expression that is especially apparent at E8.5 indicates that Lats1 and Lats2 might play specialized roles in the development of tissues of ectodermal and mesodermal origin, respectively. The observed embryonic lethality in warts/lats mutant flies contrasted with the perinatal lethality observed in a majority of Lats1−/− mice (St John et al, 1999). Our study demonstrates that the developmental requirements for Lats1 and Lats2 are strikingly different, in that Lats2 is essential for development before E12.5. Hence the embryonic expression patterns observed together with the embryonic requirement for Lats2 in contrast to Lats1 suggest that mammalian warts/lats orthologues are required during distinct developmental stages.
Differential requirements for Lats2 in developmental versus cell-intrinsic control of proliferation
The mesodermal prominence of Lats2 expression, together with the observed defects in cell growth control in lats/warts mutant flies, led to the expectation of lineage-restricted overproliferation in Lats2−/− mice. However, cellular hyperplasia was only observed to occur in a subset of Lats2−/− embryos, and was only noted in the atrial chamber of the developing heart. Furthermore, these observations contrasted to the marked ventricular hypoplasia observed in a number of embryos. These findings suggest differential requirements for Lats2 in atrial versus ventricular growth in the embryonic heart, and may explain the cardiac insufficiency observed in Lats2−/− embryos. However, the predominant defect that preceded embryonic lethality was a progressive arrest in proliferation.
The proliferation arrest in Lats2−/− embryos, but not in MEFs, indicates that Lats2 facilitates essential processes for developmental stage-specific cell cycle progression (Vidwans and Su, 2001). Lats2−/− early passage MEFs were indistinguishable from wild-type littermate control MEFs with respect to morphology, proliferation and analysis of cell cycle profiles. However, upon further passage, Lats2−/− MEFs bypass culture-induced replicative arrest and fail to exhibit the expected slow-growth phase. Normal MEFs typically arrest in response to contact inhibition of growth. Proliferation control in Lats2−/− MEFs exhibited less dependence on contact inhibition in that Lats2−/− MEF monolayers achieved higher cellular densities and exhibited the propensity to form focal overgrowths at saturated densities. These properties are strikingly reminiscent of the cell-autonomous overproliferation phenotypes observed in Drosophila with mutations in warts/lats (Bryant et al, 1993; Justice et al, 1995; Xu et al, 1995).
Impact of Lats2 on centrosome duplication and genomic integrity
Both Lats2−/− embryos and MEFs were found to exhibit propensities for centrosome amplification and genomic instability. The increased prevalence of centrosome amplification and genomic instability observed in Lats2−/− embryos and MEFs suggests that Lats2 is required to maintain correct chromosomal segregation. The subcellular localization of Lats2 to centrosomes strongly implies a role for this protein in centrosome function. Furthermore, the ability of GFP-Lats2 to suppress centrosome reduplication in the presence of HU and reverse the centrosome amplification inherent in Lats2−/− MEFs directly supports a role for this protein in a cell cycle checkpoint that regulates centrosome duplication.
Centrosomes normally replicate only once during each round of cell division, beginning near the G1/S transition and finishing by G2 phase (Doxsey, 2001; Lange, 2002). During mitosis, centrosomes facilitate the correct segregation of mitotic chromosomes through the formation of a bipolar spindle. Several studies have identified mammalian factors that regulate centrosome duplication (Meraldi et al, 1999; Okuda et al, 2000; Fisk and Winey, 2001; Chen et al, 2002). The acquisition of supernumerary centrosomes and generation of multipolar spindles during mitosis would be expected to generate conditions that facilitate the unequal segregation of chromosomes and genomic instability (Doxsey, 2001; Glotzer, 2001; Lange, 2002; Nigg, 2002). The increased prevalence of micronuclei, chromosomal defects and aneuploidy of Lats2−/− embryos and MEFs is symptomatic of structural chromosomal aberrations that result in unequal segregation of chromosomes during cell division.
