

Abstract
Unliganded (apo-) estrogen receptor α (ERα, NR3A1) is classically considered as transcriptionally unproductive. Reassessing this paradigm demonstrated that apo-human ERα (ERα66) and its N-terminally truncated isoform (ERα46) are both predominantly nuclear transcription factors that cycle on the endogenous estrogen-responsive pS2 gene promoter in vivo. Importantly, isoform-specific consequences occur in terms of poising the promoter for transcription, as evaluated by determining (i) the engagement of several cofactors and the resulting nucleosomal organization; and (ii) the CpG methylation state of the pS2 promoter. Although transcriptionally unproductive, cycling of apo-ERα66 prepares the promoter to respond to ligand, through sequentially targeting chromatin remodeling complexes and general transcription factors. Additionally, apo-ERα46 recruits corepressors, following engagement of cofactors identical to those recruited by apo-ERα66. Together, these data describe differential activities of ERα isoforms. Furthermore, they depict the maintenance of a promoter in a repressed state as a cyclical process that is intrinsically dependent on initial poising of the promoter.
Keywords: ChIP, estrogen receptor, methylation, transcription, unliganded
Introduction
Estrogens, such as 17-β estradiol (E2), are steroid hormones predominantly synthesized by ovaries that act as pivotal hormones in the physiology of the female reproductive tract and in the homeostasis of other tissues (Nilsson et al, 2001). Although protective against pathologies such as osteoporosis, Alzheimer's and cardiovascular disease, E2 has roles in the ontogeny of breast cancer (Bian et al, 2001; Jordan et al, 2001; Honjo et al, 2003). Most functions exerted by E2 are mediated by estrogen receptors (ERα and ERβ; NR3A1 and NR3A2; NR Committee, 1999) that modify, through binding ERs to their promoter region, the pattern of expression of specific target genes. This can occur either directly through interaction with cognate DNA sequences (EREs or estrogen-responsive elements) or through protein/protein interaction with other transcription factors.
ERs belong to the nuclear receptor (NR) superfamily of transcription factors (Robinson-Rechavi et al, 2003), whose sequence can be divided into six domains (A–F). The central C region is responsible for DNA sequence recognition and cooperative binding of ER dimers on EREs that contain a palindromic PuGGTCA motif separated by 3 bp (Krust et al, 1986). E2 lodges in a hydrophobic pocket defined by the C-terminal E domain (ligand binding domain (LBD)). This LBD also contains a dimerization interface that stabilizes receptor dimers. Binding of E2 induces a considerable three-dimensional remodeling of ERs, generating surfaces that can associate with intermediate transcription factors. These surfaces are included within the two transactivation functions (AFs) of ERs, located in the N- (AF1) and C-terminal (AF2) domains (Brzozowski et al, 1997; Robinson-Rechavi et al, 2003). ERα induces the ordered recruitment of a series of coactivator complexes, leading to histone acetylation, methylation, chromatin remodeling and recruitment of the basal transcription machinery (Shang et al, 2000; Belandia and Parker, 2003; Métivier et al, 2003). To date, six classes of coactivator complexes have been identified as interacting with ERα in a ligand-dependent manner: (i) members of the p160 subfamily (exemplified by SRC1 and TIF2); (ii) histone acetyl transferases (HATs) such as CBP/p300; (iii) histone arginine methyl transferases (HMTs) such as CARM1 or PRMT1; (iv) nucleosome remodeling complexes such as SWI/SNF; (v) complexes of the SMCC/TRAP/DRIP/ARC class that facilitate the activation of the polymerase II (PolII); and (vi) the AF1-specific coactivators p68 and p72 RNA helicase (Robyr et al, 2000; Watanabe et al, 2001). In contrast, ERα liganded to antiestrogens such as 4-hydroxytamoxifen (OHT) downregulates transcription. The three-dimensional changes provoked by binding of OHT induce recruitment of corepressors such as NR corepressor (NCoR) and histone deacetylase (HDAC) (Jepsen and Rosenfeld, 2002).
The MCF7 human breast cancer cell line expresses, in addition to ERα66, an isoform 46 kDa in size (ERα46), encoded by an RNA variant produced by alternative splicing (Flouriot et al, 2000). Expression of ERα46 was also reported in osteoblasts (Denger et al, 2001) and in endothelial cells, in which this protein exhibits partial membrane localization (Li et al, 2003). Functionally, ERα46 lacks the amino-terminal A/B domain present in the full-length ERα66, and consequently is devoid of AF1. Also, ERα46 suppresses the AF1 activity of ERα66 (Flouriot et al, 2000). Finally, unliganded (apo-) ERα46 may repress transcription of target genes (Métivier et al, 2002). We recently described the sequence of protein recruitment engaged by ERα66 in MCF-7 cells in the presence of E2 that is required to activate the endogenous E2-responsive pS2 gene (Métivier et al, 2003). As both isoforms exhibit distinct activities in the absence of hormone, we sought to describe the mechanisms occurring during the ligand-independent association of ERα46 and ERα66 isoforms with the pS2 promoter.
Results
Apo-ERα isoforms are predominantly nuclear proteins and differently impact the expression of the pS2 gene
Concomitant expression of ERα46 and ERα66 in MCF7 cells prevented elucidation of their individual modes of action. We therefore stably expressed each isoform in ERα-negative MDA-MB231 cells, giving rise to MDA∷ERα66 and MDA∷ERα46 cells. Immunostaining using HC20, which targets both isoforms (Figure 1A), shows that ERα46 and ERα66 are similarly distributed in these cells, residing mainly in the nucleus (Figure 1B). Functionally, expression of ERα66 or ERα46 in MDA-MB231 cells induces ligand-dependent expression of the endogenous estrogen-inducible pS2 gene similar to that observed in MCF7 cells (Figure 1C). However, in the MDA∷ERα46 cell line, the level of pS2 mRNA was three-fold lower in the absence of E2 than in MDA∷ERα66 cells (Figure 1C). Chromatin immunoprecipitation (ChIP) assays using unsynchronized cells demonstrate that the E2-induced expression of the pS2 gene reflects binding of ERα and of activated/phosphorylated RNA PolII (P-PolII) to the promoter (Figure 1D). Furthermore, the pS2 promoter contains acetylated histones (Ac-Hist), a marker of an active transcription locus (Sterner and Berger, 2000), in cells only when they express ERα (Figure 1D). The relative amounts of P-PolII and Ac-Hist present on the pS2 promoter were significantly lower in MDA∷ERα46 cells than in ERα66-expressing cells in the absence of E2, suggesting that apo-ERα46 specifically represses basal transcription of the pS2 gene.
Figure 1.
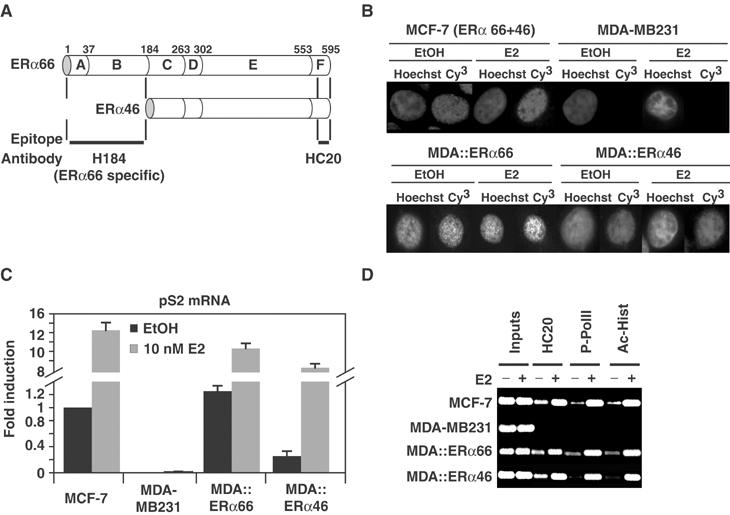
Functional properties of ERα isoforms. (A) Scheme illustrating ERα isoforms sequence and the location of the epitopes targeted by antibodies used. (B) Immunostaining of ERα isoforms with HC20. MCF-7 cells expressing both isoforms and MDA-MB231 cells expressing none or either isoforms (MDA∷ERα66 or MDA∷ERα46) were fixed after a 3 h treatment with 10−8 M estradiol (E2) or ethanol (EtOH) as vehicle control. Nuclei are visualized by Hoechst staining. (C) The expression of endogenous pS2 gene in the four cell lines was monitored by real-time PCR. After 72 h of culture in stripped media, cells were treated for 3 h with 10−8 M E2 or EtOH as vehicle control. pS2 mRNA levels were normalized against invariant GAPDH mRNA. (D) ChIP probed the recruitment of ERα, P-PolII and Ac-Hist to the pS2 promoter in unsynchronized cells sampled as in (C).
