

Abstract
The palladium-catalyzed defluorinative coupling of 1-aryl-2,2-difluoroalkenes with boronic acids is described. Broad functional group tolerance arises from a redox-neutral process via a Pd(II) active species that is proposed to undergo a β-fluoride elimination to afford the products. The monofluorostilbene products were formed with excellent diastereoselectivity (≥50:1) in all cases, which is critical, as traditional chromatographic techniques often fail to separate monofluoroalkene isomers. As a demonstration of this method’s unique combination of reactivity and functional group tolerance, a Gleevec© analogue using a monofluorostilbene as an amide isostere was synthesized.
Keywords: monofluoroalkenes, C-F bond activation, C-C coupling, palladium catalysis, difluoroalkenes
The amide bond is a ubiquitous structural motif found medicinal chemistry, and consequently much effort has been made to develop peptide bond mimics known as amide isosteres.1 Some important properties that might be conferred by an amide isosteres include enhanced stability to peptidases, increased lipophilicity, conformational stability, and improved recognition.1 Monofluoroalkenes have emerged as a useful peptide mimic that exhibits a similar steric and electronic profile to amides, and consequently have been used extensively as amide isosteres.1,2 While a wide variety of methods have been developed to access different classes of monofluoroalkenes, modular synthesis and stereoselective synthesis remain outstanding challenges.3 Moreover, traditional chromatographic techniques, including preparative HPLC, often fail to separate E and Z isomers of monofluoroalkenes, highlighting the importance of stereoselective methods.4
Hu and co-workers have recently developed Julia-Kocienski olefination procedure that utilized a selective decomposition strategy of sulfinate intermediates to isolate the E and Z isomers by extraction (Scheme 1).4 While the procedure allowed for isolation of both fluoroalkenes stereoisomers from the same reaction, the issue of selective synthesis remains unsolved. Additionally, this protocol required the synthesis of a new olefination reagent for each derivative, an impediment to modularity that is inherent to olefination strategies for monofluoroalkene synthesis.3 As an alternative, Cao and co-workers have recently reported a nickel-catalyzed Suzuki type coupling of boronic acids and difluorostyrenes that afforded monofluorostilbenes stereoselectively (Scheme 1).5 They proposed that reaction proceeds through oxidative addition of the alkene C-F bond,6,7 followed by cross-coupling with an aryl boronic acid. While a cross-coupling strategy provides the opportunity for more rapid diversification, the high temperature and reactive catalysts required for C-F bond activation led to limited functional group tolerance. We hypothesized that a mechanistically distinct cross-coupling strategy, not requiring oxidative addition into a C-F bond, would tolerate the functional groups relevant to pharmaceutical, agrochemical and material sciences, would be required.8 Herein we report the development of a palladium-catalyzed defluorinative coupling reaction to access single isomers of monofluorostilbenes under mild conditions (Scheme 1).
Scheme 1.
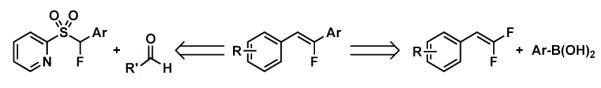
Synthesis of Monofluorostilbenes.
Our group’s recent interest in interrupted Heck reactions led us to wonder if stereoselective fluoro-olefin synthesis might be achieved via a defluorinative coupling reaction with difluoroalkenes.9-11 We envisioned a scenario as outlined in Scheme 2, wherein insertion of a difluoroalkene into a palladium aryl bond of A would lead to alkylpalladium intermediate, B. In an analogous fashion to beta-hydride elimination in the Heck reaction, β-fluoride elimination would lead to formation of the alkene product. The resulting palladium(II) species, C, could readily transmetallate with an aryl boronic acid to close the catalytic cycle. β-fluoride elimination from organometallic intermediates is not without precedent; however it is infrequently designed into catalytic transformations.12 Importantly, such a process circumvents the generation of palladium(0) and is a redox neutral process, thus not requiring the use of external oxidants or added base. These aspects would be expected to help minimize issues with functional group compatibility.
Scheme 2.