It is unclear whether the impaired cytokinesis in Lats2-deficient cells is directly caused by deregulated centrosome duplication, or whether Lats2 plays an independent role in this process. A study that analysed centrosome movement in living cells during cytokinesis suggests a requirement for centrosomes in facilitating cell separation (Piel et al, 2001); however, this finding is seemingly incompatible with observations that cytokinesis proceeds to completion in cells following laser-mediated ablation of centrosomes (Khodjakov et al, 2000). As Lats2−/− MEFs display a propensity to exhibit loss of contact inhibition of growth and defects in cell division, it is tempting to speculate that Lats2 may act as a conduit in processes linking contact inhibition of growth with cell division.
Defects in centrosome copy number, cytokinesis and the aneuploidy associated with Lats2 mutation might contribute to the impaired embryonic proliferation. Embryonic requirements for proper centrosome duplication and cytokinesis might be expected to be more stringent than for cells in culture, as centrosome-dependent pathways would be expected to impact spatial controls that govern the formation of bipolar spindles, thereby determining the plane of cell division during development (Vidwans and Su, 2001).
A potential role for Lats2 in tumour suppression
The warts/lats gene family confers tumour suppressor activity both in vertebrates (St John et al, 1999) and invertebrates (Bryant et al, 1993; Justice et al, 1995; Xu et al, 1995). Interestingly, Lats2 deficiency was found to impair cytokinesis, promote the accumulation of supernumerary centrosomes and genomic instability. Supernumerary centrosomes and multipolar mitotic spindles have been predicted to contribute to aneuploidy and cancer development (Lingle and Salisbury, 2000); however, it remains to be shown whether centrosome amplification is a cause or a consequence of aneuploidy (Doxsey, 2001; Lange, 2002; Nigg, 2002). An imbalance in chromosomal partitioning might facilitate the emergence of cells defective in various aspects of proliferation control observed. Subsequent cell divisions in the presence of unbalanced centrosome numbers would then be expected to contribute to the chromosome mis-segregation and the generation of aneuploid cell populations. Increased number of micronuclei in Lats2−/− cells might indicate the loss of whole chromosome(s) or chromosomal regions as these are eliminated from the cells as micronuclei during subsequent cell division (Therman and Susman, 1993).
Previous studies have implicated warts/lats family members as tumour suppressors; however, the underlying mechanisms remain unclear. Although mammalian Lats1/h–Warts-1 has been found to be associated with Cdk1, a direct link between tumorigenesis in Lats1/Warts-1-deficient mice and loss of Cdk1 control is unclear. Furthermore, the deregulation of Cdk1 activity as a mechanism sufficient to explain the excessive cell proliferation in Drosophila warts/lats mutants has been questioned (Weinkove and Leevers, 2000; Tapon et al, 2002). Lats2−/− embryonic lethality presently impedes direct evaluation of Lats2 as a tumour suppressor; however, the bypass of replicative arrest and loss of contact inhibition in MEFs provide direct evidence for a conserved role of Lats2 in processes linked to proliferation control. Furthermore, we demonstrate that Lats2 deficiency promotes genomic instability. The localization of Lats2 to centrosomes and its role as a negative regulator of centrosome copy number is supportive of a role for centrosome amplification as a process that facilitates genomic instability.
Materials and methods
Northern analysis and in situ hybridization
Northern blot analysis was performed with embryonic RNA blots (Clontech) using Lats1 and Lats2 cDNAs as probes. Whole-mount embryo in situ hybridization was performed as described previously (Bruneau et al, 2001). In situ hybridization of embryonic tissue sections was performed using 33P-labelled RNA probes specific for Lats1, Lats2, cardiac actin, Nkx2.5, PPARγ, and Bmp4 according to standard methodology.