Does the apo-ERα46-mediated silencing of the pS2 promoter involve DNA methylation processes?
Long-term silencing of genes is often associated with an increased CpG methylation (Bird, 2002). To question the involvement of these epigenetic modifications in the actions of apo-ERα46, we evaluated the methylation status of CpGs present within the pS2 promoter (Figure 2A). Digestion of genomic DNA isolated from the four cell lines by HpaII (does not digest methylated or hemimethylated CMeCGG) limited PCR amplification of the B, C and D fragments to varying degrees without disturbing amplification of the control A fragment (Figure 2B). While these three CpGs are likely fully methylated in MDA-MB231 cells, their methylation status is lower or even null in MCF7 and in MDA cells expressing either ERα isoform. These variations do not correlate with different proportions of cells engaged in the S or G2 phases, which are points where replication-induced methylation occurs (Urnov, 2003; data not shown). Sequencing of PCR-amplified upper strand of the pS2 promoter, following bisulfite modification of genomic DNA, demonstrates that all 19 CpGs are methylated in MDA-MB231 cells, reflecting the silent state of the pS2 promoter in these cells. In contrast, CpGs located 3′ to the −555 position exhibit a lower and similar methylation status in all cells expressing ERα isoforms (Figure 2C). Therefore, the methylation status of the pS2 promoter is not modified by the ERα46 isoform. These results provide evidence that both ERα isoforms poise the pS2 gene for expression and, overlying this effect, that apo-ERα46 represses the basal transcription of the pS2 gene through mechanisms distinct from gene silencing by CpG methylation.
Figure 2.

Evaluation of intercellular variations in MeCpG content of the pS2 promoter. (A) Schematic representation of the pS2 promoter, with significant cis-acting features and phased nucleosomes (Sewack and Hansen, 1997) highlighted. CG dinucleotides are indicated, with those within an HpaII site in gray. Regions amplified by PCR primers are also depicted. (B) Genomic DNA prepared from MDA-MB231, MCF-7 and MDA-MB231 cells stably expressing either ERα isoforms was digested by HpaII and then subject to PCR to amplify the described regions. (C) To quantify the methylation of given CpG, genomic DNA was chemically modified using bisulfite-mediated C to U transformation. The upper strand of the pS2 promoter was then specifically amplified by PCR, and the total pool of fragments was sequenced. Values are the mean±s.e.m. from data acquired in three separate experiments.
Cyclical binding of apo-ERα isoforms to the pS2 promoter has different effects
Association of liganded and apo-ERα66 with target promoters is cyclical in vivo (Reid et al, 2003). We therefore evaluated the properties of the binding of apo-ERα46 to DNA. Basal transcription of the pS2 gene in the absence of E2 occurs in MCF-7 and stably transfected MDA cells, even after 3 days of steroid deprivation (Figure 1C). Transcription factors present on the promoter limit the clear analysis of the kinetics of apo-ERα binding. We abrogated this background activity of the pS2 promoter by treating cells for 2 h with 2.5 μM α-amanitin, as described previously (Reid et al, 2003). Following the removal of α-amanitin, a maximum of 20% of endogenous pS2 promoters can be recovered by ChIP in the absence of E2 using either the ERα66-specific (H184) or HC20 antibodies in MCF7 cells (Figure 3A). Both antibodies gave similar curves with apo-ERα cycling with a periodicity of 20 min. The similarity of the curves obtained MDA∷ERα66 and in MDA∷ERα46 (Figure 3B) further demonstrates that both apo-ERα isoforms have similar cyclical DNA binding characteristics in vivo.
Figure 3.
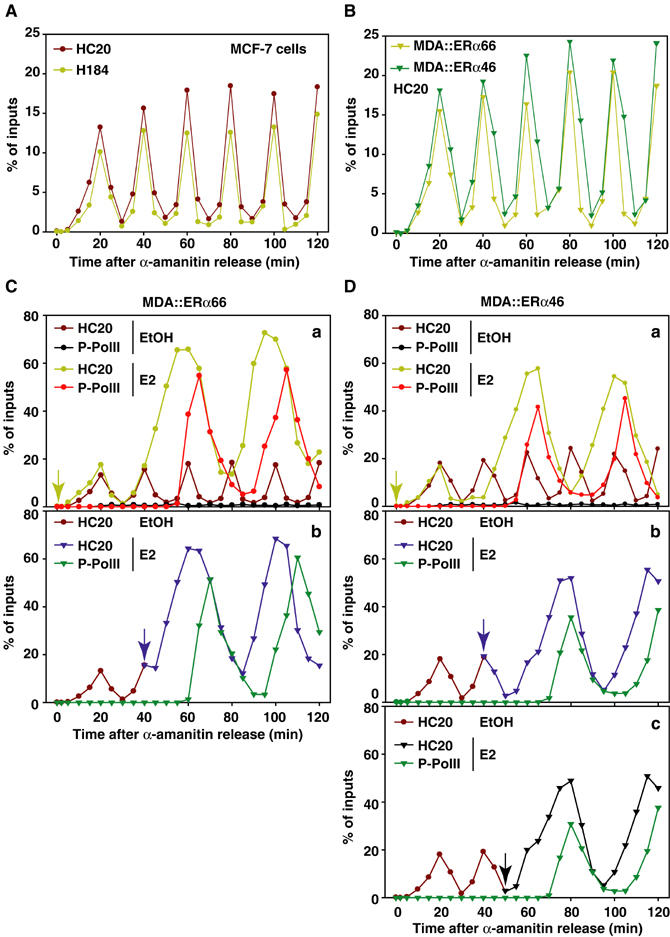
Cyclical permissiveness of the pS2 promoter for ERα46 and ERα66 bindings, with distinct functional consequences. (A) Kinetic ChIP experiments performed using H184 or HC20 antibodies. Cells were first synchronized by a 72 h deprivation of serum and then treated for 2 h with 2.5 μM α-amanitin. After washing and renewal of the media, chromatin was prepared on sampled cells with 5 min intervals. Amounts of immunoprecipitated pS2 promoter were quantified by real-time PCR. Values, expressed as the percent of inputs, are the mean of three separate experiments, and have an s.d. <2%. (B) No differences in the cycling of both ERα isoforms on the pS2 promoter are evidenced in kinetic ChIP assays performed using chromatin prepared from MDA-MB231 cells expressing either ERα isoforms. Next, experiments probed the functional impact of these cycles of ERα isoforms binding in MDA∷ERα66 (C) and MDA∷ERα46 cells (D). These assays evaluated the recruitment and activation of P-PolII in response to 10−8 M estradiol (E2) added at times indicated within the graphs (A–C of each panels).
Unlike apo-ERα66, apo-ERα46 represses transcription (Métivier et al, 2002), as illustrated by the lower expression of pS2 mRNA in MDA∷ERα46 cells in the absence of ligand (Figure 1C). We reasoned that differential poising of the pS2 promoter by the two apo-ERα isoforms may result in different dynamics in gene responses to E2. After α-amanitin synchronization, cells were thus treated with 10 nM E2 at different times and the activation of PolII on the pS2 promoter was followed. These experiments demonstrate that apo-ERα66 initiates specific events sufficient for immediate response to E2 (Figure 3C), bypassing, or equivalent, to those occurring during the first transcriptionally unproductive cycle observed in the presence of E2 (Métivier et al, 2003). In contrast, binding of apo-ERα46 slows the response to E2, as P-PolII binding is observed only if E2 is added when ERα46 has finished one of its cycles (Figure 3D). Therefore, while apo-ERα66 poises the pS2 gene for expression, apo-ERα46 inhibits pS2 gene commitment.
