

ABSTRACT
Definition of cell cycle control proteins that modify tumor cell resistance to estrogen (E2) signaling antagonists could inform clinical choice for estrogen receptor positive (ER+) breast cancer (BC) therapy. Cyclin G2 (CycG2) is upregulated during cell cycle arrest responses to cellular stresses and growth inhibitory signals and its gene, CCNG2, is directly repressed by E2-bound ER complexes. Our previous studies showed that blockade of HER2, PI3K and mTOR signaling upregulates CycG2 expression in HER2+ BC cells, and that CycG2 overexpression induces cell cycle arrest. Moreover, insulin and insulin-like growth factor-1 (IGF-1) receptor signaling strongly represses CycG2. Here we show that blockade of ER-signaling in MCF7 and T47D BC cell lines enhances the expression and nuclear localization of CycG2. Knockdown of CycG2 attenuated the cell cycle arrest response of E2-depleted and fulvestrant treated MCF7 cells. These muted responses were accompanied by sustained inhibitory phosphorylation of retinoblastoma (RB) protein, expression of cyclin D1, phospho-activation of ERK1/2 and MEK1/2 and expression of cRaf. Our work indicates that CycG2 can form complexes with CDK10, a CDK linked to modulation of RAF/MEK/MAPK signaling and tamoxifen resistance. We determined that metformin upregulates CycG2 and potentiates fulvestrant-induced CycG2 expression and cell cycle arrest. CycG2 knockdown blunts the enhanced anti-proliferative effect of metformin on fulvestrant treated cells. Meta-analysis of BC tumor microarrays indicates that CCNG2 expression is low in aggressive, poor-prognosis BC and that high CCNG2 expression correlates with longer periods of patient survival. Together these findings indicate that CycG2 contributes to signaling networks that limit BC.
KEYWORDS: Cell Cycle Arrest, CCNG2, CDK10, Estrogen Deprivation, Fulvestrant, IGF-1R, Insulin, Metformin, RAF/MEK/MAPK pathway, Tamoxifen-Resistant
Introduction
Mitogenic signaling through either estrogen (E2) activated estrogen receptors (ER) or human epidermal growth factor receptor 2 (HER2, Neu/ErbB2) drives tumor cell proliferation and growth in most primary breast cancers (BC).1,2 Indeed, approximately 75% of BC patients are initially diagnosed with ER positive (ER+) estrogen-dependent tumors.2 Thus systemic blockade of ER signaling is essential for therapeutic management of BC in the majority of patients. Selective ER modulators (SERMs), downregulators (SERDs) and E2 synthesis inhibitors (aromatase inhibitors (AIs)) form the cornerstone of available endocrine therapies.3,4 Use of the SERM tamoxifen as a first line adjuvant therapy has significantly increased 5-year survival rates and has become the standard of care for successful treatment of primary ER+ BC. However, ER+ BC tumors are comprised of a heterogeneous group of cells that will exhibit variable sensitivity to endocrine-based therapeutics over a course of treatment.3,5 Thus, despite the initial success of SERM therapy, the occurrence of tumor resistance to tamoxifen is observed in a significant proportion of patients.3,5 The intramuscularly (IM) administered SERD fulvestrant has been used as a second or third line endocrine therapy and primarily restricted to treatment of ER+ metastatic breast cancer (MBC)[6]. Recent studies have shown that high dose fulvestrant (500mg/IM) is superior to AI therapy in improving overall survival of ER+ MBC patients.7 However tumor resistance to fulvestrant eventually occurs in the majority of MBC patients. Altogether, the occurrence of de novo and acquired tumor cell resistance to SERM, SERD and AIs limits the long-term efficacy of endocrine therapy in a significant proportion (~25-50 %) of patients.2,4 Hence, identification of tumor biomarkers that can better predict tumor responsiveness to endocrine based therapeutics and the potential for acquired resistance to these drugs are needed to improve therapeutic outcomes and extend relapse free survival.
Estrogen stimulation of tumor growth is achieved in large part through ER regulation of gene expression. Cyclin G2 (CycG2) is an unconventional but well conserved cyclin paralog encoded by the estrogen-regulated gene, CCNG2.8-10 CCNG2 is negatively regulated by E2 bound ER through the recruitment of the N-CoR co-repressor complex and histone deacetylases (HDACs) to CCNG2 promoter region.10 In contrast to the prototypical cell cycle promoting cyclins, increased CycG2 expression is predominately associated with growth inhibition.8,11-17 CCNG2 transcripts are strongly upregulated during G1 and G2-phase cell cycle arrest responses to ligand activated growth inhibitory signaling, DNA damage, and various environmental stresses.8,14,17-22 Elevation of CCNG2 expression also correlates with the onset of cellular differentiation in a variety cell types including oral epithelia,13 haematopoietic cells8,23,24 and neurons.25,26 Furthermore, ectopic CycG2 expression triggers a cell-cycle arrest in numerous cell types.11,12,14 Thus the findings that basal transcription of CCNG2 is directly repressed by E2-bound ER interactions at the CCNG2 promoter suggests that inhibition of CycG2 expression promotes E2-mediated stimulation of BC cell proliferation.10
We previously reported that CycG2 overexpression blunts CDK2 activity and induces a p53-dependent G1-phase cell cycle arrest.11,12 Even moderate inducible expression of ectopic CycG2 inhibits cell cycle progression.14,27,28 Notably, reduced CCNG2 expression has been observed in some cancers, suggesting that loss of CycG2 promotes cancer development or progression.13,27,29-31 Our recent work showed that shRNA-mediated repression of CycG2 expression dampens the G2/M checkpoint arrest response of cells to the DNA-damaging chemotherapeutic doxorubicin.17 Although CCNG2 mRNA levels are increased during the G1/S-phase arrest response of E2-depleted ER+ BC cells,10 the contribution of CycG2 to the cell cycle inhibitory effects of therapeutics that blockade E2 signaling in BC cells is unknown.
BC resistance to endocrine therapy arises in part from crosstalk between the ER and growth factor receptor tyrosine kinase (RTK) signaling pathways. Elevated expression of HER2, as well as increased signaling through the insulin (IR) and insulin-like growth factor (IGF-1R) RTKs triggers hyperactivation of the pro-growth PI3K/AKT/mTOR pathway and development of endocrine therapy-resistance.2-5,32 Hyperactivation of PI3K/AKT/mTOR signaling also induces ER phosphorylation, a modification that promotes ligand-independent ER activity.32 We and others determined that signaling through HER2, IR and IGF-1R RTKs inhibits CycG2 expression and that pharmacological blockade of HER2, IR and IGF-1R signaling or direct suppression of PI3K or mTOR activity upregulates CycG2 expression coincident with induction of a G1-phase cell cycle arrest.14,15,33,34 These findings suggest that suppression of CycG2 expression is at the nexus of ER and RTK crosstalk and that downregulation of CycG2 may be a contributing factor to BC growth.
Obesity and type II diabetes with hyperinsulinemia are associated with higher risk of breast cancer and poor outcomes.35-37 The increased expression of IR isoform A (IR-A) and IGF-1R frequently observed in ER+ breast cancer tumors together with their mitogenic potential and ability to form hybrid receptors suggests a mechanism by which uncontrolled insulin levels could promote tumor growth.38-41 Compelling epidemiological and pre-clinical evidence suggests that the insulin-sensitizing biguanide metformin has anti-cancer effects.42-45 In addition to its anti-glucogenic activity, studies suggest that metformin triggers inhibition of mTOR by AMP-activated protein kinase (AMPK) and promotes cell cycle arrest.45,46 Given that stimulation of BC cell lines with insulin or IGF-1 blunts CycG2 expression,33 and that the mTOR inhibitor rapamycin increases CycG2 levels,14,34 metformin treatment could promote CycG2 expression and its associated cell cycle inhibitory activity.
Here we examine CycG2 expression and localization during BC cell responses to E2 deprivation, ER antagonism by the SERD fulvestrant and treatment with the AMPK activator metformin. We relate changes in CycG2 expression to the anti-mitogenic effects of these treatments in BC cell lines and test the consequence of CycG2 depletion to the anti-proliferative effects induced by blockade of E2 signaling and co-treatment with metformin in MCF7 cells. The effects of blunted CycG2 expression on changes in signal transduction pathways triggered by ablation of E2 signaling are evaluated, and a potential CycG2 binding partner and mechanism by which CycG2 could mediate its growth inhibitory effects are identified. Finally, we use curated microarray datasets from BC patient tumor samples and meta-analysis tools to assess the potential contribution of CycG2 to tumor growth control and patient survival outcomes. Taken together our results strongly suggest that CycG2 contributes to signaling networks that restrict BC cell proliferation and has value as a prognostic biomarker for treatment of ER+ BC.
Methods
Cell culture and treatment
MCF7 and T47D cells were obtained from American Type Culture Collection. MCF7 cells were cultured in EMEM (Gibco) supplemented with 10% heat-inactivated (HI)-FBS, 2 mM L-glutamine (RPI), 1 mM sodium pyruvate, 100 units/mL penicillin and 100 µg/mL streptomycin sulfate (Gibco), and 10 µg/mL bovine insulin (Sigma). T47D cells were cultured in RPMI1640 (Gibco) supplemented with 10% HI-FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin and 100 µg/mL streptomycin sulfate. All cultures were plated at a density of 20-30% and maintained at 50-90% confluency in a humidified chamber at 37°C with 5% CO2. For estrogen depletion experiments, plated cells were washed twice with 1x PBS (Gibco) and cultured for indicated periods of time in phenol red free MEM medium containing 10% heat inactivated charcoal-dextran (CD)-treated FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 units/mL penicillin and 100 μg/mL streptomycin sulfate, 10 μg/mL bovine insulin and 10 mM HEPES (Gibco). Re-stimulation of E2-starved cultures was achieved by addition of 10 nM estradiol (E2). For ICI and 4OHT treatments cells were seeded the day before treatment to achieve approximately 70% confluency at the point of harvest. Culture medium was replaced with ICI (100 nM) or 4OHT (100 nM) containing regular MCF7 (E2 and phenol red containing) medium for the indicated time periods.
shRNA expression constructs and establishment of stable clones
The generation of DNA constructs for expression of GFP-epitope tagged CycG2 fusion proteins (full length and truncation) in mammalian cells have been described previously.11,12 The HA tagged human CDK10 vector pReceiver-M08 was purchased from GeneCopoeia (Rockville, MD). The generation of stable cyclin G2 knockdown cell lines has been described elsewhere.17 In brief, the vector pSuper.retro.puro (Oligoengine; Seattle, WA) and oligonucleotides (Integrated DNA Technologies, IA) were ligated via BglII/HindIII cloning site. The shRNA oligonucleotide stem-loop and restriction site overhang sequences were previously described.17 Freshly established MCF7 cultures were transfected with NdeI linearized vector and clonal populations were generated by culturing with 3 µg/mL puromycin. Selected clonal populations were tested for their ability to suppress expression of exogenous and endogenous human CycG2 by immunoblot analysis.
Antibodies (source and dilutions)
The following primary antibodies, mouse anti-α-tubulin (DM1A, sc-32293, 1:20 000), goat anti-CDK10 (sc-326, 1:100), mouse anti-cyclin D1 (sc-20044, 1:6 000) were obtained from Santa Cruz Biotechnology, mouse anti-GAPDH (MAB374, 1:200 000) was obtained from Millipore, rabbit anti-β-actin (#4970, 1:2 000), anti-cRaf (#9422, 1:500), anti-phospho cRaf S338 (#9457, 1:1 000), anti-ERK (#9102, 1:2 000), anti-phospho ERK T202/204 (#9101, 1:3 000), anti-MEK (#8727, 1:1 000), anti-phospho MEK S217/221 (#9121, 1:3 000), anti-phospho Rb (#9307, 1:3 000) and mouse anti Rb (#9309, 1:2 000) were purchased from Cell Signaling, sheep anti-α-tubulin (ATN02, 1:100) and mouse anti-γ-tubulin (GTU-88, T6557, 1:400) were purchased from Cytoskeleton (Denver, CO) and Sigma, respectively, mouse anti-HA (MMS-101-P, 1:1 000) was from Covance (CA) and mouse anti-BrdU (B35141, 1:40) was from Invitrogen. HRP conjugated secondary antibodies against rabbit and mouse IgG (1:5 000) were purchased from BioRad and Jackson ImmunoResearch. Alexa 488, 568 and 660 conjugated secondary antibodies (1:1 000) were purchased from Molecular Probes/Invitrogen. The CycG2-specific antibodies 68232 (Ab1; 1:300) and 68964 (Ab2; 1:500) produced in our laboratory were affinity purified from rabbit anti-sera and tested for specificity toward CycG2 fusion proteins essentially as described.11,12,17
Immunoblot analysis
Cells were lysed in RIPA buffer (10% glycerol, 1% Nonidet P-40, 0.4% deoxycholate, 0.05% SDS, 150 mM NaCl, 10 mM EDTA, 5 mM EGTA, 50 mM Tris, pH 7.4) containing protease inhibitors (1 µg/mL pepstatin A, 1 µg/mL leupeptin, 2 µg/mL aprotinin, 200 nM phenylmethanesulfonyl fluoride) and phosphatase inhibitors (25 mM sodium fluoride, 25 mM sodium pyrophosphate, 1 mM p-Nitrophenyl-phosphate, 2 µM microcystin). Cell lysates were cleared by centrifugation at 10,000 x g to remove insoluble material. Protein concentration was measured using BCA reagent (Pierce). Protein lysates were fractionated by SDS-PAGE, blotted onto PVDF membranes and subjected to immunoblotting as previously described.11,12,17 Multiple exposures over increasing time periods were taken to ensure that all signals were in the linear range. Immunoblot signals were quantified using ImageJ (National Institutes of Health) and Adobe Photoshop CS3 software (Adobe Systems Inc.). Statistical analysis of data from 3 or more independent repeats was done using GraphPad Prism version 4.0 (GraphPad Software, Inc.). Data are shown as mean ± SE. Student's t test was used for 2-group comparison, and analysis of variance (one-way ANOVA with the Tukey and Bonferroni post hoc tests) was used for comparisons of more than 2 groups.