Targeted disruption of Lats2
A targeting strategy by use of homologous recombination in E14K ES cells was devised to create an insertional mutation resulting in the disruption of downstream kinase domains of Lats2. The targeting vector contained the neomycin gene flanked by a 0.6-kb upstream homologous region and a 6.0-kb downstream homologous region. The upstream homology short arm sequence was modified to replace sequence encoding serine 371 with a stop codon, thereby disrupting translation of the downstream carboxy-terminal kinase domain. G418-resistant ES clones were screened for homologous recombination by PCR using primers 5′-CCCAGGCTCACCAGCATCCTC-3′ and 3′-CTCACCCTCCTTACTCGACCGG-5′. Correctly targeted ES cells were further verified by Southern analysis. Four Lats2 heterozygote ES cell lines were injected into C57BL/6 blastocysts and transferred to pseudopregnant ICR females. Germline transmission was verified using Southern analysis. Genotyping of DNA isolated from ES cells, mouse tails or yolk sacs was performed by PCR using primers 5′-CGCCACCAGATGCCTATTCCA-3′ and 3′-ACCTCTGCTTCCTCCCGTCG-5′ (wild type) or primers 5′-CCCAGGCTCACCAGCATCCTC-3′ and 3′-CTCACCCTCCTTACTCGACCGG-5′ (mutant). Western blots of protein from wild-type and Lats2−/− MEFs were performed using affinity-purified antisera raised against Lats2 aa 255–273 (YGVQRSSSFQNKTPPDAYS).
Histological analysis, BrdU incorporation, TUNEL analysis and immunohistochemistry
Mouse embryos were fixed, dehydrated and processed according to standard protocols. Embryonic vascularization was analysed with anti-PECAM immunostaining. Placental differentiation was analysed by placental PPARγ expression (Barak et al, 1999). Pregnant mice were injected with 0.1 mg BrdU/g mouse weight intraperitoneally 45 min prior to collecting embryos. Sections of embryos were immunostained for BrdU as described previously (Hakem et al, 1998). TUNEL staining was performed on histological sections using an in situ cell death detection kit (Boehringer Mannheim).
Analysis of embryonic cells and derivation of embryonic fibroblasts
E10.5 embryos (judged to be alive by visual confirmation of cardiac contractions) were harvested, trypsinized and seeded in αMEM media with 10% fetal bovine serum. For immunostaining of embryonic cells, freshly trypsinized embryos were seeded on coverslips coated with poly-L-lysine and processed as described below. Initial MEF cultures were obtained in 6 cm dishes and considered at passage 0 prior to dilution. For growth curves, 3 × 105 cells/6 cm dish at passage 2 were plated and counted, before dilution to 3 × 105 cells/dish for the next passage. For saturated density of growth, 3 × 105 cells/6 cm dish at passage 2 or 3 were plated, counted and replated without dilution on the indicated days. For colony formation assays, 3 × 103 cells/6 cm dish were seeded and the number of colonies formed was enumerated. Immunostaining was performed on cells according to standard methodology with either anti-α-tubulin (FITC-conjugated, Sigma), anti-actin (Sigma), anti-phospho-histone H3 (Upstate Biotechnologies) or anti-γ-tubulin (Sigma), followed by TRITC-conjugated donkey anti-mouse or anti-rabbit secondary IgG (Jackson). Centrosomes were enumerated in cells from three pairs of wild-type or Lats2−/− sibling embryos as well as two independently derived Lats2−/− and wild-type MEF lines from sibling embryos at passage 5 following γ-tubulin immunostaining.
MEF transfection and centrosome duplication assays
A full-length Lats2 cDNA cloned from a mouse cDNA library (Origene) was modified by PCR to facilitate ligation into pEGFP-C2 (Clontech) as an EcoRI–XhoI fragment fused in-frame with the GFP coding sequence. A kinase-inactive mutant of Lats2 was created by PCR (as described in Hori et al, 2000) and inserted into pEGFP-C2 fused in-frame with GFP coding sequence. Wild-type or Lats2−/− MEFs were seeded onto coverslips in six-well tissue culture dishes and DNA constructs were transfected using Lipofectamine PLUS (Invitrogen/Life Technologies). At 3 h after transfection, cells were treated with HU (Sigma) at a final concentration of 2 mM for 36 h and processed for indirect immunofluorescence as described previously. Quantitative analysis of centrosome numbers was performed on 200–600 GFP-positive cells/transfection and each transfection was replicated in at least three separate experiments.