Recruitment of transcription factors by both ERα isoforms
MCF7 cells, which express both ERα66 and ERα46, were initially used to identify proteins recruited by both ERα isoforms to the pS2 promoter through comparison of ChIP patterns obtained on chromatin prepared from untreated MCF7 cells and from cells synchronized by α-amanitin. Without this synchronization step, ERα-independent initiating events occur, mediated by factors such as AP1 (binding site close to the ERE; Figure 2A), and are likely responsible for the basal transcription of the pS2 gene (Barkhem et al, 2002; Supplementary Figure S1). Hence, α-amanitin-induced clearance of the pS2 promoter is essential to assess kinetically the recruitment of transcription factors to the promoter mediated by apo-ERα (Reid et al, 2003; Supplementary Figure S1). Subsequent ChIP assays demonstrate that, in MCF7 cells, either apo-ERα isoform or both have the ability to promote the engagement of HATs (Tip60, GCN5 and p300, but not CBP), HMT (only PRMT1), general transcription factors (only TBP and TFIIA) and repressive complexes (Sin3 and NuRD) on the pS2 promoter (Figure 4A, compare black and gray bars). As anticipated, transcription factors such as elongator or PolII, whose presence indicates an active pS2 gene, are not recruited by apo-ERα isoforms (Figure 4A). H3 was acetylated on K14 but not dimethylated on R17; H4 was dimethylated on R3 but not acetylated on K16. This is in contrast to an active pS2 promoter organization, where all these histone residues are modified (Métivier et al, 2003).
Figure 4.
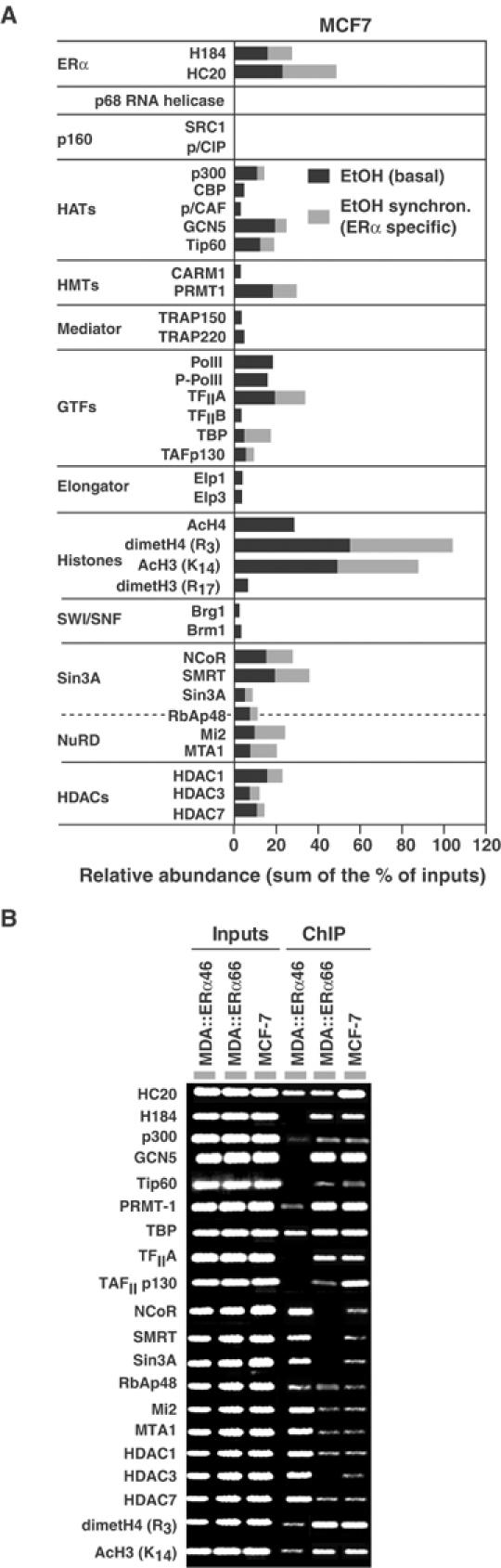
Identification of the transcription factors targeted by apo-ERα isoforms on the pS2 promoter in vivo. (A) Real-time PCR-mediated quantification of ChIP experiments performed using chromatin prepared from MCF-7 cells either treated with ethanol as vehicle control (EtOH) or firstly synchronized by α-amanitin treatment. Comparing the results obtained in both conditions enables determination of proteins directly recruited by both apo-ERα isoforms on the pS2 promoter (those after α-amanitin treatment, in gray). (B) Comparative ChIP assays using chromatin prepared from MCF-7, MDA-MB231 or MDA-MB231 cells expressing either ERα isoforms.
Comparison of ChIP patterns obtained with MDA∷ERα66, MDA∷ERα46 and MCF7 cells confirms that both apo-ERα isoforms promote engagement of HATs and PRMT1 (Figure 4B). Importantly, although TBP engages the pS2 promoter in all cell lines, recruitment of TFIIA and TAFp130 occurs only in cells that express ERα66. Conversely, components of the Sin3 repressive complex (NCoR/SMRT) are present on the pS2 promoter only in MDA∷ERα46 cells (Figure 4B). This is associated with lower acetylation of H3 K14 as compared to MDA∷ERα66 cells. Recruitment of NuRD (Mi2 and MTA1) to the pS2 promoter may not be a repressive event but reflect a clearance process similar to that we described recently (Métivier et al, 2003). Re-ChIP next elucidated the organization of transcription complexes recruited by both ERα isoforms to DNA (Figure 5A and B). Recruitment of HATs (p300, GCN5 and Tip60) and of HDACs are combinatorial, reflecting functional redundancy among these protein subfamilies. In summary, Re-ChIP identified five different protein complexes, of which two are ERα isoform specific (Figure 5C): (i) complex IV containing GCN5 together with TBP, TFIIA and TAFp130 is specific for ERα66; and (ii) the Sin3 repressive complex is specifically targeted by apo-ERα46.
Figure 5.
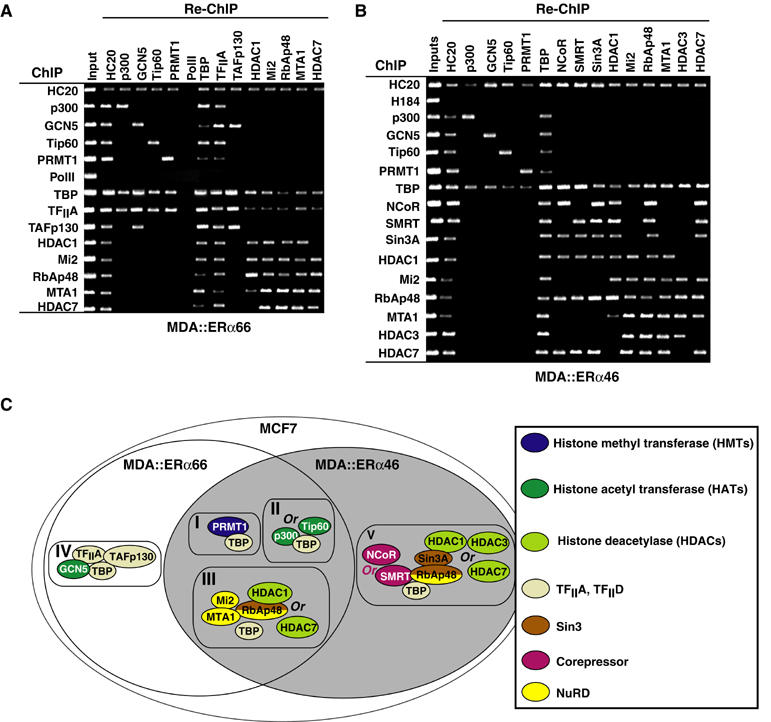
Re-ChIP screening of the factors recruited to the pS2 promoter by apo-ERα isoforms. Chromatin prepared from α-amanitin synchronized MDA-MB231 cells expressing ERα66 (A) or ERα46 (B) was subject to the ChIP procedure with the antibodies shown at the left side, and again immunoprecipitated using antibodies shown at the top of the panels. (C) Scheme illustrating the topology of the complexes recruited to the pS2 promoter, as indicated through the comprehensive Re-ChIP analysis shown above.