Immunoprecipitation
Cleared whole lysate protein (150 to 1000 μg) was precleared with 20 μL protein A agarose or protein G sepharose bead slurry including non-specific IgG and incubated at 4ºC for 1 h, to remove non-specific binding. This precleared supernatant was then incubated with 20 μL bead slurry and 1 to 10 μg indicated precipitation antibody for 16 h at 4ºC. Preclear and immunoprecipitation beads were extensively washed with lysis buffer and proteins were eluted with 20 μL 1.5x SDS loading buffer and subjected to immunoblotting as described.11,12,17
Immunofluorescence microscopy
MCF7 cells were seeded at 1.5 × 105 cells/35 mm well onto a 22 mm-square glass coverslip coated with 10 mg/mL collagen and 1 µg/mL poly-L-lysine (Sigma) 14-18 h before treatment. Coverslips were removed 72 h after initiation of treatment, rinsed with PBS and immediately fixed with 4% paraformaldehyde in PBS for 10 min at RT or ice-cold MeOH at -20°C for 5 min. PFA fixed specimens were permeabilized with 0.4% Triton-X in PBS for 20 min at RT. Specimens were stained and mounted and images were collected by confocal microscopy as described.11 In brief, cells were blocked for 2 h in blocking solution (2% Glycerol, 50 mM NH4Cl, 5% FBS, 2% goat serum in PBS) and stained with indicated primary antibodies diluted in blocking solution ON at 4ºC. Secondary antibodies and Hoechst 33342 stain were diluted in TBS containing blocking solution and incubated with the cells for 2 h at RT. Coverslips were mounted with ProLong Antifade from Molecular Probes on microscope slides and images were collected by confocal microscopy.
Cell cycle analysis by flow cytometry
The DNA content in cell cultures was assessed by single and dual parameter flow cytometry of propidium iodide stained DNA. For single parameter DNA flow cytometry cultured cells were fixed with 70% EtOH at -20°C, to preserve fluorescent signals, cells were 10 min prefixed with 0.5% PFA/10 mM EDTA in PBS following fixation with 100% methanol at -20ºC. Fixed cells were washed, and stained in PBS containing 0.25 mg/mL RNase A (Fermentas) and 50 µg/mL propidium iodide (PI, Sigma) for 30 min at RT. DNA flow cytometry was performed using a FACScan (BD) flow cytometer to collect at least 20,000 events as described.8,9,11,17 Two parameter DNA flow cytometry of BrdU labeled DNA was used to measure the percentage of cells in the population undergoing DNA synthesis. Here cells were pulse labeled with 10 μM BrdU (Sigma) for 1 h at 37°C prior methanol fixation at -20°C. DNA was denatured with 2 M HCl for 45 min at RT and neutralized by addition of 0.1 M Sodium-Borate buffer pH 8.2. BrdU labeled DNA was stained with anti-BrdU primary antibodies for 2h at RT followed by incubation with Alexa488 conjugated secondary antibodies for 1h at RT. Total DNA was then stained with PI as above and FACScan analysis was performed. Cell Cycle analysis of PldIns or BrdU labeled DNA was performed using FlowJo 8.5 and 10.1 software. For statistical analysis, one-way analysis of variance tests (one-way ANOVA with Tukey and Bonferroni post-hoc tests) was done on data from at least 3 independent experimental repeats using Prism 4.0 software (GraphPad Software, Inc., www.graphpad.com).
Meta-analysis of CycG2 expression in breast cancer tumors
Microarray analysis of CCNG2 gene expression in tumor samples from breast cancer patients was done using publically available data sets and the following online meta-analysis tools: 1) the GOBO - Gene expression-based Outcome for Breast Cancer Online software47 that curates Affymetrix U133A microarray expression data in a merged 1881-sample breast tumor data set from 11 different publicly available data sets (http://co.bmc.lu.se/gobo) and 2) the KM-plotter tool48 for breast cancer (http://kmplot.com/analysis/) that assesses Affymetrix HG-U133A, HG-U133 Plus 2.0 and HG-U133A 2.0 microarray expression data downloaded from GEO (Affymetrix microarrays only), EGA and TCGA databases. High and low quantile CCNG2 expression levels in tumor samples from specific patient cohorts were compared using Kaplan-Meier survival plots, including calculations of the hazard ratio with 95% confidence intervals and log-rank P values.
Results
Estrogen deprivation of MCF7 and T47D ER+ BC cell lines upregulates CycG2 levels during cell cycle arrest and induces accumulation of CycG2 in MCF7 nuclei
As expression of the CCNG2 gene is negatively regulated by E2-bound ER/NCoR transcription factor complexes,10 we examined the effects of E2 deprivation and re-stimulation on the expression and subcellular localization of the CycG2 protein in ER+ BC cell lines (Fig. 1). MCF7 and T47D cells were cultured in non-treated (NT) and E2-depleted (DM) media for 4 d. DNA flow cytometry (Fig. 1A) showed the expected cell cycle arrest response of MCF7 cells to E2-deprivation and re-entry after re-stimulation with 10 nM E2. Immunoblot analysis shows a 2-fold increase in CycG2 protein levels in the E2-depleted T47D and MCF7 cultures compared to non-treated controls (Fig. 1B) and a 2.5-fold downregulation of CycG2 4 h after the re-addition of 10 nM E2 to MCF7 cells (Fig. 1C). Confocal microscopy of immunostained MCF7 cells cultured in the presence (NT) versus absence of E2 (DM) shows that the bulk of CycG2 upregulated upon E2-deprivation is localized within the nucleus (Fig. 1D). Note that the basal level of CycG2 in non-treated cells is relatively low and is mainly cytoplasmic, except when localized at centrosomes of mitotic spindles (yellow dot at end of mitotic spindle). Re-stimulation of E2-depleated MCF7 cells with 10 nM E2 for 72 h resulted in an obvious reduction in the nuclear CycG2 immunosignal (Fig. 1E).
Figure 1.

CycG2 expression is modulated by estrogen signaling in breast cancer cells. (A) Left, Histogram overlays of DNA content in MCF7 cells grown in E2-depleted medium (DM, dashed line) for 4 d vs. those cultured in non-treated medium (NT, light gray fill) and E2-depleted cultures re-stimulated with 10 nM E2 for an additional 48 h (DM+E2, dark gray fill). Right, One-way ANOVA statistical analysis of cumulative DNA flow cytometry data arising from experimental conditions shown at left. (B) Immunoblot analysis of CycG2 expression in MCF7 and T47D cells depleted of estrogen (DM) for 4 d relative to control protein (GAPDH or α-tubulin and Ponceau S stain). (C) Immunoblot analysis of CycG2 expression following 4 h re-stimulation of 3 day E2-depleted MCF7 cells with (+) 10 nM E2 compared to non-treated control (-). (D) Confocal immunofluorescence micrographs of expression and localization of CycG2 in MCF7 cells cultured in E2 containing (NT) or in E2-depleted (DM) medium for 6 d. Cells were stained with antibodies to CycG2 (shown in green channel) and α-tubulin (αTub, red). (E) Confocal micrographs of CycG2 expression and localization following 72 h re-stimulation of E2-starved (DM) MCF7 cells with 10 nM E2 (DM+E2). Micrographs at top show immunosignals for CycG2 (green), lamin B (Lam B, blue; nuclear marker) and γ-tubulin (γTub, red; centrosome marker); micrographs at bottom show single channel anti-CycG2 immunosignal from above images in black and white for better contrast.
Pharmacological blockade of ER signaling upregulates CycG2 expression in MCF7 and T47D cells and increases nuclear localization of CycG2 in MCF7 cells
Given that CycG2 expression is upregulated upon E2 depletion, we tested the effect of BC therapeutics that antagonize ER signaling on CycG2 expression in ER+ BC cell lines (Fig. 2). MCF7 and T47D cells were cultured with fulvestrant (ICI) for up to 48 hours. Fulvestrant induced a 2-3-fold upregulation of CycG2 expression (Fig. 2A and B) in MCF7 cells and a 2-fold increase in T47D cells (Fig. 2B). Immunofluorescence microscopy showed that, as with E2 depletion, fulvestrant also induced CycG2 accumulation within the nuclei of MCF7 cells (Fig. 2C). Treatment of cells with 100 nM of the SERM tamoxifen (TAM) also increased CycG2 protein levels (Fig. 2D), though not to the same degree as fulvestrant. To better define the effects of SERM and SERD-mediated antagonism of estrogen signaling on CycG2 expression, cells were cultured in estrogen-depleted media followed by stimulation with E2 in the presence or absence of fulvestrant or tamoxifen. The increased CycG2 expression level (1.7-fold) induced by E2 deprivation was repressed by E2 stimulation alone while co-treatment with fulvestrant increased CycG2 by 3.8-fold and tamoxifen co-treatment upregulated CycG2 by 2.9-fold.
Figure 2.
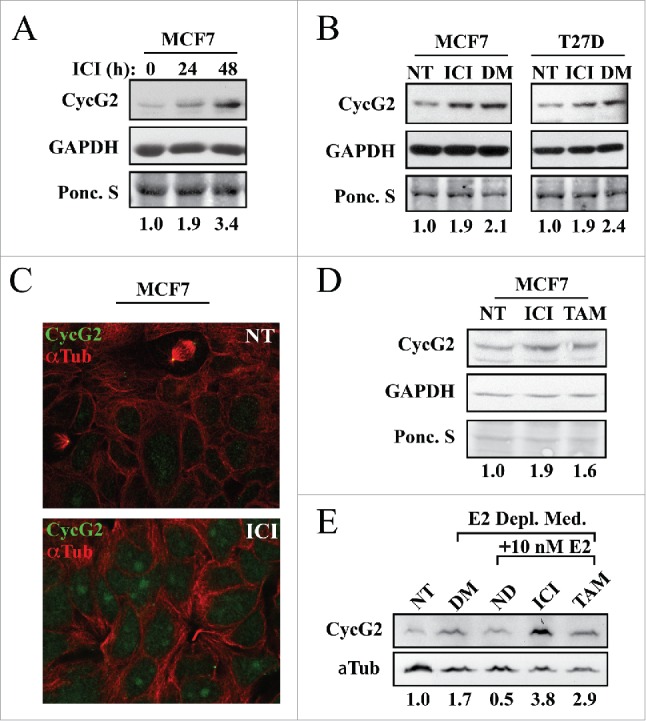
Inhibition of estrogen signaling with fulvestrant upregulates CycG2 expression in ER positive breast cancer cell lines. (A, B) MCF7 and T47D cells cultured in the presence or absence of fulvestrant (ICI, 100 nM) for 24 (A) or 48h (A, B) or absence of E2 (DM, 6 days) were assessed for changes in CycG2 expression by immunoblot analysis. (C) Confocal immunofluorescence microscopy images showing CycG2 expression (green) in fulvestrant-treated (ICI, bottom panel) and non-treated control (NT, top) MCF7 cells. Cells were stained with antibodies against CycG2 (green), and α-tubulin (α-Tub, red). (D) Immunoblot of CycG2 in MCF7 cells cultured for 48h in the absence (NT) or presence of the E2-signaling antagonists fulvestrant (ICI, 100 nM) or tamoxifen (TAM, 100 nM). (E) Immunoblot of CycG2 in E2-depleted (DM, 5 days) MCF7 cultures compared to E2-depleted cultures re-stimulated for an additional 48h with E2 alone or E2 plus ICI or TAM as indicated.