Karyotyping analysis
Mitotic MEFs were collected following colcemid treatment and processed by standard cytogenetic procedures. Slides were stained with DAPI (Sigma) and chromosome number and gross chromosomal rearrangements were determined in 50 metaphase cells per sample from each genotype (two samples per genotype were used). For in vivo studies, pregnant females were injected with colcemid (5 μg) 1 h prior to harvesting. E9.5 embryos were isolated and used to prepare single-cell suspensions, which were incubated with colcemid (100 ng/ml) for 4 h at 37°C. Cells were then processed by standard cytogenetic procedures.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank J Rossant, M Soengas and J de la Pompa for reviewing the manuscript, JM Roberts and A Jurisicova for helpful advice, and D Bouchard and A Wakeham for technical support. JPM was supported by a fellowship from the Canadian Institute of Health Research. This work was supported by Amgen Inc. and by grants from the Canadian Institute of Health Research and the National Cancer Institute of Canada to RH. BGB is supported by the Canadian Institute of Health Research and the Heart and Stroke Foundation of Ontario. MPH acknowledges the grant support from Academic Research Fund and Oncology Research Institute (OLS), National University of Singapore.
References
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL (2001) Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 12: 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR (1995) Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 130: 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM (1999) PPARy is required for placental, cardiac, and adipose tissue development. Mol Cell 4: 585–595 [DOI] [PubMed] [Google Scholar]
- Borel F, Lohez OD, Lacroix FB, Margolis RL (2002) Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc Natl Acad Sci USA 99: 9819–9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkley BR (2001) Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol 11: 18–21 [DOI] [PubMed] [Google Scholar]
- Bruneau BG, Bao ZZ, Fatkin D, Xavier-Neto J, Georgakopoulos D, Maguire CT, Berul CI, Kass DA, Kuroski-de Bold ML, de Bold AJ, Conner DA, Rosenthal N, Cepko CL, Seidman CE, Seidman JG (2001) Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol Cell Biol 18: 5099–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant PJ, Watson KL, Justice RW, Woods DF (1993) Tumour suppressor genes encoding proteins required for cell interactions and signal transduction in Drosophila. Dev Suppl 1: 239–249 [PubMed] [Google Scholar]
- Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD (2002) CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 3: 339–350 [DOI] [PubMed] [Google Scholar]
- Doxsey S (2001) Re-evaluating centrosome function. Nat Rev Mol Cell Biol 2: 688–698 [DOI] [PubMed] [Google Scholar]
- Fisk HA, Winey M (2001) The mouse Mps1p-like kinase regulates centrosome duplication. Cell 106: 95–104 [DOI] [PubMed] [Google Scholar]
- Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ (1996) 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci USA 93: 7081–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M (2001) Animal cell cytokinesis. Annu Rev Cell Dev Biol 17: 351–386 [DOI] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94: 339–352 [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Kastan MB (1994) Cell cycle control and cancer. Science 266: 1821–1828 [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Sluder G (2001) ‘It takes two to tango': understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev 15: 1167–1181 [DOI] [PubMed] [Google Scholar]
- Hirota T, Morisaka T, Nishiyama Y, Marumoto T, Tada K, Hara T, Masuko N, Inagaki M, Hatakeyama K, Saya H (2000) Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumour suppressor. J Cell Biol 149: 1073–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Takaori-Kondo A, Kamikubo Y, Uchiyama T (2000) Molecular cloning of a novel human protein kinase, kpm, that is homologous to warts/lats, a Drosophila tumor suppressor. Oncogene 19: 3101–3109 [DOI] [PubMed] [Google Scholar]
- Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ (1995) The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev 9: 534–546 [DOI] [PubMed] [Google Scholar]