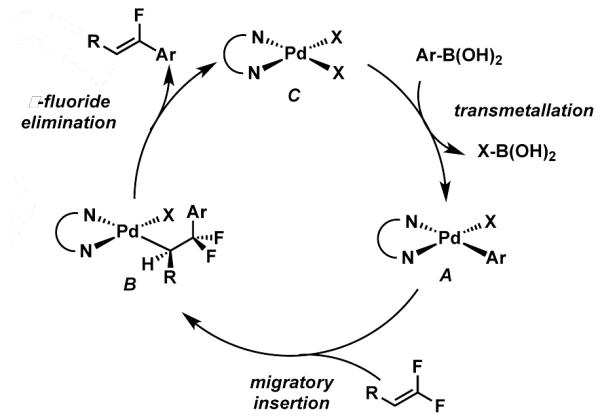
Proposed Reactivity Manifold
Difluoroalkenes are a readily accessible class of substrates.13 While a variety of methods exist for their synthesis, perhaps the most notable is through a Julia-Kocienski olefination protocol utilizing commercially available difluoromethyl 2-pyridyl sulfone (Hu’s reagent) and aldehydes or ketones, which we found to be useful for our purposes.
With a variety of difluoroalkenes and boronic acids on hand, conditions for a palladium-catalyzed defluorinative coupling were sought (see supporting information). The optimization efforts revealed palladium(II) trifluoroacetate as a palladium source, with 4,4’-di-tert-butyl-2,2’-bipyridine as a ligand in dimethyformamide (DMF) at 50 °C. The scope of the reaction, under these reaction conditions, is detailed in Scheme 3. In all cases the monofluorostilbene products were formed in ≥50:1 diastereomeric ratio. With respect to difluoroalkene, a wide variety of functional groups were tolerated including alkyl, ester, nitro, trifluoromethyl, ether, and halogen substituents, and substitution at ortho-, meta-, and para- positions of the aromatic ring. The reaction also tolerated a variety of functional groups on the boronic acid coupling partner including halogens, ester, methoxy, trifluoromethoxy, trimethylsilyl, free alcohol, and protected amine substituents, as well as substitution at ortho-, meta-, and para- positions. Some heteroarene substituted difluoroalkenes were compatible as well, albeit with extended reaction times and/or elevated temperature (Scheme 3 footnotes c and d). Both benzofuran product 3j and pyridine 3k were synthesized, increasing the diversity of molecules accessible by this method.
Scheme 3.

Scope of Defluorinative Coupling. a.) standard reaction conditions: 0.2 mmol 1, 1.05-2.0 eq 2, 10 mol% Pd(TFA)2, 11 mol% 4,4’-di-tert-butyl-2,2’-bipyridine (dtbbpy), 0.5 ml DMF, 50 °C; completion of reaction monitored by thin-layer chromatography. b.) Isolated yields c.) reaction run for 5 days. d.) reaction run at 85 °C for 4 days. e.) 47 % yield when corresponding potassium trifluoroborate salt is employed in place of boronic acid.
The reaction developed herein exhibits some interesting chemoselectivity and functional group tolerances worth further comment. First, in addition to being highly stereoselective, the reaction was also highly selective for mono-coupled products. Only in the case of 3m, did we observe over-coupled side products; however, this undesired reactivity was easily remedied by reduction of boronic acid loading to near stoichiometric quantities, and 3m was isolated in moderate yield.
Second, the reaction conditions are uniquely suited to boron-based nucleophiles including boronic acids and potassium tetrafluoroborate salts (Scheme 3 footnote e), although boronic esters react sluggishly. Interestingly, we found that arylstannanes and arylsilanes were inert under our reaction conditions both with and without added equivalents of fluoride, affording the opportunity for orthogonal cross coupling.14
Finally, a variety of functional groups were tolerated that might be useful for further synthetic manipulations. Importantly, arylhalides were tolerated under these reaction conditions, including chlorides, bromides, and iodide. Even a substrate bearing a 2-bromopyridine was tolerated. In no case was cross-coupling with these functional groups, which stands in stark contrast to the previously described nickel-catalyzed procedure which also produced competitive aryl halide cross-coupling, even with arylchlorides.5 These results together strongly suggest that our process does not involve formation of Pd(0), and supports the initial mechanistic hypothesis and demonstrates that the distinct reactivity manifold enables significant expansion of functional group tolerance.
In an effort to lend further support to the mechanistic hypothesis, difluoroacrylate 4 was subjected to reaction conditions. Analysis of the proton and fluorine NMR spectra revealed a mixture of defluorocoupled product 6, and 1,4-conjugate addition product, 7 (Scheme 4a). Formation of 7 presumably proceeds through a palladium alkyl intermediate,15 further supporting the proposal that the reaction proceeds through a migratory insertion, β-fluoride elimination pathway. Although analogous protonolysis products were not observed for the difluorostyrene substrates, observation of 7 lead us to consider whether protonolysis followed by base catalyzed elimination might be responsible for the formation of the monofluorostilbenes. To test for this possibility, 1,1-difluoro-1,2-dipheneylethane, 8, was subjected to standard reaction conditions (Scheme 4b). The corresponding monofluoroalkene, 9, was not observed, thus suggesting the β-fluoride elimination is not simply base mediated and likely occurs from a palladium alkyl intermediate.
Scheme 4.
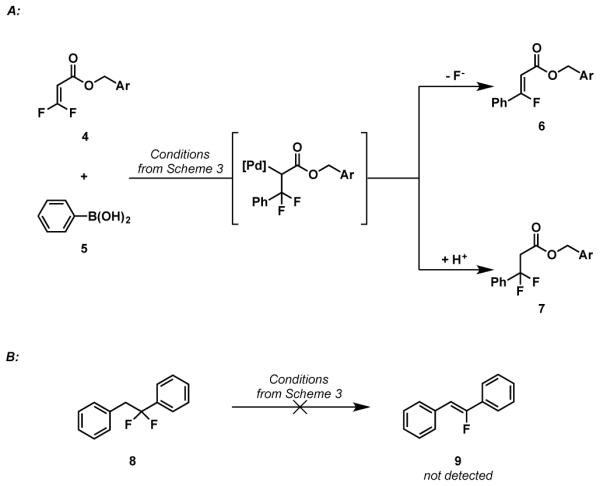
Mechanistic Probes. Ar = p-Br-C6H4
In order to demonstrate the utility of this palladium catalyzed defluorinative coupling the methodology was applied to the synthesis of a Gleevec® isostere. Gleevec© is a tyrosine kinase inhibitor, and the first of its class to be approved for the treatment of a variety of cancers.16 Gleevec© contains a benzamide moiety, in its structure (Scheme 5), that we sought substitute with a monofluoroalkene moiety. Crystallographic studies suggest that the benzamide moity adopts a trans- geometry in the enzyme binding site, making our stereoselective method particularly useful for accessing a conformationally locked analogue of Gleevec©.17 From commercially available 3-bromo-4-methyl-benzaldehyde, difluoroalkene 11 was synthesized using Hu’s reagent, and then subjected to the standard reaction conditions in presence of 4-formylphenylboronic acid. Gratifyingly, the defluoro-coupled product 13 was obtained in 57% yield. Reductive amination with 1-methylpiperazine followed by palladium catalyzed Buchwald-Hartwig coupling with commercially available 4-(3-pyridinyl)-2-pyrimidine amine afforded the monofluoroalkene derivative of Gleevec®, 10, in 2 additional steps (See supporting information for details).18 Critical to the synthesis was the compatibility of functional groups such as the benzaldehyde moiety and the aryl bromide to the palladium-catalyzed coupling.
Scheme 5.
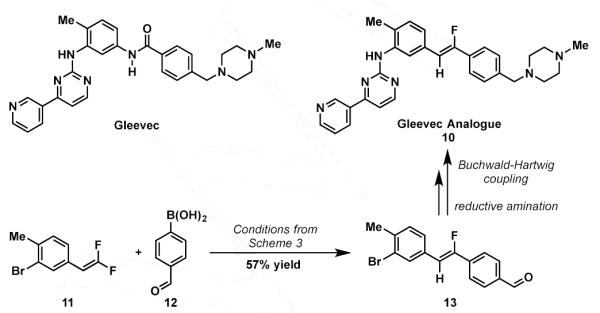
Synthesis of Gleevec® Analogue.
In conclusion, the palladium-catalyzed defluorinative coupling of 1-aryl-2,2-difluoroalkenes with boronic acids to afford a monofluoroalkenes with high diasteroselectivity is described. The reaction is proposed to proceed through a rare example of a beta-fluoride elimination of a palladium(II) intermediate in a catalytic process. This distinct mechanistic manifold and the resulting mild reaction conditions allow for the incorporation of variety of synthetically useful functional groups into the coupling partners. The utility of this method and the monofluoroalkene building blocks was demonstrated by the synthesis of a Gleevec® amide isostere.
Supplementary Material
Footnotes
Supporting information for this article is given via a link at the end of the document
[**] We gratefully acknowledge NIGMS (R35 GM118190) for financial support. Omkar Kulkarni and Dr. Gabriel Schafer are thanked for the synthesis of some starting materials.