Stable RNAi-mediated knockdown of CycG2 expression blunts the cell cycle-arrest response of MCF7 cells to estrogen-deprivation and fulvestrant-mediated blockade of ER signaling
To assess the contribution of CycG2 to the cell cycle inhibitory effects of E2 signaling blockade, stable MCF7 clones expressing 2 different CCNG2-targeting shRNAs (1-B, ID3) were tested for their ability to inhibit CycG2 expression (Fig. 3A and B) and modify the cycle arrest response to E2-deprivation (Fig. 3C–F). We previously established the ability of these CCNG2-targeting shRNAs to block ectopic overexpression of CycG2 and stably knockdown (KD) endogenous CycG2 in MCF7 cells.17 Cultures of MCF7 WT, and CycG2 KD as well as non-silencing control shRNA (NSC) clones were cultured in regular or E2-depleted media (DM) for 6 d. Immunoblot analysis of lysates from these cultures verified the shRNA-mediated silencing of CycG2 expression and blockade of E2-deprivation-induced upregulation of CycG2 in the CycG2 KD clones (Fig. 3A and B). Importantly, flow cytometry analysis of total DNA content in the E2-depleted and untreated cultures indicated that shRNA-mediated repression of CycG2 upregulation significantly reduced the cell cycle arrest response to E2-withdrawl (Fig. 3C-F). Compared to the E2-depleted MCF7 WT and NSC control cultures, cultures of the E2-deprived CycG2 KD clones exhibited a decreased percentage of cells accumulated in G1-phase and a larger percentage of cells in S-phase (Fig. 3C and D). Two-parameter DNA flow cytometry of BrdU-pulse labeled DNA in the E2-depeted vs. non-treated cultures revealed that the percentage of cells synthesising DNA was greater in the E2-depleted CycG2 KD clone cultures compared to the E2-depleted control (WT, NSC) cultures (Fig. 3E and F).
Figure 3.
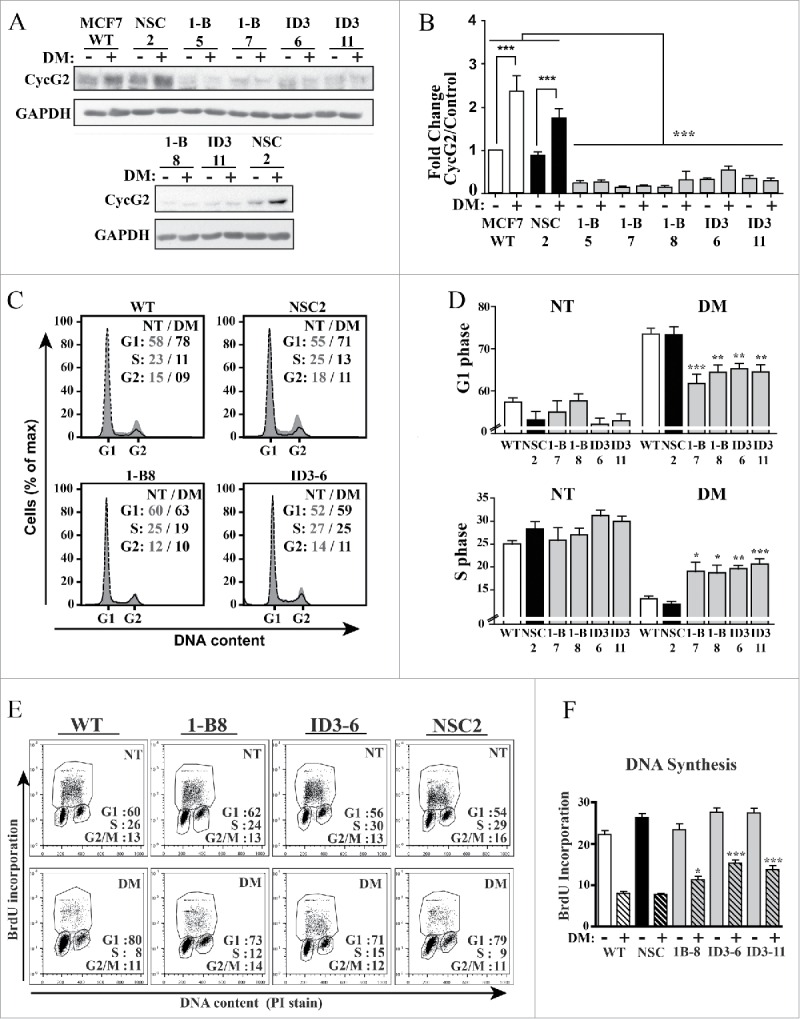
Stable silencing of CycG2 expression attenuates the cell cycle arrest response of MCF7 cells to estrogen withdrawal. (A) Immunoblot of endogenous CycG2 in MCF7 clones expressing CCNG2-targeting shRNAs (1-B and ID3) or a non-silencing control shRNA (NSC) cultured with (+) or without (-) estrogen depletion (DM). Numbers underneath shRNA denominators refer to clone number for the respective stably transfected cell line. (B) One-way ANOVA statistical analysis of CycG2 expression data from > 3 replicate experiments quantified by immunoblotting as in A. (C) Histogram overlays of DNA content in MCF7 control and CycG2 KD clones cultured in E2-depleted (black line, DM days) or normal media (gray area, NT). (D) One-way ANOVA statistical analysis of G1- and S-phase cell cycle distribution of single parameter DNA flow cytometry data analyses similar to that shown in C. (E) Two-parameter flow cytometry analysis of BrdU-labeled DNA in E2-depleted (DM) compared to non-treated (NT) CycG2 KD clones and controls. (F) One-way ANOVA analysis of BrdU incorporation and cell cycle distribution data as shown in E. ***p = < 0.001; **p = < 0.01; *p = < 0.05.
Given that knockdown of CycG2 promoted cell cycle progression of E2-deprived MCF7 cells and that fulvestrant-mediated blockade of ER signaling strongly upregulated CycG2 expression, we tested the consequence of CycG2 loss on fulvestrant-induced inhibition of MCF7 cell proliferation (Fig. 4). The CycG2 KD clones and controls were cultured +/− fulvestrant for 48h. Immunoblot analysis confirmed the 2-3-fold upregulation of CycG2 in treated MCF7 WT and NSC control cultures and the lack of fulvestrant-mediated induction of CycG2 in the KD clones (Fig. 4A and B). Flow cytometry analysis of PI-stained total (Fig. 4C and D) and BrdU-labeled DNA (Fig. 4E and F) showed that the fulvestrant-mediated G1-phase arrest response and inhibition of DNA synthesis observed in MCF7 WT and NSC control cells was significantly blunted in the CycG2 KD clones. The decreased sensitivity of the CycG2 KD cells to the cell cycle inhibitory effects of E2-signaling blockade indicates that E2 promotes cell cycle progression, in part, through suppression of CycG2 expression.
Figure 4.
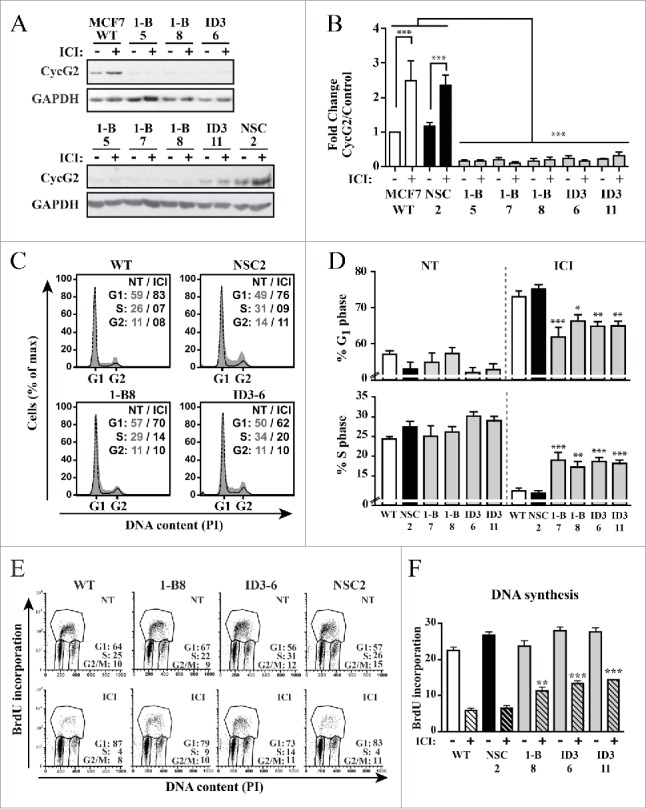
Silencing CycG2 expression reduces the cell cycle inhibitory effects of fulvestrant-mediated ER signaling blockade. (A) Immunoblots of CycG2 levels in indicated MCF7 CycG2 KD clones and controls (NSC, WT) following 48 h culture in the presence (+) or absence (-) of fulvestrant (ICI, 100 nM). (B) One-way ANOVA statistical analysis of CycG2 expression data from >3 replicate experiments quantified by immunoblotting as in A. (C) Representative histogram overlays of DNA content in parental MCF7 control (WT, NSC) and CycG2 KD clones cultured in normal (gray area, NT) or ICI (black line) containing medium for 48h). (D) Statistical analysis (one-way ANOVA) of G1- and S-phase DNA distribution data of flow cytometry analyses as shown in C. (E) Two-parameter flow cytometry analysis of BrdU-labeled DNA in CycG2 KD clones and WT and NSC controls cultured with (ICI) or without (NT) fulvestrant. (F) One way ANOVA analysis of BrdU data from >3 repeats of experiments shown in E. ***p = < 0.001; **p = < 0.01; *p = < 0.05.
Loss of CycG2 blunts fulvestrant-induced dephosphorylation of the retinoblastoma protein Rb and the MAP kinases MEK1 and ERK1/2 and downregulation of c-RAF
E2-activated ER mitogenic signaling induces inhibitory hyperphosphorylation of the retinoblastoma pocket protein Rb via G1-phase cyclin/CDK complexes, a pathway key for progression past the G1/S-phase checkpoint and initiation of DNA synthesis in BC cells.49-51 We examined the expression of total and phosphorylated forms of Rb (pRb) in lysates from non- and fulvestrant-treated MCF7 control and CycG2 knockdown cells (Fig. 5A). As expected, fulvestrant treatment diminished phosphorylation of Rb on S780 in WT MCF7 cells (lane 2) compared to pRb levels in the untreated control cells (lane 1). In contrast, fulvestrant treatment of the CycG2 KD clones results in a more moderate reduction in hyperphosphorylated pRb (lanes 3 through 10) compared to treated WT. These results are supported by the banding patterns observed in the immunoblots for total Rb; the relative abundance of slower migrating hyper-phosphorylated forms of the Rb doublet are more strongly reduced relative to untreated controls in fulvestrant-treated MCF7 WT than in the CycG2 KD clones. In addition, untreated CycG2 KD clones appeared to have an overall higher pRb levels compared to untreated WT. Comparable results are seen for the reduction of S780 phosphorylated pRb in E2-depleted vs. untreated cell cultures (Fig. 5B). The S780 pRB levels in E2-deprived CycG2 KD clones did not reach the low pRB level achieved by E2-withdrawl from WT cells.
Figure 5.