- Kamikubo Y, Takaori-Kondo A, Uchiyama T, Hori T (2003) Inhibition of cell growth by conditional expression of kpm, a human homologue of Drosophila warts/lats2 tumor suppressor. J Biol Chem 278: 17609–17614 [DOI] [PubMed] [Google Scholar]
- Khodjakov A, Cole RW, Oakley BR, Rieder CL (2000) Centrosome-independent mitotic spindle formation in vertebrates. Curr Biol 10: 59–67 [DOI] [PubMed] [Google Scholar]
- Lange BMH (2002) Integration of the centrosome in cell cycle control, stress response and signal transduction pathways. Curr Opin Cell Biol 14: 35–43 [DOI] [PubMed] [Google Scholar]
- Li Y, Pei J, Xia H, Ke H, Wang H, Tao W (2003) Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 22: 4398–4405 [DOI] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL (2002) Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA 99: 1978–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL (1998) Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci USA 95: 2950–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Salisbury JL (2000) The role of the centrosome in the development of malignant tumors. Curr Top Dev Biol 49: 313–329 [DOI] [PubMed] [Google Scholar]
- Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD (1992) Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 70: 923–935 [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA (2002) Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 21: 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2–cyclin A. Nat Cell Biol 1: 88–93 [DOI] [PubMed] [Google Scholar]
- Minn AJ, Boise LH, Thompson CB (1996) Expression of Bcl-xL and loss of p53 can cooperate to overcome a cell cycle checkpoint induced by mitotic spindle damage. Genes Dev 10: 2621–2631 [DOI] [PubMed] [Google Scholar]
- Mitelman F (1971) The chromosomes of fifty primary Rous rat sarcomas. Hereditas 69: 155–186 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2: 21–32 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2002) Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2: 1–11 [DOI] [PubMed] [Google Scholar]
- Nishiyama Y, Hirota T, Morisaki T, Hara T, Marumoto T, Iida S, Makino K, Yamamoto H, Hiraoka T, Kitamura N, Saya H (1999) A human homolog of Drosophila warts tumor suppressor, h-warts, localized to mitotic apparatus and specifically phosphorylated during mitosis. FEBS 459: 159–165 [DOI] [PubMed] [Google Scholar]
- Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K (2000) Nucleophosmin/B23 is a target of CDK/cyclin E in centrosome duplication. Cell 103: 127–140 [DOI] [PubMed] [Google Scholar]
- Piel M, Nordberg J, Euteneuer U, Bornens M (2001) Centrosome-dependent exit of cytokinesis in animal cells. Science 291: 1550–1553 [DOI] [PubMed] [Google Scholar]
- St John MAR, Tao W, Fei X, Fukumoto R, Carcangi ML, Brownstein DG, Parlow AF, McGrath J, Xu T (1999) Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet 21: 182–186 [DOI] [PubMed] [Google Scholar]
- Tao W, Zhang S, Turenchalk GS, Stewart RA, St John MA, Chen W, Xu T (1999) Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet 21: 177–181 [DOI] [PubMed] [Google Scholar]
- Tapon N, Harvey KF, Bell DW, Wahrer DCR, Shiripo TA, Haber DA, Hariharan IK (2002) Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110: 467–478 [DOI] [PubMed] [Google Scholar]
- Therman E, Susman M (1993) Human Chromosomes, Structure, Behaviour and Effects. New York, NY: Springer-Verlag [Google Scholar]
- Vidwans SJ, Su TT (2001) Cycling through development in Drosophila and other metazoa. Nat Cell Biol 3: E35–E39 [DOI] [PubMed] [Google Scholar]
- Weinkove D, Leevers SJ (2000) The genetic control of organ growth: insights from Drosophila. Curr Opin Genet Dev 10: 75–80 [DOI] [PubMed] [Google Scholar]
- Xu T, Wang W, Zhang S, Stewart RA, Yu W (1995) Identifying tumor suppressors in genetics mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121: 1053–1063 [DOI] [PubMed] [Google Scholar]
- Yabuta N, Fujii T, Copeland NG, Gilbert DJ, Jenkins NA, Nishiguchi H, Endo Y, Toji S, Tanaka H, Nishimune Y, Nojima H (2000) Structure, expression, and chromosome mapping of LATS2, a mammalian homologue of the Drosophila tumour suppressor gene lats/warts. Genomics 63: 263–270 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures