

Abstract
Active changes in mitochondrial structure and organization facilitate cellular homeostasis. Because aberrant mitochondrial dynamics are implicated in a variety of human diseases, their assessment is potentially useful for diagnosis, therapy, and disease monitoring. Because current techniques for evaluating mitochondrial morphology are invasive or necessitate mitochondria-specific dyes, their clinical translation is limited. We report that mitochondrial dynamics can be monitored in vivo, within intact human skin by relying entirely on endogenous two-photon–excited fluorescence from the reduced metabolic coenzyme nicotinamide adenine dinucleotide (NADH). We established the sensitivity of this approach with in vivo, fast temporal studies of arterial occlusion-reperfusion, which revealed acute changes in the mitochondrial metabolism and dynamics of the lower human epidermal layers. In vitro hypoxic-reperfusion studies validated that the in vivo outcomes were a result of NADH fluorescence changes. To demonstrate the diagnostic potential of this approach, we evaluated healthy and cancerous human skin epithelia. Healthy tissues displayed consistent, depth-dependent morphological and mitochondrial organization patterns that varied with histological stratification and intraepithelial mitochondrial protein expression. In contrast, these consistent patterns were absent in cancerous skin lesions. We exploited these differences to successfully differentiate healthy from cancerous tissues using a predictive classification approach. Collectively, these results demonstrate that our label-free, automated, near real-time assessments of mitochondrial organization—relying solely on endogenous contrast—could be useful for accurate, noninvasive in vivo diagnosis.
INTRODUCTION
Mitochondria undergo trafficking, fusion, and fission, creating continuously changing networks to support mitochondrial function and accommodate cellular homeostasis (1). Aberrant mitochondrial dynamics and the corresponding changes in mitochondrial organization are associated with a variety of human pathologies, including neurodegenerative (2–4), metabolic (5, 6), cardiovascular (7, 8), and neoplastic (9, 10) diseases. In this context, the mitochondrial machinery is an attractive therapeutic target for a broad set of diseases. Compounds that modulate mitochondrial dynamics protect cardiac tissue against ischemia/ reperfusion injury (11), attenuate neurodegeneration (4, 12), and reduce lung cancer cell proliferation (9) and breast cancer metastasis (10) in experimental disease models. Because of these associations with disease, it would be beneficial to monitor the organization of mitochondria. To date, most studies investigating mitochondrial morphology rely on invasive methods, such as scanning electron microscopy, mitochondria-specific dyes, or genetically engineered expression of fluorescent proteins (13–15). These measurement techniques cannot be used in humans, especially in a clinical setting, because they require tissue excision and processing or contrast agents that are not approved for human use.
Multiphoton microscopy (MPM) is uniquely placed to address these limitations. MPM is a noninvasive imaging method capable of providing subcellular resolution in dense, optically scattering tissue, obviating the need for tissue excision (16). MPM signals can be generated from a variety of intrinsic tissue chromophores, facilitating multidimensional tissue contrast without the use of exogenous labels (17). Of special interest for mitochondria is the reduced metabolic co-enzyme nicotinamide adenine dinucleotide (NADH). NADH is intrinsically fluorescent and plays a key role in cellular energy metabolism. Moreover, images of NADH fluorescence predominantly reflect contributions from the bound form of NADH that resides primarily within mitochondria, owing to its increased fluorescent yield in that state (18). The spatial distribution of bright autofluorescent features in two-photon– excited fluorescence (TPEF) NADH images of cells correlates with the spatial distribution of traditional mitochondrial dyes in vitro (19). Thus, NADH imaging could be a label-free approach to monitor the state of mitochondria and their organization in vivo. In addition, multiphoton technology is being integrated in endoscopes (20) and compact portable tomographic systems (21), making MPM ideally suited for clinical translation of mitochondrial monitoring.
Recent in vivo clinical MPM studies of nevi monitoring and detection of melanoma or basal cell carcinoma (BCC) have established the first diagnostic MPM image criteria (22–24). In the case of melanoma detection, these criteria rely on quantitative manual tracing analysis of lesion-specific features of interest, such as dendrite density, and assessment of the endogenous TPEF NADH intensity variations (23). In the case of BCC lesions, detection is based on the qualitative demonstration of important diagnostic features, such as the presence of nests of basaloid cells in the papillary and upper reticular dermis (24). Other optical techniques, such as optical coherence tomography and confocal reflectance microscopy, also rely on specialized morphological criteria for skin lesion evaluation (25, 26). The specificity of these criteria increases their usefulness for differential diagnosis yet limits their application to particular lesion types and requires advanced histopathological knowledge for proper identification and evaluation. In contrast to the focus of these approaches on morphological characteristics, assessment of mitochondrial organization offers a more nuanced metric of both structure and function. Metabolism is often altered in a diseased state (27), and mitochondria are key regulators of cellular homeostasis; therefore, markers related to mitochondrial dynamics may be useful for multiple diseases, as well as for early disease states where traditional morphological hallmarks may not yet be evident. Furthermore, information from such markers could complement that from morphological criteria, potentially enhancing diagnostic performance in ambiguous or challenging cases.
Previous evaluations of in vivo NADH MPM images focused exclusively on intensity-based assessments (22–24). These can be related to biochemical metabolic alterations but not necessarily to mitochondrial organization. Recently, we demonstrated that Fourier-based analysis is well suited to evaluate high-resolution NADH MPM images to assess intracellular texture in cell monolayers and three-dimensional (3D) engineered tissues (28, 29). In these studies, mitochondrial clustering was reliably quantified in cell layers with distinct nuclear-to-cytoplasmic ratios and used as a sensitive marker of metabolic changes in the relative levels of oxidative phosphorylation, glycolysis, and glutaminolysis, which often occur at the onset of cancer and may serve as diagnostic biomarkers or to guide therapy (28, 29).