Kinetics of recruitment of transcription factors by apo-ERα isoforms
Following ERα and PRMT1 engagement on the pS2 promoter, Tip60 and p300 are recruited together (Figure 6A). Re-ChIP analysis shows that these HATs are present in different complexes (Figure 5), suggesting that both proteins are alternatively recruited to different sets of pS2 promoters. These data also confirm that PRMT1 methylates H4 R3 (Wang et al, 2001). Of the 20 factors evaluated, only TAFp130, p300, GCN5 and Tip60 associate with the pS2 promoter with a frequency identical to ERα66 (Figure 6Aa and b). In contrast, the pS2 promoter remains permissive for the association of TBP and TFIIA for about 1 h, with GCN5 association slightly increased when TBP and TFIIA load onto the promoter (Figure 6Aa and b). This suggests that GCN5 is required for the recruitment of TBP and TFIIA, or vice versa. ChIP analysis with a resolution of 1 min confirms these conclusions and further shows that PRMT1 is the first protein recruited to the pS2 promoter by either ERα isoforms. This is then followed by p300, Tip60 and GCN5 (Figure 6Ac). This analysis also shows that TBP is recruited 3–4 min before TFIIA. In MDA∷ERα46, the kinetics and sequence of association of a few of these proteins with the pS2 promoter were different, even when probed at 1 min intervals (data not shown). Notably, recruitment of TBP occurred on fewer pS2 promoters than in MDA∷ERα66 cells (Figure 6Ad and e). The most striking difference between the actions of apo-ERα isoforms is the specific recruitment of Sin3 repressors by apo-ERα46 (Figure 6B). As summarized in Figure 6C, both apo-ERα isoforms recruit, with similar kinetics, transcriptional activators such as HATs and HMTs. However, following these steps, where the pS2 promoter becomes permissive for transcriptional activity, ERα46 specifically recruits repressive complexes.
Figure 6.
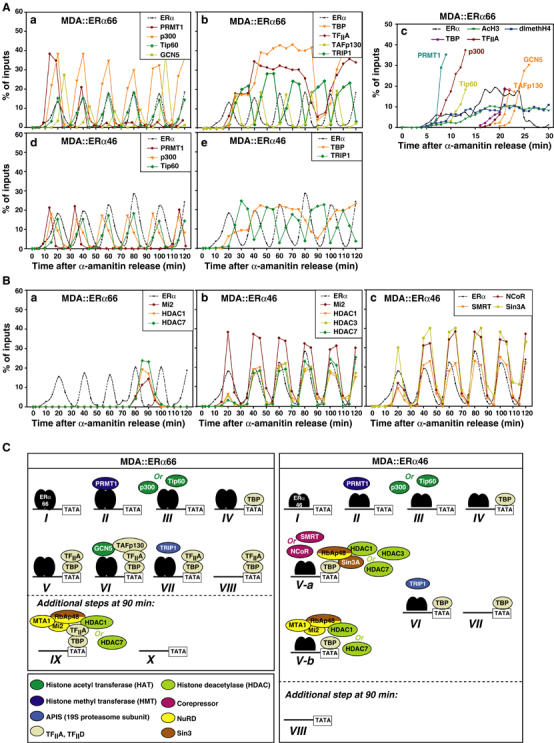
Kinetics of the recruitment of transcription factors directed by apo-ERα isoforms on the pS2 promoter. (A, B) Kinetic ChIP experiments were performed and expressed as in Figure 4, using antibodies and chromatin prepared from synchronized MDA-MB231 cells expressing either ERα isoforms, as indicated within the graphs. (C) Scheme summarizing the sequence of recruitment of transcription factors directed by ERα66 and ERα46 onto the pS2 promoter. The numbering reflects the order of engagement of the different complexes onto the promoter. In the case of ERα46, V-a and V-b are combinatorial steps.
In the presence of E2, cyclic clearance of the pS2 promoter of transcription factors is generated by (i) proteasome-mediated degradation of ERα through recruitment of the APIS complex (Reid et al, 2003); and (ii) recruitment of HDACs-SWI/SNF and NuRD complexes (Métivier et al, 2003). Kinetic analysis of the recruitment of NuRD and TRIP1 (a component of the APIS complex) in MDA∷ERα66 cells indicates that NuRD may be involved in the removal of TFIIA and TBP from the pS2 promoter, while the APIS complex might cyclically remove ERα66 from the pS2 promoter (Figure 6Ab, e and B). Unfortunately, cyclical recruitment of NuRD and Sin3 that occurs in MDA∷ERα46 cells precludes a similar description of the action of these complexes in the generation of ERα46 cycles. However, recruitment of TRIP1 at the end of the cycles driven by apo-ERα46 (Figure 6Ae) indicates that, as for apo-ERα66, APIS is key in clearing ERα from the pS2 promoter.
Apo-ERα induces changes in the nucleosomal organization of the pS2 promoter
We next verified the consequences of engagement of HATs, HMT and HDACs on chromatin organization within the pS2 promoter. In MDA∷ERα66 cells, histones H3 and H4 become, respectively, acetylated or methylated to constant levels following recruitment of p300 or Tip60 and PRMT1 to the pS2 promoter (Figures 6A, C and 7A). In contrast, in MDA∷ERα46 cells, acetylation of H3 K14 parallels ERα46 binding, with dimethylation of H4 R3 exhibiting a more complex pattern. Repressors specifically recruited by apo-ERα46 may thus impact on the acetylation levels of H3 K14. In ERα-positive cells, ChIP assays performed on mononucleosomes generated by micrococcal nuclease (MNAse) digestion of chromatin show both nucleosomes NucE and NucT (Sewack and Hansen, 1997; Figure 7B) to include dimethylated H4. In contrast, acetylation of H3 K14 only occurs on NucT (Figure 7C). The position of NucE and NucT vary during cycles directed by ERα isoforms (Figure 7D). In ERα-positive cells, stabilization of both nucleosomes in the preferred positions described by Sewack and Hansen (1997) is first observed at 20 min (A and C). In both MDA∷ERα66 and MDA∷ERα46 cells, the region encompassing the TATA box becomes accessible to MNAse (compare the C, H, I and J products in Figure 7D) when TBP associates with the promoter (20 min but not at 90 min; Figure 6A). At 90 min, the J fragment, which is digested in MDA∷ERα66 and MCF7 cells, is protected in MDA∷ERα46 cells. This indicates that NucT is more flexible within MDA∷ERα46 than in MDA∷ERα66 and MCF7 cells, where NucT protects only the 5′ extremity, as revealed by amplification of the I fragment (Figure 7D). NucE also has dynamic flexibility in its position encompassing the 3′ end of the sequence, as the F but not G fragment is protected from MNAse in all cells. NucE adopts a fixed position in MCF7 and in MDA∷ERα66 cells at 30 and 90 min, when the pS2 promoter is free of ERα. In contrast, NucE has a higher flexibility in MDA∷ERα46 cells at these time points. Re-ChIP assays on mononucleosomes demonstrate that, at 20 min, dimethylated H4 R3 and acetylated H3 K14 are present together in the same pool of NucT (Figure 7E). It is only in MDA∷ERα66 that an additional dimethylation of H4 R3 appears within NucE at 40 min (Figure 7E). Moreover, it is only in MDA∷ERα46 cells that levels of acetylated H3 K14 in NucT cycle functionally reflect the sequential associations of HATs and HDACs (Figures 6 and 7D). Conclusions from these experiments are presented in Figure 7F.
Figure 7.