References
- [1].Taguchi T, Yanai H. In: Fluorine in Medicinal Chemistry and Chemical Biology. Ojima I, editor. Wiley-Blackwell; Chichester, U.K.: 2009. pp. 257–290. [Google Scholar]
- [2].a) Sciotti RJ, Pliushchev M, Wiedman PE, Balli D, Flamm R, Nilius AM, Marsh K, Stolarik D, Jolly R, Ulrich R, Djuric SW. Biorog. Med. Chem. Lett. 2002;12:2121–2123. doi: 10.1016/s0960-894x(02)00352-9. [DOI] [PubMed] [Google Scholar]; b) Asahina Y, Iwase K, Iinuma F, Hosaka M, Ishizaki T. J. Med. Chem. 2005;48:3194–3202. doi: 10.1021/jm0402061. [DOI] [PubMed] [Google Scholar]; c) Couve-Bonnaire S, Cahard D, Pannecoucke X. Org. Biomol. Chem. 2007;5:1151–1157. doi: 10.1039/b701559c. [DOI] [PubMed] [Google Scholar]; d) Edmondson SD, Wei L, Xu J, Shang J, Xu S, Pang J, Chaudhary A, Dean DC, He H, Leiting B, Lyons KA, Patel RA, Patel SB, Scapin G, Wu JK, Beconi MG, Thornberry NA, Weber AE. Bioorg. Med. Chem. Lett. 2008;18:2409–2413. doi: 10.1016/j.bmcl.2008.02.050. [DOI] [PubMed] [Google Scholar]; e) Oishi S, Kamitani H, Kodera Y, Watanabe K, Kobayashi K, Narumi T, Tomita K, Ohno H, Naito T, Kodama E, Matsuoka M, Fujii N. Org. Biomol. Chem. 2009;7:2872–2877. doi: 10.1039/b907983a. [DOI] [PubMed] [Google Scholar]; f) McKinney BE, Urban JJ. J. Phys. Chem. A. 2010;114:1123–1133. doi: 10.1021/jp9094535. [DOI] [PubMed] [Google Scholar]; g) Jakobsche CE, Choudhary A, Miller SJ, Raines RT. J. Am. Chem. Soc. 2010;132:6651–6653. doi: 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Osada S, Sano S, Ueyama M, Chuman Y, Kodama H, Sakaguchi K. Biorog. Med. Chem. 2010;18:605–611. doi: 10.1016/j.bmc.2009.12.005. [DOI] [PubMed] [Google Scholar]
- [3].a) Landelle G, Bergeron M, Turcotte-Savard M-O, Paquin J-F. Chem. Soc. Rev. 2011;40:2867–2908. doi: 10.1039/c0cs00201a. For reviews on the synthesis of monofluoroalkenes see: [DOI] [PubMed] [Google Scholar]; b) Yanai H, Taguchi T. Eur. J. Org. Chem. 2011:5939–5954. [Google Scholar]
- [4].Zhao Y, Jiang F, Hu J. J. Am. Chem. Soc. 2015;137:5199–5203. doi: 10.1021/jacs.5b02112. [DOI] [PubMed] [Google Scholar]
- [5].Xiong Y, Huang T, Ji X, Wu J, Cao S. Org. Biomol. Chem. 2015;13:7389–7392. doi: 10.1039/c5ob01016k. S. [DOI] [PubMed] [Google Scholar]
- [6].a) Amii H, Uneyama K. Chem. Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. For a reviews of C-F bond activation see: [DOI] [PubMed] [Google Scholar]; b) Ahrens T, Kohlmann J, Ahrens M, Braun T. Chem. Rev. 2015;115:931–972. doi: 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]
- [7].Ohashi M, Saijo H, Shibata M, Ogoshi S. Eur. J. Org. Chem. 2013:443–447. For examples of alkene C-F oxidative addition adducts with palladium and nickel see: [Google Scholar]
- [8].Fuchibe K, Morikawa T, Shigeno K, Fujita T, Ichikawa J. Org. Lett. 2015;17:1126. doi: 10.1021/ol503759d. [DOI] [PubMed] [Google Scholar]
- [9].Talbot EPA, de Fernandes TA, McKenna JM, Toste FD. J. Am. Chem. Soc. 2014;136:4101–4104. doi: 10.1021/ja412881j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nelson HM, Williams BD, Miro J, Toste FD. 2015;137:3213–3216. doi: 10.1021/jacs.5b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) He Y, Yang Z, Thornbury RT, Toste FD. 2015;137:12207–12210. doi: 10.1021/jacs.5b07795. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miro J, del Pozo C, Toste FD, Fustero S. Angew. Chem. Int. Ed. DOI: 10.1002/anie.201603046. [Google Scholar]
- [12].a) Miura T, Ito Y, Murakami M. Chem. Lett. 2008;37:1006–1007. For examples involving rhodium see see: [Google Scholar]; b) Tian P, Feng C, Loh T-P. Nat. Commun. 2015;6:7472. doi: 10.1038/ncomms8472. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ichitsuka T, Fujita T, Arita T, Ichikawa J. Angew. Chem. Angew. Chem. Int. Ed. 2014;2014;12653:7694–7698. 7564–7568. doi: 10.1002/anie.201402695. For examples involving nickel see: [DOI] [PubMed] [Google Scholar]; d) Ichitsuka T, Fujita T, Ichikawa J. ACS Catal. 2015;5:5947–5950. [Google Scholar]; e) Heitz W, Knebelkamp A. Makromol. Chem. Rapid Commun. 1991;12:69–75. For examples involving palladium see: [Google Scholar]; f) Saeki T, Takashima Y, Tamao K. Synlett. 2005;11:1771–1774. [Google Scholar]; g) Ichikawa J, Nadano R, Ito N. Chem. Commun. 2006:4425–4427. doi: 10.1039/b610690k. [DOI] [PubMed] [Google Scholar]; h) Ichikawa J, Sakoda K, Mihara J, ito N. J. Fluorine Chem. 2006;127:489–504. [Google Scholar]; i) Hu M, He Z, Gao B, Li L, Ni C, Hu J. J. Am. Chem. Soc. 2013;135:17302–17305. doi: 10.1021/ja409941r. For examples involving copper see: [DOI] [PubMed] [Google Scholar]; j) Zhang Z, Zhou Q, Yu W, Li T, Wu G, Zhang Y, Wang J. Org. Lett. 2015;17:2474–2477. doi: 10.1021/acs.orglett.5b00980. [DOI] [PubMed] [Google Scholar]; k) Kikushima K, Sakaguchi H, Sayo H, Ohashi M, Ogoshi S. Chem. Lett. 2015;44:1019–1021. [Google Scholar]
- [13].a) Zhao Y, Huang W, Zhu L, Hu J. Org. Lett. 2010;12:1444–1447. doi: 10.1021/ol100090r. [DOI] [PubMed] [Google Scholar]; b) Chelucci G. Chem. Rev. 2012;112:1344–1462. doi: 10.1021/cr200165q. [DOI] [PubMed] [Google Scholar]; c) Gao B, Zhao Y, Hu M, Ni C, Hu J. Chem. Eur. J. 2014;20:7803–7810. doi: 10.1002/chem.201402183. [DOI] [PubMed] [Google Scholar]; d) Hu M, Ni C, Li L, Han X, Hu J. J. Am. Chem. Soc. 2015;137:14496–14501. doi: 10.1021/jacs.5b09888. [DOI] [PubMed] [Google Scholar]
- [14].a) Cordovilla C, Bartolome C, Martinez-Ilarduya JM, Espinet P. ACS Catal. 2015;5:3040–3053. For a recent review on cross-coupling with organotin reagents see: [Google Scholar]; b) Sore HF, Galloway WRJD, Spring DR. Chem. Soc. Rev. 2012;41:1845–1866. doi: 10.1039/c1cs15181a. For recent reviews on cross-coupling with organosilicon reagents see: [DOI] [PubMed] [Google Scholar]; c) Denmark SE, Ambrosi A. Org. Process Res. Dev. 2015;19:982–994. doi: 10.1021/acs.oprd.5b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kikushima K, Holder JC, Gatti M, Stoltz BM. J. Am. Chem. Soc. 2011;133:6902–6905. doi: 10.1021/ja200664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Capdeville R, Buchdunger E, Zimmerman J, Matter A. Nat. Rev. Drug Discovery. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]; b) Deininger M, Buchdunger E, Druker BJ. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- [17].Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- [18].Maiti D, Fors BP, Henderson JL, Nakamura Y, Buchwald SL. Chem. Sci. 2011;2:57–68. doi: 10.1039/C0SC00330A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.