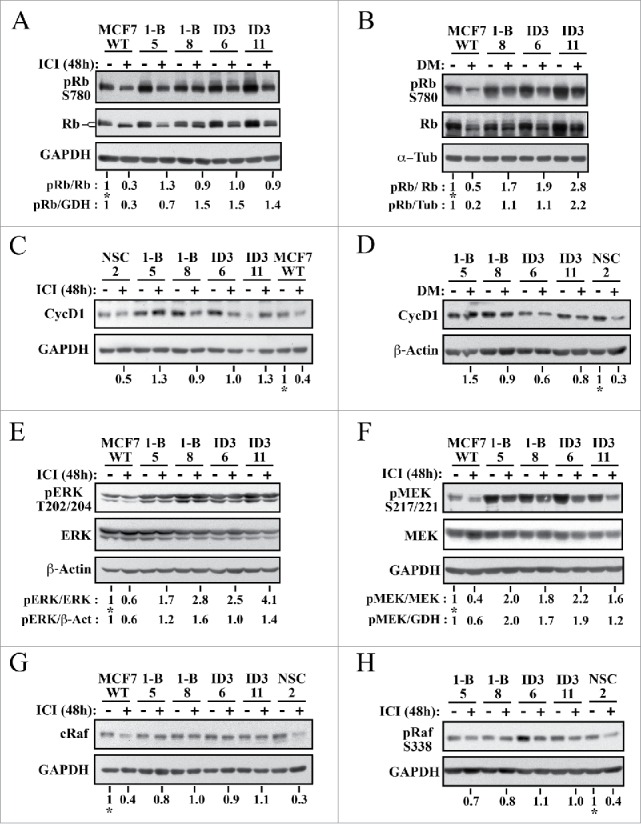
Silencing CycG2 expression blunts fulvestrant-induced dephosphorylation of Rb, inhibition of MEK/ERK signaling, and downregulation of Raf1. Immunoblot analysis of the indicated cell cycle regulatory and MAP kinase pathway proteins in MCF7 control (WT, NSC) and CycG2 KD (1-B, ID3) clones cultured with (+) or without (-) fulvestrant (ICI, 100 nM) for 48 h (A, C, E-H) or (B,D) E2-depletion (DM). Immunoblots for (A, B) S780 pRb and total Rb, (C, D) CycD1, E) T202/204 pERK 1/2, total ERK, (F) S217/221 pMEK1/2 and total MEK, (G) total cRaf, and (G) S338 p-cRaf are shown atop the indicated loading controls (GAPDH, α-tubulin, or β-actin). Numbers below lanes in blot panels indicate relative expression levels normalized to level in non-treated no-knockdown control (MCF7 WT or NSC control set at 1, marked with an *). Band intensity quantification is relative to amount of either loading control (GAPDH (GDH), α-tubulin (Tub) or β-actin (β−Act)) or total Rb, ERK, or MEK as indicated.
Aberrantly elevated expression of cyclin D1 promotes early G1-phase CDK inactivation of Rb in ER+ BC and inhibition of ER signaling is known to decrease cyclin D1 expression.51-53 We compared cyclin D1 expression levels in control and CycG2 KD MCF7 cells cultured in the presence and absence of fulvestrant (Fig. 5C). Consistent with our observations for S780-pRb, cyclin D1 levels were diminished by fulvestrant treatment of WT and NSC cultures (compare lanes 11 to 12 and 1 to 2) but appeared more moderately reduced in the treated CycG2 KD clones (lanes 3 through 10). Similar results were found for the effect of E2-withdrawal on cyclin D1 levels. As expected E2-deprivation led to a clear reduction in cyclin D1 levels in the NSC control, but had an overall more muted effect on cyclin D1 expression in the CycG2 KD clones (Fig. 5D).
ER+ BC cell resistance to endocrine therapy often arises from increased growth factor receptor (e.g. IGF-1R, HER2) activation of downstream PI3K/mTOR and RAF/MEK/ERK mitogenic signaling pathways that ultimately promote phosphorylation-mediated inhibition of Rb.54,55 As E2 signaling activates and anti-E2 therapeutics inhibit the RAF/MEK/ERK pathway and cyclin D/CDK4/6 phospho-inactivation of Rb is associated with activated ERK,51-53,56 we examined relative expression of activated forms of ERK1/2 and MEK1/2 in fulvestrant treated cultures of MCF7 WT and CycG2 KD cells. Immunoblot analysis of T202/Y204 pERK (Fig. 5E) as well as S217/221 pMEK1 expression (Fig. 5F) indicated that MEK/ERK activity was overall higher in the CycG2 KD clones. Moreover fulvestrant treatment did not as strongly reduce T202/Y204 pERK levels in the CycG2 KD cultures compared to WT cultures (Fig. 5E). In addition S217/221 pMEK1 levels of the fulvestrant treated CycG2 KD clones did not drop to that observed for treated WT cells. Similar results are seen for S217/221 pMEK1 levels in cultures of E2-depleted WT compared to CycG2 KD cells (data not shown). Given the apparent higher levels of phospho-activated MEK/ERK MAP kinases in the CycG2 KD clones we looked next at the expression levels of the proximal upstream activating kinase, cRaf in control and CycG2 KD cell populations (Fig. 5G). Fulvestrant treatment strongly blunted cRaf levels in control cell cultures, but did not result in such an obvious reduction of cRaf expression in the CycG2 KD clones. In agreement, a more reduced expression of S338 phosphorylated (activated) cRaf was observed in the fulvestrant treated control culture compared to the treated CycG2 KD cultures (Fig. 5H). In sum, KD of CycG2 promotes sustained inhibitory phosphorylation of Rb during blockade of ER signaling and continued expression of cyclin D1, cRaf and phospho-activated isoforms of ERK/MEK kinases despite repression of ER activity.
CycG2 associates and colocalizes with the negative regulator of c-RAF expression, CDK10
Our findings indicate that suppression of CycG2 upregulation blunts fulvestrant-mediated downregulation of cRaf expression (Fig. 5G and H). ER+ BC cell resistance to blockade of E2-signaling has been linked to decreased expression of the cyclin-dependent kinase, CDK10.57 The authors found that RNAi-mediated ablation of CDK10 promotes cell cycle progression, activates MAP kinase signaling and upregulates cRAF expression.57 Though CDK10 was shown to bind to and inhibit Ets2-mediated transcriptional activation of cRaf, no cyclin binding partner for this CDK had been identified.57 Here we explored the possibility that CycG2 is a CDK10 interacting protein. First we tested whether overexpressed CycG2 can interact with CDK10 in U2OS cells, using this cell line because of the ease and high efficiency by which it can be transiently transfected (Fig. 6). HA-tagged CDK10 was expressed together with GFP-tagged full length or C-terminal truncated forms of CycG2. The CycG2 constructs 1-187 and 1-160 are absent the C-terminus and some to most of the predicted 5 α-helices (α1′-α5′) of the cyclin fold bundle-repeat but maintain the conserved 5 α-helices (α1-α5 ) of the N-terminal cyclin box domain that contains key AA residues essential for cyclin-CDK interactions.8,58-60 In contrast the 1-140 construct lacks an additional 20 amino acids extending from the last 9 AAs in the predicted α5 helix of the cyclin box to the first 3 AAs of α1′, a key cyclin-CDK interfacing region that includes the highly conserved glutamic acid (Glu295 in cyclin A) critical for the cyclin fold and CDK interaction (Fig. 6A).8,58-60 Anti-CycG2, –HA and -GFP antibodies were used to immunoprecipitate (IP) the ectopically expressed proteins and the fractionated immunocomplexes were immunoblotted with the reciprocal antibody. Immunoblotting indicated that the full length and 1-160 and 1-187 truncated CycG2 constructs including the entire ‘Cyclin Box’ domain co-IP'd with HA-CDK10, but that the 1-140 CycG2 construct lacking key conserved CDK2 interfacing residues did not efficiently co-IP with HA-CDK10 (Fig. 6B). This evidence that CycG2GFP and HA-CDK10 interact is further bolstered by the colocalization of these 2 proteins in transfected cells (Fig. 6C). In contrast to the smooth nucleus-restricted anti-HA signal seen in cells not expressing CycG2GFP (Fig. 6C, top left corner), HA-CDK10 coexpressed with CycG2GFP is distributed throughout the cell, exhibiting a clear overlap and colocalization with the CycG2GFP fluorescence signal in both the cytoplasm and at distinct foci within the nucleus (Fig. 6C). This redistribution of a substantial fraction HA-CDK10 from the nucleus to the cytosol upon ectopic expression of CycG2GFP is consistent with CycG2 binding and thereby redistributing CDK10. In addition, ectopically expressed CycG2GFP is mostly cytosolic under basal conditions.11,12 However, when co-expressed with CDK10, CycG2GFP shows a striking colocalization with CDK10 at nuclear foci (Fig. 6C, lower right corner), a pattern similar to the foci observed for endogenous CycG2 in E2-depleted and fulvestrant-treated MCF7 cells (Figs. 1 and 2).
Figure 6.
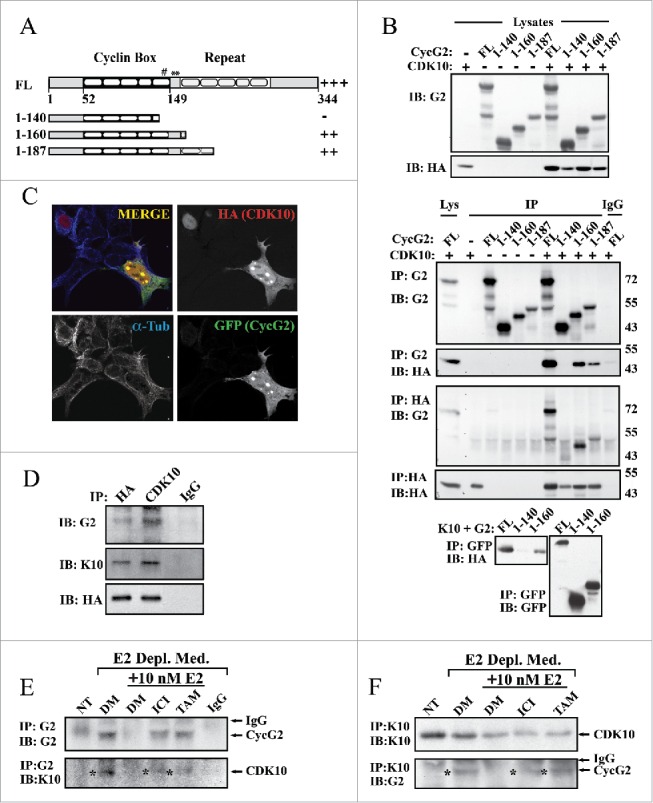
CycG2 associates with CDK10. (A) Schema showing the sequence predicted α-helical regions in the conserved Cyclin Box domain and c-terminal bundle repeat of full length (FL) CycG2 and the relative extent of the regions remaining in each expression construct used in B. The # symbol depicts the glutamate residue in CycG2 equivalent to the well conserved Glu295 of cyclin A critical for the cyclin fold and CDK interactions; * indicates the CycG2 residues conserved with the cyclin A CDK-interacting residues Asp305 and Leu306. (B) Top, Immunoblots of whole lysates from the indicated transfected U2OS cultures used for the co-immunoprecipitation studies of CycG2-GFP and HA-CDK10 complexes. Below, Immunoblots of indicated immunoprecipitates isolated from lysates of transfected U2OS cells. (C) Confocal immunofluorescence micrographs of CycG2-GFP (green) and HA-CDK10 (red) co-expressed in transfected U2OS cells and cytoskeleton marker α-tubulin (blue). Single channel micrographs of the individual fluorescence signals are shown in black and white for better contrast while the merged 3 channel image at the bottom shows the areas of colocalization (yellow). (D) Immunoblots of endogenous CycG2 coimmunoprecipitated from HA-CDK10 transfected U2OS cells by antibodies against HA (E, F) Immunoprecipitation of endogenous CycG2 or CDK10 from lysates of MCF7 cultures depleted of E2 for 5 d (DM), with or without E2 re-stimulation (E2) in the presence or absence fulvestrant (ICI,) or tamoxifen (TAM) for an additional 48h. NT = non-treated control.
We next tested whether endogenous CycG2 could be co-precipitated with either exogenous or endogenous CDK10 (Fig. 6D-F). Anti-CDK10 and -HA antibodies not only IP ectopically expressed CDK10 in singly transfected U2OS cells, but also co-IP endogenous CycG2 (Fig. 6D). Next we used the anti-CycG2 and anti-CDK10 antibodies to IP the endogenous proteins from untreated control (NT), E2-depleted (DM) or drug-treated MCF7 cells (Fig. 6E-F). As before (see Fig. 2), anti-CycG2 IB revealed an upregulation of CycG2 in MCF7 cells subjected to E2-signaling blockade, but not in untreated controls or cells re-stimulated by E2 supplementation after E2-withdrawal (DM +E2). Importantly reprobing of the anti-CycG2 IPs with anti-CDK10 antibodies showed co-IP of an ~39 kDa CDK10 positive band only in samples from the E2-depleted or drug treated (fulvestrant or tamoxifen) cultures. Reciprocal IP of CDK10 from lysates of both treated and untreated control cultures pulled down an anti-CDK10 positive band in each sample, but reprobing with anti-CycG2 antibodies showed co-IP of CycG2 only after inhibition of E2 signaling (Fig. 6F). These results suggest an association between endogenous CycG2 and CDK10 in MCF7 cells undergoing cell cycle arrest in response to inhibition of E2-signaling. As knockdown of either CycG2 (herein) or CDK1057 promotes cell cycle progression, MAP kinase signaling and cRAF expression, CycG2 could function in concert with CDK10 to modulate these processes.
CycG2 expression is enhanced by metformin and required for the metformin-enhanced arrest response to co-treatment with fulvestrant
CycG2 expression is repressed by HER2, IGF-1R and IR mitogenic signaling,14,33 growth factor receptors known to promote acquired resistance of ER+ BC to estrogen antagonizing therapeutics.2-4 Given that the anti-diabetic drug metformin inhibits mTOR and antagonizes BC cell growth in vitro61-64 and patients taking metformin have reduced BC recurrence,42-44 we investigated the effects of metformin on CycG2 expression in ER+ BC cell lines. As expected culture of MCF7 cells in the presence of 1 mM metformin for 48 h inhibits DNA synthesis and triggers a G1-phase cell cycle arrest (Fig. 7A). Moreover, a 48 h treatment with metformin induced a 3-fold CycG2 upregulation, similar to (though not as high) as MCF7 cells cultured in the absence of added insulin (Fig. 7B, right panels). We found that culturing MCF7 with metformin for 24 h also upregulated CycG2 expression to a similar extent as treatment with fulvestrant (Fig. 7B, left panels) and inhibited cell cycle progression (data not shown), though to a lesser extent than seen with exposure to the drug for 48 h.
Figure 7.
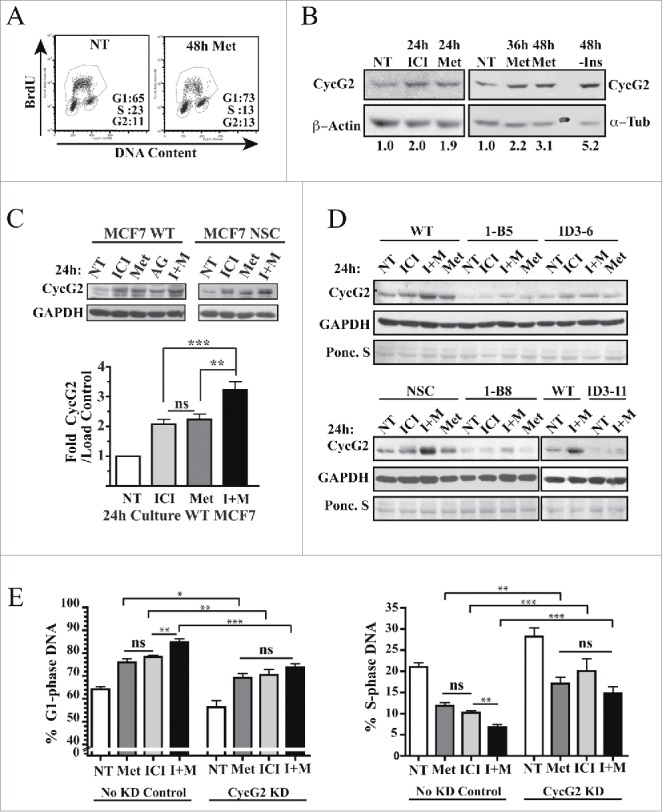
CycG2 contributes the cell cycle inhibitory effects induced by metformin. (A) Two parameter DNA flow cytometry analysis of DNA synthesis (BrdU incorporation) and cell cycle status of metformin treated cultures of MCF7 WT cells. (B) Immunoblot of CycG2 vs. loading control signals (α-tubulin, β-actin) in MCF7 WT cells cultured in normal medium (NT) or in the presence of metformin (Met,1 mM), or fulvestrant (ICI, 100 nM) or in the absence of insulin (-Ins) (C) Top, representative immunoblots show metformin (Met) enhancement of fulvestrant (ICI)-induced CycG2 expression in MCF7 control (WT, NSC) cultures I + M = fulvestrant + metformin; AG = 10 μM IGF-1R inhibitor AG1024. Bottom, statistical analysis of CycG2 expression in MCF7 WT cells treated for 24 hours with either metformin, fulvestrant or both agents. (D) CycG2 expression in CycG2 KD clones (1-B, ID3) treated for 24 hours with either metformin, fulvestrant or both agents compared to similarly treated MCF7 controls. (E) Statistical analysis of (one-way ANOVA) bar graphs of G1- and S-phase DNA distribution data. MCF7 control and CycG2 KD cultures that were treated for 24 h with metformin or fulvestrant alone or in combination. ***p = < 0.001; **p = < 0.01; *p = < 0.05.
Next we tested whether metformin could enhance the effects of fulvestrant on CycG2 expression in MCF7 control cell cultures and what consequence this co-treatment has on the cell cycle arrest response of CycG2 KD clones compared to controls (Fig. 7C and D). As before, culture of MCF7 WT cells for 24 h in the presence of either metformin or fulvestrant resulted in a similar 2-fold increase in CycG2 expression (Fig. 7C). Importantly, simultaneous co-treatment with both drugs further enhanced CycG2 expression to ~3.5-fold over non-treated cultures in WT cells but had minimal effects on CycG2 levels in the KD clones (Fig. 7C and D). Consistent with these results and the idea that CycG2 promotes cell cycle arrest, culture of MCF7 controls with this drug combination induced a stronger cell cycle arrest response than either drug treatment alone (Fig. 7E). In contrast, the same level of an arrest response to these therapeutics, either alone or in combination, is not observed in CycG2-KD cultures (Fig. 7E).
Lower CCNG2 expression is associated with aggressive and higher grade BC, and correlates with shorter time to relapse and distance metastasis free survival
As our results indicate that elevation of CycG2 expression promotes cell cycle inhibition of BC cells responding to E2-antagonizing therapeutics, we investigated the clinical significance of CCNG2 expression levels in BC tumors (Fig. 8). We used GOBO47 and KM-plotter tool48 software to search publically available microarray databases and analyze CCNG2 expression in BC tumor samples. Among the BC tumors stratified on the basis of their molecular subtypes, we found that CCNG2 levels are highest in the less aggressive Luminal A subtype while it is lowest in the more aggressive Basal BC tumor subtype associated with poorer clinical outcomes (Fig. 8A). Similar results are found for CCNG2 levels in BC tumors stratified on the basis of pathological grade, with the highest CCNG2 levels present in the lowest grade tumors (Grade 1) associated with the best prognosis for clinical outcome (Fig. 8B). To better estimate how CCNG2 transcript levels might help predict patient survival we performed Kaplan-Meier analysis. CCNG2 abundance in resected tumors was compared to the length of distant metastasis free (DMFS) and relapse free (RFS) survival periods of patient populations stratified on the basis of receptor expression and treatment with E2-antagonizing therapeutics (Fig. 8 C, D). Tumors (ER+ or unclassified) from BC patient populations with the longer DMFS periods exhibit the highest quartile level of CCNG2 expression (Fig. 8C). Among the 313 patients with ER+ and progesterone receptor positive (PR+) disease spread to lymph nodes (LN+), those with longer periods of RFS had primary tumors with the highest CCNG2 mRNA levels (HR 0.44, logrank P= 5e-04; Fig. 8D, left panel). Importantly, analysis of expression data from ER+ tumor samples from the 1172 patients who had been treated systemically with any endocrine-based therapy, showed those with tumors expressing higher levels of CCNG2 exhibited the highest probability of long periods of RFS (HR = 0.58, logrank P = 5.8e-04; Fig. 8D, right panel). A similar cohort of endocrine therapy treated patients whose tumors exhibit high levels of CDK10 mRNA have a comparable likelihood of long periods of RFS (HR = 0.71, logrank P = 8.9e-07) over the same time period (KM plot not shown). In contrast, RFS Kaplan-Meier analyses of tumor CCND1 and CCNE1 expression in patients who received any endocrine therapy indicate that the higher tumor levels of either predicts poor outcomes for these patients (HR = 1.51, logrank P = 7.4e-09 and HR = 1.62, logrank P = 1.9e-11, respectively; KM plots not shown). Finally, the probability of longer periods of DMFS for 947 endocrine therapy treated patients who did not receive chemotherapy was considerably better when the BC tumors had the highest CCNG2 mRNA levels (HR = 0.57, logrank P = 9.7e-05, KM plot not shown).
Figure 8.
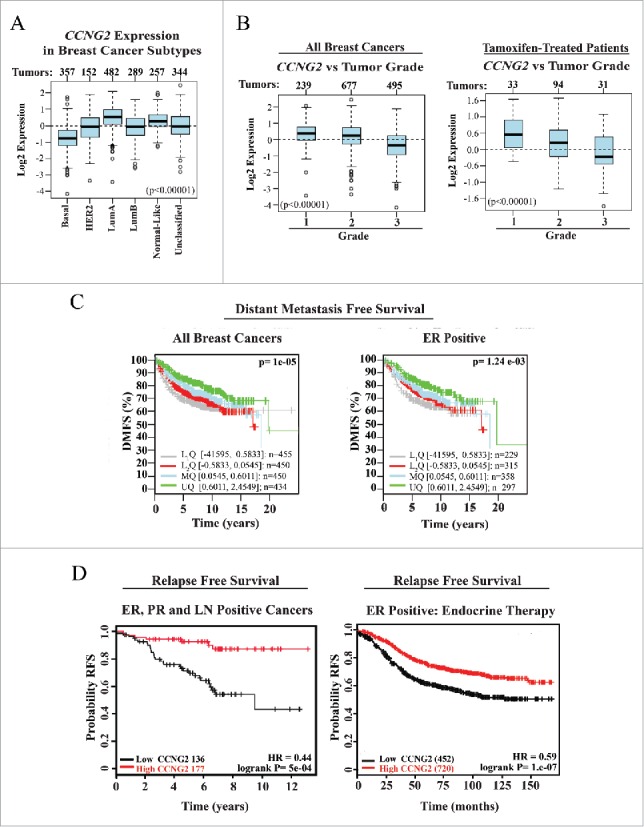
Meta-analysis of CCNG2 expression in breast cancer (BC) tumor types and its correlation with relapse free (RFS) and distant metastasis free (DMFS) survival in breast cancer patients. (A, B) Box-plots assessing CCNG2 expression in BC subtype (A) or grade classified BC tumor tissues (B) from all or tamoxifen-treated (B, right) patients (numbers of sample size at top of box-plot panels). (C, D) Kaplan-Meier survival curves estimating probability of survival for patients with tumors expressing high or low levels of CCNG2 (stratified on basis of tumor characteristics and therapy). HR = Hazard ratio. Data were assembled from online microarray databases and analyzed using the GOBO - Gene expression-based Outcome for Breast cancer Online software (http://co.bmc.lu.se/gobo) and the KM plotter tool for breast cancer (http://kmplot.com/analysis/). PR = progesterone receptor, LN = lymph node.
Discussion
Consistent with previous reports that CCNG2 transcription is directly repressed by E2-bound ER co-repressor complex,10 we show here 1) that during the response to E2-withdrawal CycG2 protein expression is upregulated up to 3-fold and accumulates in the nucleus; and 2) that re-stimulation of E2-depleted cells with E2 reduces CycG2 expression and reverses the nuclear accumulation (Fig. 1). Moreover, we also now show that CycG2 expression is enhanced upon pharmacological blockade of E2 signaling by the SERM tamoxifen and ER antagonist fulvestrant (Fig. 2), the latter triggering a strong overall increase in CycG2 and accumulation within the nucleus similar to the distribution pattern seen upon E2-depletion. This pattern of nuclear CycG2 immunostaining is distinctly different from the more punctual distribution we observed upon DNA damage-induced upregulation of CycG2 within MCF10a nuclei.17 Significantly, we found that shRNA mediated KD of CycG2 reduces the G1-phase arrest response of MCF7 cells to both E2-depletion and fulvestrant-mediated antagonism of ER signaling (Figs. 3 and 4). In contrast to MCF7 controls, CycG2 KD clones treated with fulvestrant or depleted of E2 exhibit an increased number of cells with an S-phase content actively synthesising DNA. This result indicates that the growth inhibitory properties of anti-estrogen therapeutics could be mediated, in part, through upregulation of CycG2 expression.
As our findings indicate that elevation in CycG2 abundance is required to enforce the G1-phase cell cycle arrest response triggered by blockade of E2/ER signaling, loss of CycG2 expression may contribute to the development of ER+ BC tumor cell resistance to these therapeutics. This idea is further supported by our meta-analysis of curated tumor sample microarray database sets (Fig. 8). We found that the higher levels of CCNG2 expression correlated with lower grade neoplasms among all BC types and in particular among the ER+ tumors, including those from tamoxifen-treated BC patients. Using Kaplan-Meier analysis of CCNG2 transcript levels in ER+ BC tumor samples we found both reduced relapse-free and distant-metastasis free survival time periods in patients with tumors exhibiting lower levels of CCNG2 mRNA compared to those patient populations whose tumors had the highest levels of CCNG2 expression (Fig. 8). Additional analysis of CCNG2 in ER+ tumors from anti-estrogen therapy treated patients clearly showed that those patients with higher CCNG2 expression exhibited longer periods of RFS (Fig. 8). This increased probability of a long RFS period is similar to that found for an equivalent patient cohort with tumors expressing high levels of CDK10 but in stark contrast to the low probability of a similarly long RFS period for BC patients with tumors expressing high levels of cyclins D1 or E1 mRNAs (see results above).
Estrogen and growth factor-induced mitogenic signaling leads to phosphorylation and thereby inactivation of Rb by CDKs.49,55 When Rb is in the hypophosphorylated state, it binds to and sequesters the transcription factor E2F, thereby attenuating the expression of E2F-regulated genes that are required for entry into S-phase. E2 induced mitogenic signaling stimulates CDK phosphorylation of Rb, allowing E2F to drive the expression of proliferation promoting genes.65,66 Thus antagonism of ER signaling inhibits phospho-inactivation of Rb.51-53 Consistent with the blunting effects CCNG2 KD on the cell cycle arrest response of MCF7 cells to blockade of ER activity (Figs. 3 and 4), Rb phosphorylation in fulvestrant-treated and E2-deprived CCNG2 KD clones was clearly elevated relative to the underphosphorylated state of Rb in similarly treated WT and non-silencing shRNA control MCF7 populations (Fig. 5A and B). Rb-inactivating hyperphosphorylation is achieved through activated CDK4/6 and CDK2 kinases. E2 induces activation of these kinases in part through upregulation of the CDK4/6 regulatory partner, cyclin D1.65,66 In some ER+ BC, overstimulation of CDK4/6 activity results in part from aberrantly elevated expression of cyclin D1, and in HER2+ BC, receptor stimulation of PI3K/Akt and MAP kinase activity stimulates D-type cyclin expression and concomitant Rb inactivation.67,68 We found that, in contrast to the decrease in cyclin D1 observed in WT and shRNA controls responding to E2/ER inhibiting treatments, cyclin D1 levels in the CCNG2 KD clones is not as potently repressed under these conditions (Fig. 5C and D). MAPK activity induces cyclin D1 transcription through regulation of AP1 (Fos/Jun) complexes that stabilize cyclin D1 mRNA and/or protein.69 Early work had shown that overexpression of cyclin D1 expression promotes tamoxifen resistance by abolishing the reliance on E2 signaling.70 Since then multiple models of endocrine-resistance indicate ER-independent stimulation of cyclin D1 expression through activation of the MAPK pathway downstream of elevated growth factor signaling.55,71 CycG2 KD clones exhibiting diminished arrest responses, increased Rb phosphorylation and elevated cyclin D1 protein expression despite E2/ER antagonism. In addition, KD showed sustained expression of cRaf and phospho-activated forms of the MAPK pathway kinases, MEK1 and ERK1/2 (Fig. 5E–H). These results suggest that the loss of CycG2 expression promotes the development of resistance to E2/ER antagonizing therapeutics by allowing sustained cRaf/MEK/ERK MAPK pathway activation of cyclin D1 expression and thus continued phospho-inactivation of Rb.
A previous RNAi screen for determinants of tamoxifen resistance linked the orphan CDK, CDK10 to the cRaf/MEK/ERK MAPK signaling pathway in MCF7 cells.57 CDK10 had already been known to bind and repress the transcription factor Ets2.72 Silencing CDK10 relieves its repressive effect on Ets2 and thereby enhances cRaf expression and downstream signaling through the MEK/ERK pathway, ultimately leading to cyclin D1 upregulation, Rb inactivation and cell cycle progression.57 Although CDK10 exhibits clear homology to CDKs, a corresponding estrogen-controlled regulatory cyclin in MCF7 cells had not been identified. CycG2 is a CRM1-dependent nucleocytoplasmic shuttling protein that when ectopically expressed is largely localized to the cytosol and centrosome,12 whereas CDK10 is localized to the nucleus. We determined that CycG2 associates with CDK10 and when overexpressed, CDK10 and CycG2 co-localize in a distinct pattern within the nucleus and to a lesser extent in the cytosol (Fig. 6A-C). This association requires an intact CycG2 N-terminal cyclin box region (Fig. 6A and B). Coimunoprecipitation of endogenous CycG2 associated with endogenous CDK10 is dependent on blockade of E2/ER activity. Given that both CDK10 KD57 and CycG2 KD (Fig. 5) promotes cRaf expression and enhances phosphorylation of MEK1/2 and ERK1/2, the impaired cell cycle arrest response to ER inhibition by CycG2 KD could be due to decreased CycG2 binding to and modulation of CDK10 activity. Accordingly CycG2-mediated regulation of CDK10 activity would likely be dampened in estrogen-responsive endocrine-therapy resistant ER+ BCs.
CycG2 is known to associate with active PP2A complexes and requires its C-terminal region beyond the cyclin box to bind and interact with PP2A subunits.11,12,27,73 Recent work indicates that CDK10 is the target of Pin1-mediated ubiquitination, the Pin1 recognition and binding depending on phosphorylation of CDK10 at Thr133.74 Our preliminary studies (data not shown) indicate that CycG2 can bind both CDK10 and PP2A at the same time. It is thus possible that CycG2 recruits PP2A to dephosphorylate CDK10 and thereby inhibits Pin1-mediated degradation of CDK10, ultimately leading to suppression of cRaf. Alternatively, CycG2 may more directly participate in CDK10 regulation of cRaf. Early work showed that kinase dead CDK10 is able to suppress Ets2 transactivation, indicating that CDK10 mediated inhibition of Ets2 was independent of any potential CDK10 kinase activity.72 The authors proposed that CDK10 interaction with Ets2 might interfere with the transcription factors ability to interact with the basal transcription machinery or co-activators. Ets2 activity is positively regulated by phosphorylation of Thr72 in its N-terminus, however the phosphatase that dephosphorylates this site is not described.75,76 Thus it is possible that CycG2/CDK10 complexes recruit PP2A to dephosphorylate and inactivate Ets2. A recent study showed that cyclin M, a cyclin deficient in patients with STAR syndrome, binds to and induces CDK10 kinase activity toward Ets2 in vitro.77 The authors link Ets2 phosphorylation on serines 220 and 225 to its MG132-sensitive degradation and show increased CDK10 expression in STAR patient-derived lymphoblastoid cells deficient in cyclin M. We found that KD of CycG2 in MCF7 cells subjected to blockade of E2-signaling phenocopies the deficiency in cell cycle arrest response, sustained hyperphosphorylation of Rb, and enhanced expression of phospho-activated ERK1/2, MEK1/2 and total cRaf previously reported for siRNA KD of CDK10.57 Further work will be needed to determine whether the impact of CycG2 expression on the RAF/MAPK pathway reflects its ability to interact with CDK10 and how this interaction influences CDK10 modulation of Ets2 activity.
In addition to its regulation by E2-bound ERs, the CCNG2 gene is transcriptionally activated by the FOXO family of transcription factors and repressed by growth factor receptor stimulation of the PI3K/AKT pathway that inhibits FOXO transcriptional activity.15 We previously determined that dampening of HER2 signaling in HER2 positive BC lines via the PI3K inhibitor Ly294002, mTOR inhibitor rapamycin, or HER2-targeting monoclonal antibody trastuzumab results in CycG2 upregulation and accumulation within the nucleus.14 We have also shown that stimulation of IGF-1 and insulin receptors downregulates and that withdrawal of insulin upregulates CCNG2/CycG2 expression in various cell lines, including MCF7 cells.33 It is well established that IGF-1R and IR can form hybrid receptors that bind all 3 receptor family ligands IGF-I, IGF-II and insulin.38-41 Moreover, increased expression of IGF-1R and IR receptors are frequently observed in BC.39,78,79 The IR in BC cells (including MCF7) frequently contains the embryonic isoform IR-A, which is known to elicit strong mitogenic signals.38,80,81 IR-A containing receptors bind not only insulin but also IGF-II and pro-insulin with high affinity, IR-A exhibiting a 1.7-fold higher affinity for insulin than IR-B and faster recycling than IR-B.78 It is thought that mitogenic signaling through hybrid IGF-IR/IR-A receptors triggered by insulin, not just IGF-I and IGF-II, plays a role in promoting BC growth in hyperinsulinemic type II diabetes patients and may promote resistance to E2/ER targeting in a subset of BC.40,82 Thus IGF-1R and IR receptor-mediated repression of CycG2 expression could be a factor contributing to the mitogenic effects of IGF-IR/IR-A hybrids in endocrine therapy resistant BC.
Epidemiological and clinical evidence indicates that metformin inhibits breast cancer.42-45 Laboratory studies indicate that metformin activates the mTOR antagonizing kinase AMPK and dampens PI3K/AKT/mTOR mitogenic signaling.46,61-64 Thus there is considerable interest in the potential use of metformin in combinatorial adjuvant therapy.45,83-86 We tested the effects of 1 mM metformin on CycG2 expression and cell cycle progression of MCF7 cells (Fig. 7). As predicted, metformin inhibited DNA synthesis, induced a G1-phase cell cycle arrest and significantly upregulated CycG2 expression. Moreover, co-treatment of cells with fulvestrant and metformin resulted in a stronger increase in CycG2 expression then treatment with either agent alone (Fig. 7C and D). In contrast, similar treatments of CycG2 KD clones with metformin did not result in a substantial increase in CycG2 expression or as potent a cell cycle arrest. Importantly, we found that metformin/fulvestrant combination treatment boosted the cell cycle arrest response of MCF7 WT cells beyond what is observed for treatment with a single agent. Moreover, the enhanced arrest response induced by co-treatment with both agents is greatly diminished in CycG2 KD cultures. Taken together our results suggest that assessment of CycG2/CCNG2 expression levels could inform the anti-mitogenic potential of metformin-containing combinatorial therapies targeting ER+ breast cancers.
Conclusion
To improve upon stratification of clinical risk and selection of therapy for ER+ BC additional prognostic and predictive tumor biomarkers for tumor characterization are needed. Our findings point to CycG2 as an important estrogen-controlled cell cycle inhibitory protein that helps restrain signaling through the RAF/MAPK pathway to Rb. As CycG2 expression is also downregulated by HER2, IGF-1R, and IR activity but upregulated by drugs that inhibit PI3K/AKT/mTOR signaling downstream of these receptors14,15,33,34 (Fig. 7), CycG2 expression could be a valuable predictive and prognostic biomarker to help assess the potential development of ER+ BC tumor resistance to adjuvant therapeutics and the overall likelihood of patient long term survival outcomes.
Abbreviations
- 4OHT
tamoxifen
- AMPK
AMP activated protein kinase
- BrdU
5-bromo-2′-deoxyuridine
- BC
Breast cancer
- CDK
cyclin dependent kinase
- Cyc
cyclin
- DMFS
distant metastasis free survival
- E2
estrogen/estradiol
- EGFR
epidermal growth factor receptor
- ER
estrogen receptor
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- HA
hemagglutinin
- HER2
human epidermal growth factor receptor 2
- ICI
fulvestrant
- IGF-1R
insulin-like growth factor 1 receptor
- IB
immunoblot
- IP
immunoprecipitation
- IR
insulin receptor
- KD
knockdown
- MAPK
mitogen activated protein kinase
- MEK
MAPK/ERK kinase
- MET
metformin
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositol 3-kinase
- PP2A
protein phosphatase 2A
- RFS
relapse free survival
- SERD
selective estrogen receptor downregulators
- SERM
selective estrogen receptor modulators
- TF
transcription factor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Johannes Hell, Colleen Sweeny, Jason Hatakeyama, and Jessica Wald of UC Davis and Ms. Catalina Simion (UC Davis) for critical reading of the manuscript. We express our gratitude to Colleen Cowan for technical assistance and Justin Fishbaugh and Gene Hess of the Holden Comprehensive Cancer Center Flow Cytometry Facility (University of Iowa), and Carol Oxford of the UC Davis Cancer Center Flow Cytometry Facility (funded by the UC Davis CCSG awarded by the National Cancer Institute (NCI P30CA093373)) for expert technical support.
Funding
Portions of this work was supported, in part, by USAMRMC/CDMRP (DOD) Grants BC045656 and TS080043 and a pilot grant awarded to MCH through the National Institutes of Health Program Project Grant P20CA103672 (University of Iowa Cancer and Aging Program's NIA/NCI program).
Ethics
Meta-analysis of CCNG2 gene expression in publically available microarray database sets of fully-anonymised human BC tumor samples was done using the online meta-analysis tools, GOBO47 (Gene expression-based Outcome for Breast Cancer Online tool that curates publicly available Affymetrix U133A microarray datasets, http://co.bmc.lu.se/gobo), and KM-plotter48 (assesses Affymetrix HG-U133A, HG-U133 Plus 2.0 and HG-U133A 2.0 microarray data downloaded from GEO EGA and TCGA databases (http://kmplot.com/analysis/)).
Authors’ contributions
MZ carried out experiments in Figs. 1-7, analyzed data, helped prepare figures of data and wrote portions of the manuscript; AAD carried out key early preliminary work, developed reagents, obtained some of the experimental data shown in Figs. 1 and 2 and helped in preparation of figures and writing; MSD was responsible for some of the experimental work shown in Figs. 1, 2, and 7 and helped write and prepare the manuscript; T. Patriarchi carried out immunoblotting for some data in Figs. 1, 2 and 7. MCH designed experiments, analyzed data, prepared figures and wrote the manuscript. All authors read, gave feedback and approved the final manuscript.
ORCID
Tommaso Patriarchi http://orcid.org/0000-0001-9351-3734
References
- [1].Toss A, Cristofanilli M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res 2015; 17:60; PMID:25902832; http://dx.doi.org/ 10.1186/s13058-015-0560-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Patani N, Martin LA. Understanding response and resistance to oestrogen deprivation in ER-positive breast cancer. Mol Cell Endocrinol 2014; 382(1):683-94; PMID:24121024; http://dx.doi.org/ 10.1016/j.mce.2013.09.038 [DOI] [PubMed] [Google Scholar]
- [3].Johnston SR. Enhancing Endocrine Therapy for Hormone Receptor-Positive Advanced Breast Cancer: Cotargeting Signaling Pathways. J Natl Cancer Inst 2015; 107(10); PMID:26251289; http://dx.doi.org/ 10.1093/jnci/djv212 [DOI] [PubMed] [Google Scholar]
- [4].Hart CD, Migliaccio I, Malorni L, Guarducci C, Biganzoli L, Di Leo A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat Rev Clin Oncol 2015; 12(9):541-52; PMID:26011489; http://dx.doi.org/ 10.1038/nrclinonc.2015.99 [DOI] [PubMed] [Google Scholar]
- [5].Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol 2015; 12(7):381-94; PMID:25895611; http://dx.doi.org/ 10.1038/nrclinonc.2015.73 [DOI] [PubMed] [Google Scholar]
- [6].Ciruelos E, Pascual T, Arroyo Vozmediano ML, Blanco M, Manso L, Parrilla L, Munoz C, Vega E, Calderon MJ, Sancho B et al. The therapeutic role of fulvestrant in the management of patients with hormone receptor-positive breast cancer. Breast 2014; 23(3):201-8; PMID:24589524; http://dx.doi.org/ 10.1016/j.breast.2014.01.016 [DOI] [PubMed] [Google Scholar]
- [7].Ellis MJ, Llombart-Cussac A, Feltl D, Dewar JA, Jasiowka M, Hewson N, Rukazenkov Y, Robertson JF. Fulvestrant 500 mg Versus Anastrozole 1 mg for the First-Line Treatment of Advanced Breast Cancer: Overall Survival Analysis From the Phase II FIRST Study. J Clin Oncol 2015; 33(32):3781-7; PMID:26371134; http://dx.doi.org/ 10.1200/JCO.2015.61.5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Horne MC, Donaldson KL, Goolsby GL, Tran D, Mulheisen M, Hell JW, Wahl AF. Cyclin G2 is up-regulated during growth inhibition and B cell antigen receptor-mediated cell cycle arrest. J Biol Chem 1997; 272(19):12650-61; PMID:9139721; http://dx.doi.org/ 10.1074/jbc.272.19.12650 [DOI] [PubMed] [Google Scholar]
- [9].Horne MC, Goolsby GL, Donaldson KL, Tran D, Neubauer M, Wahl AF. Cyclin G1 and cyclin G2 comprise a new family of cyclins with contrasting tissue-specific and cell cycle-regulated expressions. J Biol Chem 1996; 271(11):6050-61; PMID:8626390; http://dx.doi.org/ 10.1074/jbc.271.11.6050 [DOI] [PubMed] [Google Scholar]
- [10].Stossi F, Likhite VS, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen-occupied estrogen receptor represses cyclin G2 gene expression and recruits a repressor complex at the cyclin G2 promoter. J Biol Chem 2006; 281(24):16272-8; PMID:16608856; http://dx.doi.org/ 10.1074/jbc.M513405200 [DOI] [PubMed] [Google Scholar]
- [11].Bennin DA, Don AS, Brake T, McKenzie JL, Rosenbaum H, Ortiz L, DePaoli-Roach AA, Horne MC. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B' subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J Biol Chem 2002; 277(30):27449-67; PMID:11956189; http://dx.doi.org/ 10.1074/jbc.M111693200 [DOI] [PubMed] [Google Scholar]
- [12].Arachchige Don AS, Dallapiazza RF, Bennin DA, Brake T, Cowan CE, Horne MC. Cyclin G2 is a centrosome-associated nucleocytoplasmic shuttling protein that influences microtubule stability and induces a p53-dependent cell cycle arrest. Exp Cell Res 2006; 312(20):4181-204; PMID:17123511; http://dx.doi.org/ 10.1016/j.yexcr.2006.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kim Y, Shintani S, Kohno Y, Zhang R, Wong DT. Cyclin G2 dysregulation in human oral cancer. Cancer Res 2004; 64(24):8980-6; PMID:15604262; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-1926 [DOI] [PubMed] [Google Scholar]
- [14].Le X-F, Arachchige Don AS, Mao W, Horne MC, Bast J, RC. Roles of human epidermal growth factor receptor 2, c-jun NH2-terminal kinase, phosphoinositide 3-kinase, and p70 S6 kinase pathways in regulation of cyclin G2 expression in human breast cancer cells. Mol Cancer Ther 2007; 6(11):2843-57; PMID:18025271; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-0109 [DOI] [PubMed] [Google Scholar]
- [15].Martinez-Gac L, Marques M, Garcia Z, Campanero MR, Carrera AC. Control of Cyclin G2 mRNA Expression by Forkhead Transcription Factors: Novel Mechanism for Cell Cycle Control by Phosphoinositide 3-Kinase and Forkhead. Mol Cell Biol 2004; 24(5):2181-89; PMID:14966295; http://dx.doi.org/ 10.1128/MCB.24.5.2181-2189.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu G, Bernaudo S, Fu G, Lee DY, Yang BB, Peng C. Cyclin G2 is degraded through the ubiquitin-proteasome pathway and mediates the antiproliferative effect of activin receptor-like kinase 7. Mol Biol Cell 2008; 19(11):4968-4979; PMID:18784254; http://dx.doi.org/ 10.1091/mbc.E08-03-0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zimmermann M, Arachchige Don AS, Donaldson MS, Dallapiazza RF, Cowan CE, Horne MC. Elevated Cyclin G2 expression intersects with DNA damage checkpoint signaling and is required for a potent G2/M checkpoint arrest response to doxorubicin. J Biol Chem 2012; 287(27):22838-53. PMID: 22589537; 12198119 10.1074/jbc.M112.376855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gajate C, An F, Mollinedo F. Differential cytostatic and apoptotic effects of ecteinascidin-743 in cancer cells. Transcription-dependent cell cycle arrest and transcription-independent JNK and mitochondrial mediated apoptosis. J Biol Chem 2002; 277(44):41580-9; PMID:12198119; http://dx.doi.org/ 10.1074/jbc.M204644200 [DOI] [PubMed] [Google Scholar]
- [19].Hinman RM, Bushanam JN, Nichols WA, Satterthwaite AB. B Cell Receptor Signaling Down-Regulates Forkhead Box Transcription Factor Class O 1 mRNA Expression via Phosphatidylinositol 3-Kinase and Bruton's Tyrosine Kinase. J Immunol 2007; 178(2):740-7; PMID:17202334; http://dx.doi.org/ 10.4049/jimmunol.178.2.740 [DOI] [PubMed] [Google Scholar]
- [20].Wykoff CC, Pugh CW, Harris AL, Maxwell PH, Ratcliffe PJ. The HIF pathway: implications for patterns of gene expression in cancer. Novartis Found Symp 2001; 240:212-25; PMID:11727931; http://dx.doi.org/ 10.1002/0470868716.ch15 [DOI] [PubMed] [Google Scholar]
- [21].Thomas KC, Sabnis AS, Johansen ME, Lanza DL, Moos PJ, Yost GS, Reilly CA. Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J Pharmacol Exp Ther 2007; 321(3):830-8; PMID:17332266; http://dx.doi.org/ 10.1124/jpet.107.119412 [DOI] [PubMed] [Google Scholar]
- [22].Fu G, Peng C. Nodal enhances the activity of FoxO3a and its synergistic interaction with Smads to regulate cyclin G2 transcription in ovarian cancer cells. Oncogene 2011; 30(37):3953-66; PMID:21532621; http://dx.doi.org/ 10.1038/onc.2011.127 [DOI] [PubMed] [Google Scholar]
- [23].Chen J, Yusuf I, Andersen HM, Fruman DA. FOXO transcription factors cooperate with delta EF1 to activate growth suppressive genes in B lymphocytes. J Immunol 2006; 176(5):2711-2721; PMID:16493026; http://dx.doi.org/ 10.4049/jimmunol.176.5.2711 [DOI] [PubMed] [Google Scholar]
- [24].Fang J, Menon M, Kapelle W, Bogacheva O, Bogachev O, Houde E, Browne S, Sathyanarayana P, Wojchowski DM. EPO modulation of cell-cycle regulatory genes, and cell division, in primary bone marrow erythroblasts. Blood 2007; 110(7):2361-70; PMID:17548578; http://dx.doi.org/ 10.1182/blood-2006-12-063503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Arai Y, Pulvers JN, Haffner C, Schilling B, Nusslein I, Calegari F, Huttner WB. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat Commun 2011; 2:154; PMID:21224845; http://dx.doi.org/ 10.1038/ncomms1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Castro DS, Martynoga B, Parras C, Ramesh V, Pacary E, Johnston C, Drechsel D, Lebel-Potter M, Garcia LG, Hunt C et al. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev 2011; 25(9):930-45; PMID:21536733; http://dx.doi.org/ 10.1101/gad.627811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mourgues L, Imbert V, Nebout M, Colosetti P, Neffati Z, Lagadec P, Verhoeyen E, Peng C, Duprez E, Legros L et al. The BMI1 polycomb protein represses cyclin G2-induced autophagy to support proliferation in chronic myeloid leukemia cells. Leukemia 2015; 29(10):1993-2002; PMID:25925206; http://dx.doi.org/ 10.1038/leu.2015.112 [DOI] [PubMed] [Google Scholar]
- [28].Kasukabe T, Okabe-Kado J, Honma Y. Cotylenin A, a new differentiation inducer, and rapamycin cooperatively inhibit growth of cancer cells through induction of cyclin G2. Cancer Sci 2008; 99(8):1693-8; PMID:18754885; http://dx.doi.org/ 10.1111/j.1349-7006.2008.00867.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ito Y, Yoshida H, Uruno T, Nakano K, Takamura Y, Miya A, Kobayashi K, Yokozawa T, Matsuzuka F, Kuma K et al. Decreased expression of cyclin G2 is significantly linked to the malignant transformation of papillary carcinoma of the thyroid. Anticancer Res 2003; 23(3B):2335-8; PMID:12894512 [PubMed] [Google Scholar]
- [30].Bogni A, Cheng C, Liu W, Yang W, Pfeffer J, Mukatira S, French D, Downing JR, Pui CH, Relling MV. Genome-wide approach to identify risk factors for therapy-related myeloid leukemia. Leukemia 2006; 20(2):239-46; PMID:16341039; http://dx.doi.org/ 10.1038/sj.leu.2404059 [DOI] [PubMed] [Google Scholar]
- [31].Marshall GM, Gherardi S, Xu N, Neiron Z, Trahair T, Scarlett CJ, Chang DK, Liu PY, Jankowski K, Iraci N et al. Transcriptional upregulation of histone deacetylase 2 promotes Myc-induced oncogenic effects. Oncogene 2010; 29(44):5957-68; PMID:20697349; http://dx.doi.org/ 10.1038/onc.2010.332 [DOI] [PubMed] [Google Scholar]
- [32].Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol 2011; 29(33):4452-61; PMID:22010023; http://dx.doi.org/ 10.1200/JCO.2010.34.4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Svendsen AM, Winge SB, Zimmermann M, Lindvig AB, Warzecha CB, Sajid W, Horne MC, De Meyts P. Down-regulation of cyclin G2 by insulin, IGF-I (insulin-like growth factor 1) and X10 (AspB10 insulin): role in mitogenesis. Biochem J 2014; 457(1):69-77; PMID:24059861; http://dx.doi.org/ 10.1042/BJ20130490 [DOI] [PubMed] [Google Scholar]
- [34].Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, Afonja O, Horne MC, Tanaka T, Duan E et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci U S A 2009; 106(19):7840-45; PMID:19416884; http://dx.doi.org/ 10.1073/pnas.0901854106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer 2011; 18(4):R125-147; PMID:21606157; http://dx.doi.org/ 10.1530/ERC-11-0074 [DOI] [PubMed] [Google Scholar]