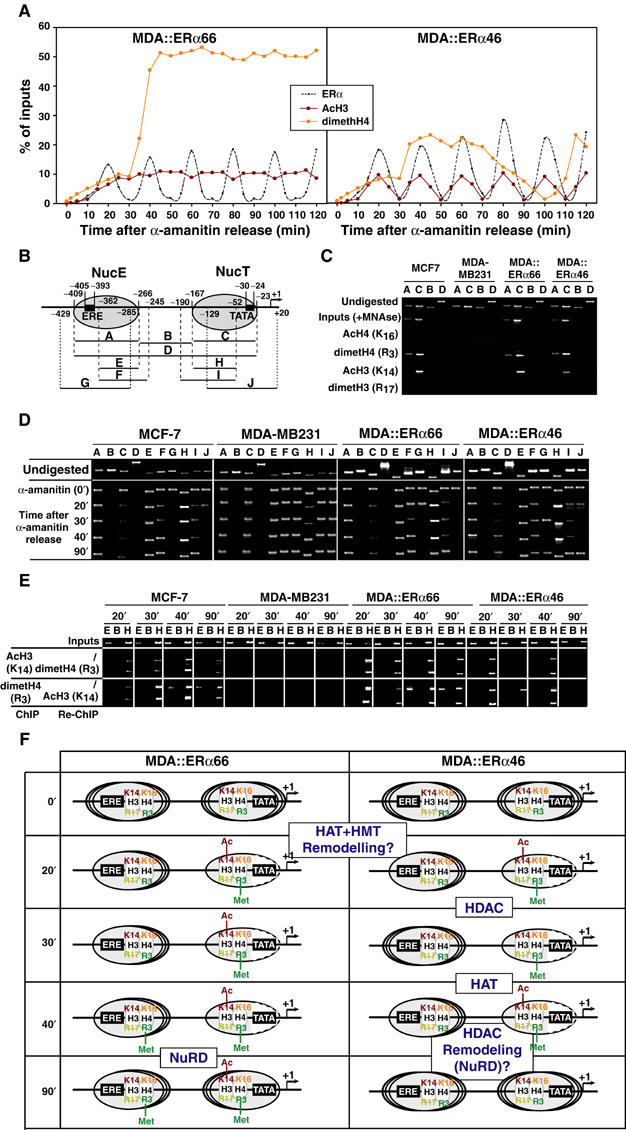
Dynamic chromatin modifications within the pS2 promoter. (A) Kinetic ChIP experiments were performed as depicted in Figure 4, using antibodies and chromatin prepared from synchronized MDA-MB231 cells expressing either ERα isoforms, as indicated within the graphs. (B) Schematic representation of the pS2 promoter, with phased nucleosomes and regions amplified by PCR primers depicted. (C) Mononucleosomes prepared from indicated cells were used in ChIP assays detecting modified histones in both NucE and NucT. The absence of amplification using the B and D primer pairs controls proper digestion. (D) Fluctuations in NucE and NucT positions on the pS2 promoter were assessed through PCR amplification of mononucleosomes using the A to J primer pairs. (E) Following α-amanitin synchronization, mononucleosomes were prepared at times indicated and subjected to ChIP and Re-ChIP procedures using the indicated antibodies. Use of the E and H primer pairs for DNA amplification showed the localization of the modified histones within either NucE or NucT. (F) Scheme summarizing the dynamic histone modifications and nucleosomal organization of the pS2 promoter during ERα isoforms cycles. Dashed lines in NucT reflect the increased accessibility of the TATA box surrounding region for MNase digestion. The recruitment events that are hypothesized to correlate with the observed nucleosome modifications are boxed and highlighted.
Discussion
In addition to a predominant 66 kDa protein, the ERα gene also produces a 46 kDa isoform, lacking N-terminal domains, that has the ability to repress transcription in the absence of ligand (Flouriot et al, 2000; Métivier et al, 2002). This prompted us to determine the sequence of recruitment of transcription factors engaged by both apo-ERα isoforms in vivo. Cyclical engagements of apo-ERα isoforms at the pS2 promoter have a periodicity of 20 min, are transcriptionally unproductive and differ in their influence on pS2 promoter commitment.
Apo-ERα66: achieving commitment of the pS2 promoter
The expression of ERα is necessary to determine pS2 gene activity (Figure 1). Correspondingly, if apo-ERα66 is allowed to achieve one cycle of DNA binding prior to the addition of E2, rapid (10 min) recruitment of PolII to the pS2 promoter occurs. This indicates that apo-ERα66 induces chromatin modifications sufficient to permit subsequent transcription of the pS2 gene in the presence of liganded ERα66. Induction of transcriptional competence by apo-ERα66 involves the cyclical engagement of HATs and HMTs that acetylate and methylate H3 K14 and H4 R3, respectively. These events impact on the phased nucleosomal structure of the pS2 promoter, with the nucleosome associated with the TATA box (NucT) subject to two different events. First, NucT becomes stabilized into an ‘active' position corresponding to that described by Sewack and Hansen (1997) followed by a disruption of DNA–histone contacts around the TATA box (Figure 7). SWI/SNF complexes stabilize the position of NucT in the presence of ligand (Métivier et al, 2003). As these complexes are not engaged on the pS2 promoter in the absence of E2 (Figure 4), apo-ERα66 may mobilize another remodeling complex, involved in the unfolding of large-scale chromatin structures described by Nye et al (2002). Stabilization of NucT in favored positions may also be an allosteric consequence of ERα binding, with this engagement stabilizing the overall equilibrium of NucT positions in the cell population towards the preferred positions. Second, dissymmetric flexibility of NucT occurs when TBP is cleared from the pS2 promoter, probably under the influence of NuRD. Recruitment of certain general transcription factors by apo-ERα66 also accounts for the poising of the pS2 gene for transcription. As described for the HNF4α gene (Hatzis and Talianidis, 2002), once prepared for transcription, the pS2 promoter has recruited TBP and TFIIA. Importantly, TAFp130 is targeted to the pS2 promoter with a similar frequency as ERα66 but not TBP, which persists over successive cycles. In addition to its role in relieving the inhibition mediated by TAFp250 and in the recruitment of the mediator complex, TAFp130 may thus play additional roles (Guermah et al, 2001).
Apo-ERα46: promoting repression of the pS2 promoter
A repressive role for apo-ERα46 was suggested by the observation that, in the absence of E2, the levels of pS2 mRNA were three-fold lower in MDA∷ERα46 cells than in MDA∷ERα66 cells. ChIP assays demonstrated that apo-ERα46 specifically recruits Sin3 and NuRD repressive complexes to the pS2 promoter. Although exhibiting a similar cycling frequency, the integration of the area covered by the respective binding curves of either apo-ERα isoforms indicates that around 20% more pS2 promoters are engaged in binding apo-ERα46. Within a single cycle of pS2 association with apo-ERα, this difference indicates that, stochastically, binding of ERα46 to DNA persists longer than ERα66 does, reflecting the recruitment of additional repressive complexes. The Sin3 complex is recruited when ERα66 is liganded with OHT (Jepsen and Rosenfeld, 2002). However, deletion of the entire N-terminal region of the receptor, as occurs in the ERα46 isoform, prevents the A domain to hinder the interaction surface for corepressors in the absence of E2 (Métivier et al, 2002). Additionally, this deletion abrogates the AF1 activity, generally accepted as a function that can be active or activated in the absence of E2 (Shao and Lazar, 1999). The low basal pS2 gene activity observed in MDA∷ERα46 cells may therefore result from a combination of an active repressive process and a passive mechanism caused by the deletion of AF1. However, treating MDA∷ERα46 cells with ICI182,780, an antagonist that inhibits ERα binding to DNA (Fawell et al, 1990), restored the basal mRNA level of pS2 observed in MCF7 and MDA∷ERα66 cells to 80% (data not shown). Correspondingly, the deletion of the AF1 domain may be responsible for 20% of the loss of basal pS2 expression seen in MDA∷ERα46 cells.
These active repressive actions of apo-ERα46 do not involve stable long-term remodeling of chromatin exemplified by the inherited methylation of cytosines within the dinucleotide CpG (Bird, 2002). Instead, apo-ERα46 induces unstable chromatin modifications through the combinatorial engagement of Sin3 and NuRD complexes onto the pS2 promoter that deacetylate K14 of H3 of NucT and that achieve cyclic, asymmetric positioning of NucE. As with activating complexes (Métivier et al, 2003), alternative and combinatorial mobilization of proteins with a similar function governs stochastic mechanisms that affect transcriptional competence. Our Re-ChIP experiments confirm the existence in vivo and on DNA of biochemically defined alternative repressive complexes. Indeed, Sin3 complexes containing either NCoR or SMRT can recruit either HDAC1, 3 or 7 (Heinzel et al, 1997; Kao et al, 2000; Guenther et al, 2001). NuRD either contained HDAC1 or 7, but never HDAC3, as demonstrated previously (Zhang et al, 1999; Downes et al, 2000). In contrast to a sequential association of Sin3 and NuRD directed in vivo by OHT-bound ERα (Liu and Bagchi, 2004), Sin3 and NuRD complexes are combinatorially recruited by apo-ERα46. This implies that OHT-bound ERα directs additional events to those engaged by apo-ERα46, such as the recruitment of additional cofactors.
New insights into a ‘transcriptional clock'?
We recently proposed that a ‘transcriptional clock' synchronizes sequential waves of promoter accessibility for given cofactors to dictate an ordered sequence of events that initiate, achieve and then limit the transcriptional process. Cycles of apo-ERα isoforms binding to DNA give further insight into these mechanisms. First, comparison of the cycles directed by apo-ERα isoforms and those observed in the presence of E2 (Métivier et al, 2003) indicates that chromatin remodeling events affecting NucT are conserved. An initial stabilization into an ‘active' conformation, followed by disruption of DNA–histone contacts around the TATA box occurs. The asymmetric flexibility of NucT that specifically occurs in MDA∷ERα66 cells correlates with acetylation of H3 K14 and dimethylation of H4 R3. Dimethylation of H4 R3 destabilizes NuRD association with chromatin (Zegerman et al, 2002). NucT H4 R3 dimethylation in MDA∷ERα66 may therefore limit the extent of NuRD action, which would otherwise result in a symmetric flexibility in NucT position as observed in MDA∷ERα46. Correspondingly, dimethylation of H4 R3 on NucE, phased in association with the ERE of the pS2 promoter, which specifically occurs in MDA∷ERα66 cells, may explain the asymmetric 3′ flexibility seen only in these cells. In the cycles described in this report, H4 K16 and H3 R17 neither become, respectively, acetylated nor dimethylated. This notwithstanding, variations in NucE positioning observed in MDA∷ERα46 at 30 min are likely generated by other histone acetylation events, as NucE variations are dependent on HDAC recruitment. However, these specific variations in NucE position observed in MDA∷ERα46 do not affect the permissiveness of the pS2 promoter to ERα, as both apo-ERα isoforms cycle with a periodicity of 20 min. Other examples of mechanisms linking the progress into the ordered recruitment of transcriptional complexes and chromatin modifications arise from the flexibility of NucE and NucT positions. Indeed, the continued binding of TBP and the presence of modified histones in MDA∷ERα66 may stabilize nucleosomes in their favored positions. In contrast, the repeated presence of NuRD and HDACs in MDA∷ERα46 may make the stabilized positioning more dependent on the presence of ERα and TBP. The transition between apo- and E2-liganded cycles (45 min frequency) relies on liganded ERα-mediated disruption of histone–DNA contacts near the ERE (Métivier et al, 2003). The exact role of the successive asymmetric and symmetric positioning of NucE observed in MDA∷ERα46 has therefore to be determined.
Interplay between protein recruitment and modification may generate additional marks that define the position of the ‘transcriptional clock'. As the apo-ERα cycles are transcriptionally unproductive, at least some phases of the ‘clock' are not dependent on the activation of PolII. In particular, TBP has a central role in synchronizing the cycles of events mediated by both apo-ERα isoforms. Indeed, the pS2 promoter recruits TBP to become poised for transcription, and cycles of TBP binding on the pS2 promoter persist over three cycles of ERα association. This implies that the presence of TBP does not strictly depend on the engagement of an initiating transactivation, in contrast to processes determined in yeast (Katan-Khaykovich and Struhl, 2002). Finally, although a number of repressive mechanisms influence the binding of TBP to the TATA box (Gaston and Jayaraman, 2003), the cyclical repression provoked by apo-ERα46 appears to be TBP independent as neither deacetylation of H3 K14 present within NucT nor the asymmetric flexibility of NucE affect the presence of TBP on the pS2 promoter. Moreover, the initial recruitment of a few general transcription factors signifies that a specific event takes place when ERα is liganded to E2, leading to the recruitment of the complete transcriptional machinery. An as yet unknown mechanism therefore controls the engagement of TFIIB and PolII: if this event does not occur, the cycle terminates and apo-ERα is cleared from the promoter. As proposed by Malik et al (1998), the PC4 component of the USA complex may be involved in this restriction of transcriptionally unproductive cycles. Recruitment of the proteasome through APIS, as described previously for ERα66-mediated cycling (Reid et al, 2003), would also offer such a mechanism. However, mechanisms inducing APIS action to limit transcriptional activation remain unclear. Furthermore, the proteasome-mediated degradation of apo-ERα46 in the cycles we describe has to be confirmed. The importance of TBP in the pS2 promoter cycles also raises questions with regard to the behavior of TATA−/CCAAT+ promoters that exhibit specific properties in terms of activator persistency and HAT recruitments (Caretti et al, 2003).
α-Amanitin versus normal conditions, physiological importance of these cycles
Although key in the elucidation of the cyclical binding of apo-ERα isoforms onto DNA, the use of α-amanitin may bias the application of some of our findings to physiological conditions. The first issue relies on the exact role ERα has in the generation of the basal activity of the pS2 gene, which is abrogated by α-amanitin and reappears 2 h after α-amanitin release. Although necessary (Figure 1), the expression of ERα is unlikely to be directly involved in the processes leading to the activation of PolII in the absence of ligand. Indeed, apo-ERα does not have the structural capacity to recruit directly transcriptional activators such as proteins of the TRAP/DRIP family (Kang et al, 2002). Rather, our data indicate that apo-ERα induces, through binding to its responsive element and by limited cofactor interactions, a chromatin environment that is permissive for transcription to occur. This remodeling allows other transcription factors to induce transcription of the pS2 gene. In accordance with experiments performed by others (Barkhem et al, 2002), we determined these factors to include at least AP1 (Supplementary Figure S1). Furthermore, binding of apo-ERα and AP1 occurs on a different set of promoters (Supplementary Figure S1). Concordantly, the PolII signal appearing 2 h after α-amanitin treatment is kinetically independent of ERα. It follows that, in normal conditions, AP1 might act after that apo-ERα recruited some basal cofactors and induced a chromatin organization of the pS2 promoter adequate for AP1 actions. The functional synergism between ERα and AP1 observed when ERα is bound to estrogen (Barkhem et al, 2002) or when AP1 activity is stimulated (Bauer et al, 2002) would then be a separate mechanism. To gain further mechanistic details into this synergism, Re-ChIP experiments following the respective bindings of stimulated AP1 and E2-bound ERα need to be performed. On the other hand, the engagement of apo-ERα proteins onto promoters provides the cell with mechanisms that enable a rapid adaptation of transcription to variations in the level of E2. As transcription can be initiated by apo-ERα through phosphorylation events originating from growth factors signaling (Shao and Lazar, 1999), the interaction between apo-ERα and chromatin facilitates crosstalk between ERα and phosphorylation pathways. The nature and kinetics of events underlying this switch between transcriptionally unproductive and productive apo-ERα activities now have to be determined.