- [36].Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer–mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol 2014; 10(8):455-65; PMID:24935119; http://dx.doi.org/ 10.1038/nrendo.2014.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Poloz Y, Stambolic V. Obesity and cancer, a case for insulin signaling. Cell Death Dis 2015; 6:e2037; http://dx.doi.org/ 10.1038/cddis.2015.381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 1999; 19(5):3278-88; PMID:10207053; http://dx.doi.org/ 10.1128/MCB.19.5.3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, Siddle K, Goldfine ID, Belfiore A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res 1999; 5(7):1935-44; PMID:10430101 [PubMed] [Google Scholar]
- [40].Fox EM, Miller TW, Balko JM, Kuba MG, Sanchez V, Smith RA, Liu S, Gonzalez-Angulo AM, Mills GB, Ye F et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res 2011; 71(21):6773-84; PMID:21908557; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Malaguarnera R, Belfiore A. The insulin receptor: a new target for cancer therapy. Front Endocrinol 2011; 2:93; PMID:22654833; http://dx.doi.org/ 10.3389/fendo.2011.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dowling RJ, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR, Leong WL, Escallon JM, Reedijk M, Goodwin PJ et al. Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res 2015; 17:32; PMID:25849721; http://dx.doi.org/ 10.1186/s13058-015-0540-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Engels CC, de Glas NA, Sajet A, Bastiaannet E, Smit VT, Kuppen PJ, Seynaeve C, van de Velde CJ, Liefers GJ. The influence of insulin-like Growth Factor-1-Receptor expression and endocrine treatment on clinical outcome of postmenopausal hormone receptor positive breast cancer patients: A Dutch TEAM substudy analysis. Mol Oncol 2016; 10(4):509-16; PMID:26706833; http://dx.doi.org/ 10.1016/j.molonc.2015.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Heckman-Stoddard BM, Gandini S, Puntoni M, Dunn BK, DeCensi A, Szabo E. Repurposing old drugs to chemoprevention: the case of metformin. Semin Oncol 2016; 43(1):123-33; PMID:26970131; http://dx.doi.org/ 10.1053/j.seminoncol.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L et al. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget 2016; 7(26):40767-40780; PMID:27004404; 24393785 10.18632/oncotarget.8194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 2014; 10(3):143-56; PMID:24393785; http://dx.doi.org/ 10.1038/nrendo.2013.256 [DOI] [PubMed] [Google Scholar]
- [47].Ringner M, Fredlund E, Hakkinen J, Borg A, Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS ONE 2011; 6(3):e17911; PMID:21445301; http://dx.doi.org/ 10.1371/journal.pone.0017911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013; 8(12):e82241; http://dx.doi.org/ 10.1371/journal.pone.0082241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res 2014; 16(3):207; PMID:25223380; http://dx.doi.org/ 10.1186/bcr3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem 2006; 97(2):261-74; PMID:16267837; http://dx.doi.org/ 10.1002/jcb.20690 [DOI] [PubMed] [Google Scholar]
- [51].Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer 2003; 10(2):179-86; PMID:12790780; http://dx.doi.org/ 10.1677/erc.0.0100179 [DOI] [PubMed] [Google Scholar]
- [52].Watts CK, Brady A, Sarcevic B, deFazio A, Musgrove EA, Sutherland RL. Antiestrogen inhibition of cell cycle progression in breast cancer cells in associated with inhibition of cyclin-dependent kinase activity and decreased retinoblastoma protein phosphorylation. Mol Endocrinol 1995; 9(12):1804-13; PMID:8614416 [DOI] [PubMed] [Google Scholar]
- [53].Watts CK, Sweeney KJ, Warlters A, Musgrove EA, Sutherland RL. Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res Treat 1994; 31(1):95-105; PMID:7981461; http://dx.doi.org/ 10.1007/BF00689680 [DOI] [PubMed] [Google Scholar]
- [54].Nardone A, De Angelis C, Trivedi MV, Osborne CK, Schiff R. The changing role of ER in endocrine resistance. Breast 2015; 24 Suppl 2:S60-66; PMID:26271713; http://dx.doi.org/ 10.1016/j.breast.2015.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lange CA, Yee D. Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr Relat Cancer 2011; 18(4):C19-24; PMID:21613412; http://dx.doi.org/ 10.1530/ERC-11-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 1996; 271(34):20608-16; PMID:8702807; http://dx.doi.org/ 10.1074/jbc.271.34.20608 [DOI] [PubMed] [Google Scholar]
- [57].Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt AN, Crook T, Lord CJ, Ashworth A. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell 2008; 13(2):91-104; PMID:18242510; http://dx.doi.org/ 10.1016/j.ccr.2008.01.001 [DOI] [PubMed] [Google Scholar]
- [58].Noble ME, Endicott JA, Brown NR, Johnson LN. The cyclin box fold: protein recognition in cell-cycle and transcription control. Trends Biochem Sci 1997; 22(12):482-7; PMID:9433129; http://dx.doi.org/ 10.1016/S0968-0004(97)01144-4 [DOI] [PubMed] [Google Scholar]
- [59].Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex [see comments]. Nature 1995; 376(6538):313-20 [DOI] [PubMed] [Google Scholar]
- [60].Lees EMaH E. Sequences within the conserved cyclin box of human cyclin A are sufficient for binding to and activation of cdc2 kinase. Mol Cell Biol 1993; 13:1194-201; PMID:8423786; http://dx.doi.org/ 10.1128/MCB.13.2.1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 2007; 67(22):10804-12; PMID:18006825; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2310 [DOI] [PubMed] [Google Scholar]
- [62].Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 2009; 8(1):88-96; PMID:19106626; http://dx.doi.org/ 10.4161/cc.8.1.7499 [DOI] [PubMed] [Google Scholar]
- [63].Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat 2010; 123(1):271-9; PMID:20135346; http://dx.doi.org/ 10.1007/s10549-010-0763-9 [DOI] [PubMed] [Google Scholar]
- [64].Hu T, Chung YM, Guan M, Ma M, Ma J, Berek JS, Hu MC. Reprogramming ovarian and breast cancer cells into non-cancerous cells by low-dose metformin or SN-38 through FOXO3 activation. Scientific Rep 2014; 4:5810; PMID:25056111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem 1997; 272(16):10882-94; PMID:9099745; http://dx.doi.org/ 10.1074/jbc.272.16.10882 [DOI] [PubMed] [Google Scholar]
- [66].Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M, Beato M, Sica V, Bresciani F, Weisz A. 17beta-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene 1996; 12(11):2315-24; PMID:8649771 [PubMed] [Google Scholar]
- [67].Casimiro MC, Velasco-Velazquez M, Aguirre-Alvarado C, Pestell RG. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin Investig Drugs 2014; 23(3):295-304; PMID:24387133; http://dx.doi.org/ 10.1517/13543784.2014.867017 [DOI] [PubMed] [Google Scholar]
- [68].Zhang Q, Sakamoto K, Wagner KU. D-type Cyclins are important downstream effectors of cytokine signaling that regulate the proliferation of normal and neoplastic mammary epithelial cells. Mol Cell Endocrinol 2014; 382(1):583-92; PMID:23562856; http://dx.doi.org/ 10.1016/j.mce.2013.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci 2008; 121(Pt 23):3853-7; PMID:19020303; http://dx.doi.org/ 10.1242/jcs.039131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wilcken NR, Prall OW, Musgrove EA, Sutherland RL. Inducible overexpression of cyclin D1 in breast cancer cells reverses the growth-inhibitory effects of antiestrogens. Clin Cancer Res 1997; 3(6):849-54; PMID:9815758 [PubMed] [Google Scholar]
- [71].Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer 2005; 12 Suppl 1:S47-59; PMID:16113099; http://dx.doi.org/ 10.1677/erc.1.00993 [DOI] [PubMed] [Google Scholar]
- [72].Kasten M, Giordano A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene 2001; 20(15):1832-8; PMID:11313931; http://dx.doi.org/ 10.1038/sj.onc.1204295 [DOI] [PubMed] [Google Scholar]
- [73].Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005; 437(7062):1173-8; PMID:16189514; http://dx.doi.org/ 10.1038/nature04209 [DOI] [PubMed] [Google Scholar]
- [74].Khanal P, Yun HJ, Lim SC, Ahn SG, Yoon HE, Kang KW, Hong R, Choi HS. Proyl isomerase Pin1 facilitates ubiquitin-mediated degradation of cyclin-dependent kinase 10 to induce tamoxifen resistance in breast cancer cells. Oncogene 2012; 31(34):3845-56; PMID:22158035; http://dx.doi.org/ 10.1038/onc.2011.548 [DOI] [PubMed] [Google Scholar]
- [75].McCarthy SA, Chen D, Yang BS, Garcia Ramirez JJ, Cherwinski H, Chen XR, Klagsbrun M, Hauser CA, Ostrowski MC, McMahon M. Rapid phosphorylation of Ets-2 accompanies mitogen-activated protein kinase activation and the induction of heparin-binding epidermal growth factor gene expression by oncogenic Raf-1. Mol Cell Biol 1997; 17(5):2401-12; PMID:9111309; http://dx.doi.org/ 10.1128/MCB.17.5.2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Yang BS, Hauser CA, Henkel G, Colman MS, Van Beveren C, Stacey KJ, Hume DA, Maki RA, Ostrowski MC. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol 1996; 16(2):538-47; PMID:8552081; http://dx.doi.org/ 10.1128/MCB.16.2.538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Guen VJ, Gamble C, Flajolet M, Unger S, Thollet A, Ferandin Y, Superti-Furga A, Cohen PA, Meijer L, Colas P. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome. Proc Natl Acad Sci U S A 2013; 110(48):19525-30; PMID:24218572; http://dx.doi.org/ 10.1073/pnas.1306814110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev 2009; 30(6):586-623; PMID:19752219; http://dx.doi.org/ 10.1210/er.2008-0047 [DOI] [PubMed] [Google Scholar]
- [79].Frasca F, Pandini G, Vigneri R, Goldfine ID. Insulin and hybrid insulin/IGF receptors are major regulators of breast cancer cells. Breast Dis 2003; 17:73-89; PMID:15687679; http://dx.doi.org/ 10.3233/BD-2003-17108 [DOI] [PubMed] [Google Scholar]
- [80].Sacco A, Morcavallo A, Pandini G, Vigneri R, Belfiore A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology 2009; 150(8):3594-3602; PMID:19443570; http://dx.doi.org/ 10.1210/en.2009-0377 [DOI] [PubMed] [Google Scholar]
- [81].Rajapaksha H, Forbes BE. Ligand-Binding Affinity at the Insulin Receptor Isoform-A and Subsequent IR-A Tyrosine Phosphorylation Kinetics are Important Determinants of Mitogenic Biological Outcomes. Front Endocrinol 2015; 6:107; PMID:26217307; http://dx.doi.org/ 10.3389/fendo.2015.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M, Orr ZS, Caspi A, Tzukerman M, LeRoith D. Highly specific role of the insulin receptor in breast cancer progression. Endocr Relat Cancer 2015; 22(2):145-57; PMID:25694511; http://dx.doi.org/ 10.1530/ERC-14-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ma J, Guo Y, Chen S, Zhong C, Xue Y, Zhang Y, Lai X, Wei Y, Yu S, Zhang J et al. Metformin enhances tamoxifen-mediated tumor growth inhibition in ER-positive breast carcinoma. BMC Cancer 2014; 14:172; PMID:24612549; http://dx.doi.org/ 10.1186/1471-2407-14-172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Berstein LM, Yue W, Wang JP, Santen RJ. Isolated and combined action of tamoxifen and metformin in wild-type, tamoxifen-resistant, and estrogen-deprived MCF-7 cells. Breast Cancer Res Treat 2011; 128(1):109-17; PMID:20683653; http://dx.doi.org/ 10.1007/s10549-010-1072-z [DOI] [PubMed] [Google Scholar]
- [85].Lau YK, Du X, Rayannavar V, Hopkins B, Shaw J, Bessler E, Thomas T, Pires MM, Keniry M, Parsons RE et al. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014; 5(21):10503-17; PMID:25361177; http://dx.doi.org/ 10.18632/oncotarget.2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wahdan-Alaswad R, Harrell JC, Fan Z, Edgerton SM, Liu B, Thor AD. Metformin attenuates transforming growth factor beta (TGF-beta) mediated oncogenesis in mesenchymal stem-like/claudin-low triple negative breast cancer. Cell Cycle 2016; 15(8):1046-59; PMID:26919310; http://dx.doi.org/ 10.1080/15384101.2016.1152432 [DOI] [PMC free article] [PubMed] [Google Scholar]