The second question relies on the ERα46-specific repressive events, the mechanistic description of which required an initial closure of the promoter induced by the α-amanitin treatment. Therefore, ERα46-specific repressive processes are maintaining rather than generating the repressed state of the pS2 promoter. To rule out definitely a potential active role for ERα46 in generating a repressed pS2 promoter, experiments using inducible expression of ERα46 will have to be carried out. The observation that apo-ERα46 directs cyclical poising/inhibition of the pS2 promoter is one of the main outcomes of this work and introduces dynamic notions to the mechanisms underlying the maintenance of a promoter in a repressed state: (i) they require ERα46 to poise initially the pS2 promoter for transcription through recruitment of factors similar to those engaged through ERα66; and (ii) these mechanisms are subsequently cyclical. Generalization of this observation to other full-length NRs and other transcription factors remains to be determined. Divergent influences of the apo-ERα isoforms on transcription of target genes are confirmed by whole-genome analysis of gene expression in breast cells (Penot et al, manuscript in preparation), and were observed in bone cells (Denger et al, 2001; McDougall et al, 2003). Critical deficiencies in ERα−/− mice expressing de facto an ERα46-like isoform (Koš et al, 2002) have made it apparent that AF-1 is the major transactivation function of ERα (Pendaries et al, 2002), and especially in differentiated cells (Mérot et al, 2004). Specific roles of ERα46 may be restricted to effects on the uterine response and endothelial production of NO through actions at the membrane level (Li et al, 2003). However, cell-specific regulation of ERα66/ERα46 ratio would also result in differential outcomes in terms of ERα-mediated transcription in both the absence and presence of hormone. Examples in normal and neoplastic tissues are yet restricted to the observation that the expression of the ERα46 in breast tumors and uterus is associated with estrogen refractoriness (Faye et al, 1986; Jozan et al, 1991). Verification of this issue is therefore a critical step to consider now.
Materials and methods
Reagents and antibodies
α-Amanitin, MNAse, hydroxyquinone, Na bisulfite and estradiol were purchased from Sigma (Taufkirchen, Germany), Taq polymerase was from Roche (Mannheim, Germany) and HpaII from New England Biolabs (Frankfurt, Germany). Antibodies were either gifts or purchased from Advanced Immunochemicals (Freiburg, Germany), Affiniti Research (Exeter, UK), Santa Cruz (Heidelberg, Germany) or Upstate Biotechnology (Buckingham, England) (Métivier et al, 2003). Oligonucleotides were synthesized by MWG GmbH (Ebersberg, Germany).
Cell culture and immunobiochemistry
Cells were maintained in DMEM (Sigma) supplemented with 5% fetal calf serum (FCS, Sigma) and antibiotics (Roche) at 37°C under 5% CO2. MDA∷MB231 cells stably expressing either ERα isoforms were created using pCDNA3.1Hygro (Invitrogen, Karlsruhe, Germany) constructs. Clones were grown in DMEM containing 5% charcoal-stripped serum, antibiotics and hygromycin (0.8 mg/ml). For imaging, cells were seeded on glass-bottomed dishes (MatTek Corp., Ashland, MA, USA), and nuclei stained with Hoechst33342 (Molecular Probes, Eugene, OR, USA). ERα isoforms were localized after cell fixation with methanol and acetone by an HC20 staining revealed with a Cy3-labeled antibody (Molecular Probes).
Methylation analysis
Genomic DNA was prepared as described by Nelson and Krawetz (1992). The methylation status of CCGG sites was analyzed through digestion of 5 μg of DNA with 10 U of HpaII for 2 h at 37°C. Once purified, digested DNA was subject to PCR using the following primer sets: A5′, TGATTCTCCTGACTTACCCTCC; A3′, CACGCTGTAATCCCAACACTTT; B5′, ATGGGCTTCATGAGCTCC; B3′, AGGGTAAATACTGTACTCAC; C5′, CAAGATGACCTCACCACATG; C3′, CGCAGATCACCTTGTTCTCCAT; D5′, TCCTTAGGCAAATGTTATCTAACG; and D3′, GAGCGTTAGATAACATTTGCC. Bisulfite modification was performed on 5 μg of DNA denatured in 0.3 M NaOH in 20 μl for 30 min at 37°C. A measure of 280 μl of a 6.24 M urea/4 M bisulfite solution and 2 μl of 100 mM hydroxyquinone was then added before an overnight incubation at 50°C. Modified DNA was purified using the Qiaex II gel extraction kit (Qiagen, Hilden, Germany), denatured in 0.3 M NaOH for 5 min and precipitated. DNA was then subjected to nested PCR using the following primers: 5′a, TTAGAGTAGTTAGGATTATAGG; 5′b, GTTATGTTGGTTAGGT; and 3′, TTACCTCCTCTCTACTCCAAA. PCR products were purified using the Qiaquik PCR purification kit (Qiagen), and sequenced.
PCRs
Sequences of the oligonucleotides used are: −701, TGATTCTCCTGACTTAACCTCC; −517, CACGCTGTAATCCCAACACTTTG; −350, GTTGTCAGGCCAAGCCTTTT; −30, GAGCGTTAGATAACATTTGCC; −430, ATTAGCTTAGGCCTAGAC; −409, ATGGGCTTCATGAGCTCC; −362, CCACTGTTGTCAGGCCAAGC; −285, CTGCAGAAGTGATTCATA; −266, AGGGTAAATACTGTACTCAC; −245, TACTCATATCTGAGAGGCCCT; −266, CTGGCGGGAGGGCCTCTCAGA; −167, CTGAGGGATCTGAGATTCA; −190, GAAAGAGGGACTTTCTGAATC; −167, CAAGATGACCTCACCACATG; −129, TCTGTCTATCAGCAAAT; −52, GCGTTAGATAACATT; −23, GGATTTTATAGGGCAG; +20, TTGCCTCCTCTCTGCTCCAAA; pS2-5′, ATGGCCACCATGGAGAACAA; pS2-3′, TAAAACAGTGGCTCCTGGCG; GAPDH-5′, TCTGGTAAAGTGGATATTGTTG; and GAPDH-3′, GATGGTGATGGGATTTCC. Quantitative PCRs were performed using a SmartCycler (Eurogentec, Seraing, Belgium) or an ABI Prism® 7000 apparatus (Applied Biosystems).
ChIPs and Re-ChIPs
ChIP assays using 2 × 106 cells synchronized by 3 days of culture in DMEM/2% dextran–charcoal-treated FCS cells and then treated with 2.5 μM α-amanitin for 2 h followed by exposure to 10−8 M E2 or ethanol were performed as described (Métivier et al, 2003).
Preparation of mononucleosomes
Mononucleosomes were prepared from 5 × 105 cells by MNAse digestion following the method presented previously (Métivier et al, 2003).
Supplementary Material
Supplementary Figure S1
Acknowledgments
We are grateful to Dr F Fuller-Pace and Pr J Svejstrup for their generous gifts of antibodies. This work was supported by an EMBO fellowship (RM), the ‘Region de Bretagne' (GP) and by funding from EMBO and EMBL.
References
- Barkhem T, Haldosén LA, Gustafsson JA, Nilsson S (2002) Transcriptional synergism on the pS2 gene promoter between a p160 coactivator and ERα depends on the coactivator subtype, the type of ERE, and the promoter context. Mol Endocrinol 16: 2571–2581 [DOI] [PubMed] [Google Scholar]
- Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T (2002) Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep 31: 39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belandia B, Parker MG (2003) Nuclear receptors: a rendezvous for chromatin remodeling factors. Cell 114: 277–280 [DOI] [PubMed] [Google Scholar]
- Bian Z, Nilsson S, Gustafsson JA (2001) Selective estrogen receptor modulators and coronary heart disease. Trends Cardiovasc Med 11: 196–202 [DOI] [PubMed] [Google Scholar]
- Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 [DOI] [PubMed] [Google Scholar]
- Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, hman L, Greene GL, Gustafson JÅ, Carlquist M (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389: 753–757 [DOI] [PubMed] [Google Scholar]
- Caretti G, Salsi V, Vecchi C, Imbriano C, Mantovani R (2003) Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J Biol Chem 278: 20435–30440 [DOI] [PubMed] [Google Scholar]
- Denger S, Reid G, Koš M, Flouriot G, Parsch D, Brand H, Korach KS, Sonntag-Buck V, Gannon F (2001) ERα gene expression in human primary osteoblasts evidence for the expression of two receptor proteins. Mol Endocrinol 15: 2064–2077 [DOI] [PubMed] [Google Scholar]
- Downes M, Ordentlich P, Kao HY, Alvarez JG, Evans RM (2000) Identification of a nuclear domain with deacetylase activity. Proc Natl Acad Sci USA 97: 10330–10335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawell SE, White R, Hoare S, Sydenham M, Page M, Parker MG (1990) Inhibition of estrogen receptor–DNA binding by the ‘pure' antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc Natl Acad Sci USA 87: 6883–6887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye JC, Fargin A, Bayard F (1986) Dissimilarities between the uterine estrogen receptor in cytosol of castrated and estradiol-treated rats. Endocrinology 118: 2276–2283 [DOI] [PubMed] [Google Scholar]
- Flouriot G, Brand H, Denger S, Métivier R, Koš M, Reid G, Sonntag-Buck V, Gannon F (2000) Identification of a new isoform of the human estrogen receptor-α (hER-α) that is encoded by distinct transcripts and that is able to repress hER-α activation function. EMBO J 19: 4688–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston K, Jayaraman PS (2003) Transcriptional repression in eukaryotes: repressors and repression mechanisms. Cell Mol Life Sci 60: 721–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA (2001) The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 21: 6091–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermah M, Tao Y, Roeder RG (2001) Positive and negative TAF(II) functions that suggest a dynamic TFIID structure and elicit synergy with traps in activator-induced transcription. Mol Cell Biol 21: 6882–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I (2002) Dynamics of enhancer-promoter communication during differentiation-induced gene activation. Mol Cell 10: 1467–1477 [DOI] [PubMed] [Google Scholar]
- Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG (1997) A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 387: 43–48 [DOI] [PubMed] [Google Scholar]
- Honjo H, Iwasa K, Fushiki S, Hosoda T, Tatsumi H, Mihara M, Hirasugi Y, Oida M, Kariya K, Kikuchi N, Kawata M (2003) Estrogen and non-feminizing estrogen for Alzheimer's disease. Endocr J 50: 361–367 [DOI] [PubMed] [Google Scholar]
- Jepsen K, Rosenfeld MG (2002) Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci 115: 689–698 [DOI] [PubMed] [Google Scholar]
- Jordan VC, Gapstur S, Morrow M (2001) Selective estrogen receptor modulation and reduction in risk of breast cancer, osteoporosis, and coronary heart disease. J Natl Cancer Inst 93: 1449–1457 [DOI] [PubMed] [Google Scholar]
- Jozan S, Julia AM, Carretie A, Eche N, Maisongrosse V, Fouet B, Marques B, David JF (1991) 65 and 47 kDa forms of estrogen receptor in human breast cancer: relation with estrogen responsiveness. Breast Cancer Res Treat 19: 103–109 [DOI] [PubMed] [Google Scholar]
- Kang YK, Guermah M, Yuan CX, Roeder RG (2002) The TRAP/Mediator coactivator complex interacts directly with estrogen receptors α and β through the TRAP220 subunit and directly enhances estrogen receptor function in vitro. Proc Natl Acad Sci USA 99: 2642–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HY, Downes M, Ordentlich P, Evans RM (2000) Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev 14: 55–66 [PMC free article] [PubMed] [Google Scholar]
- Katan-Khaykovich Y, Struhl K (2002) Dynamis of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev 16: 743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koš M, Denger S, Reid G, Korach KS, Gannon F (2002) Down but not out? A novel protein isoform of the estrogen receptor alpha is expressed in the estrogen receptor alpha knockout mouse. J Mol Endocrinol 29: 281–286 [DOI] [PubMed] [Google Scholar]
- Krust A, Green S, Argos P, Kumar V, Walter P, Bornert JM, Chambon P (1986) The chicken oestrogen receptor sequence: homology with v-erbA and the human oestrogen and glucocorticoid receptors. EMBO J 5: 891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Haynes MP, Bender JR (2003) Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA 100: 4807–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XF, Bagchi MK (2004) Recruitment of distinct chromatin modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem 279: 15050–15058 [DOI] [PubMed] [Google Scholar]
- Malik S, Guermah M, Roeder RG (1998) A dynamic model for PC4 coactivator function in RNA polymerase II transcription. Proc Natl Acad Sci USA 95: 2192–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall KE, Perry MJ, Gibson RL, Colley SM, Korach KS, Tobias JH (2003) Estrogen receptor-α dependency of estrogen's stimulatory action on cancellous bone formation in male mice. Endocrinology 144: 1994–1999 [DOI] [PubMed] [Google Scholar]
- Mérot Y, Métivier R, Penot G, Manu D, Saligaut C, Gannon F, Pakdel F, Kah O, Flouriot G (2004) The relative contribution exerted by AF-1 and AF-2 transactivation functions in ERα transcriptional activity depends upon the differentiation stage of the cell. J Biol Chem 279: 26184–26191 [DOI] [PubMed] [Google Scholar]
- Métivier R, Penot G, Hubner MR, Reid G, Brand H, Koš M, Gannon F (2003) ERα directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751–763 [DOI] [PubMed] [Google Scholar]
- Métivier R, Stark A, Flouriot G, Hubner MR, Brand H, Penot G, Manu D, Denger S, Reid G, Koš M, Russell RB, Kah O, Pakdel F, Gannon F (2002) A dynamic structural model for ERα activation by ligands, emphasizing the role of interactions between distant A and E domains. Mol Cell 10: 1019–1032 [DOI] [PubMed] [Google Scholar]
- Nelson JE, Krawetz SA (1992) Purification of cloned and genomic DNA by guanidine thiocyanate/isobutyl alcohol fractionation. Anal Biochem 207: 197–201 [DOI] [PubMed] [Google Scholar]
- Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA (2001) Mechanisms of estrogen action. Physiol Rev 81: 1535–1565 [DOI] [PubMed] [Google Scholar]
- NR Committee (1999) A unified nomenclature system for the nuclear receptor superfamily. Cell 97: 161–163 [DOI] [PubMed] [Google Scholar]
- Nye AC, Rajendran RR, Stenoien DL, Mancini MA, Katzenellenbogen BS, Belmont AS (2002) Alteration of large-scale chromatin structure by estrogen receptor. Mol Cell Biol 22: 3437–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, Bayard F, Arnal JF (2002) The AF-1 activation-function of ERα may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci USA 99: 2205–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F (2003) Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell 11: 695–707 [DOI] [PubMed] [Google Scholar]
- Robinson-Rechavi M, Garcia EG, Laudet V (2003) The nuclear receptor superfamily. J Cell Sci 116: 585–586 [DOI] [PubMed] [Google Scholar]
- Robyr D, Wolffe AP, Wahli W (2000) Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol Endocrinol 14: 329–347 [DOI] [PubMed] [Google Scholar]
- Sewack GF, Hansen U (1997) Nucleosome positioning and transcription-associated chromatin alterations on the human estrogen-responsive pS2 promoter. J Biol Chem 272: 31118–31129 [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103: 843–852 [DOI] [PubMed] [Google Scholar]
- Shao D, Lazar MA (1999) Modulating nuclear receptor function: may the phos be with you. J Clin Invest 103: 1617–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL (2000) Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64: 435–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD (2003) Chromatin as a tool for the study of genome function in cancer. Ann NY Acad Sci 983: 5–21 [DOI] [PubMed] [Google Scholar]
- Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, Zhang Y (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293: 853–857 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Yanagisawa J, Kitagawa H, Takeyama K, Ogawa S, Arao Y, Suzawa M, Kobayashi Y, Yano T, Yoshikawa H, Masuhiro Y, Kato S (2001) A subfamily of RNA-binding DEAD-box proteins acts as an estrogen receptor alpha coactivator through the N-terminal activation domain (AF-1) with an RNA coactivator, SRA. EMBO J 20: 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zegerman P, Canas B, Pappin D, Kouzarides T (2002) Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J Biol Chem 277: 11621–11624 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 13: 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1