Here, we have used MPM to characterize mitochondrial organization and dynamic responses to environmental conditions within human tissues in vivo, relying entirely on endogenous fluorescence from NADH. To illustrate the potential translational impact of this method, we also characterize in vivo the overall and depth-dependent differences in mitochondrial organization between healthy and cancerous human skin epithelia.
RESULTS
Epidermal strata respond differently to ischemic hypoxia
Different epidermal layers are differentially sensitive to vascular oxygen (30). Therefore, we first evaluated the acute changes in mitochondrial metabolism in vivo within the human epidermis by fast induction of ischemic hypoxia and reperfusion via arterial occlusion and release. We followed changes in two distinct cellular layers: one within 20 to 25 μm of the skin surface, below the stratum corneum, and a deeper one, within 35 to 40 mm of the skin surface, closer to the stratum basale and proximal to the dermal microvasculature (Fig. 1A). We examined each layer separately over time by acquiring an NADH TPEF intensity image every 10 s before, during, and after arterial occlusion, as previously described (30). Each phase lasted 3 min.
Fig. 1. Endogenous NADH TPEF intensity and mitochondrial dynamics changes in response to arterial occlusion and reperfusion.
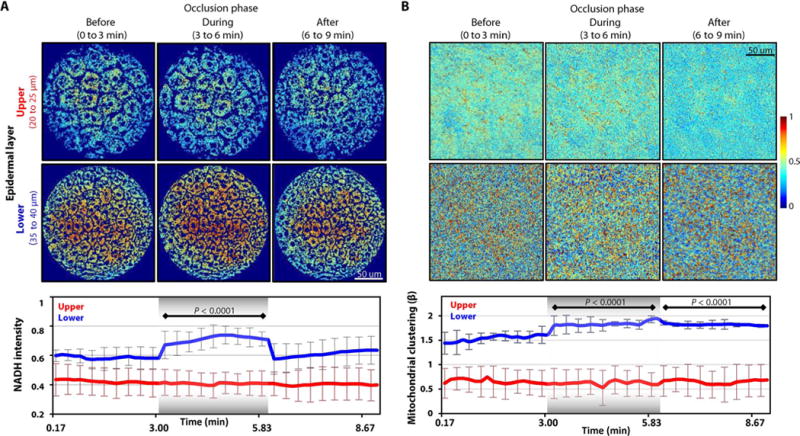
(A) Top: Representative scaled in vivo NADH intensity TPEF images for upper and lower human skin layers before, during, and after arterial occlusion. Each epidermal layer was imaged every 10 s for 3 min in each phase. Bottom: The means ± SEM values of NADH intensity extracted from each NADH image of the upper and lower layers (n = 3 independent experiments for each layer). Values extracted during and after the occlusion are compared to those acquired before the occlusion. (B) Top: Corresponding processed cloned NADH intensity images for the upper and lower human skin layers shown in (A). Bottom: The means ± SEM values of the mitochondrial clustering extracted from each cloned image (n = 3 independent experiments for each layer). Values extracted during and after the occlusion are compared to those acquired before the occlusion. For all panels, image heat maps reflect varying TPEF intensities; shaded regions within the bottom panels identify the occlusion phase. P values were determined by mixed-model analyses of variance (ANOVAs) with post hoc Bonferroni correction.
Through automated image processing, the nuclear and interstitial spaces were identified and removed from the images to allow calculations from the relevant cytoplasmic space only (fig. S1A). These images, along with the corresponding, time-dependent, mean NADH intensity profiles calculated from the measurements acquired at each depth from all subjects (Fig. 1A), revealed divergent metabolic responses in the epidermal layers. (NADH intensity is an indicator of the reducing power of a cell.) The upper cell layers were completely unaffected by the vascular occlusion, whereas the lower layers evidenced a response, an almost instantaneous NADH increase as the occlusion occurred. The intensity levels remained stably elevated throughout the duration of the ischemia and rapidly returned to baseline when the arterial flow was reinstated (after 3 min of occlusion) (Fig. 1A). These outcomes agree with previous human and mouse studies (30, 31), suggesting that the different skin layers have different metabolic dependence on vascular oxygenation.
We further evaluated the mitochondrial dynamics in each epidermal layer. Mitochondrial organization can be evaluated from NADH intensity images through a Fourier-based approach (28, 29) that capitalizes on the fact that the NADH fluorescence intensity yield is increased when NADH is enzymatically bound. The method provides a metric of mitochondrial organization, because most of the cellular bound NADH is contained within the mitochondria. To extract this metric, we computed the power spectral density (PSD) from the 2D Fourier transform of each NADH intensity image. A simple inverse power law decay expression of the form R(k) = Ak−β was then fit to the PSD, with the magnitude of the exponent β being indicative of the levels of mitochondrial clustering (28, 29). Increased clustering values represented more fragmented/fissioned mitochondrial formations.
Before the extraction of information related to mitochondrial organization, we eliminated the image signal voids created by the nuclear and interstitial space removal by digitally cloning the isolated cytoplasmic intensity fluctuations into the voids (Fig. 1B and fig. S1C). This procedure is essential for the sensitivity of the method to mitochondrial organization features, which is maintained independent of the size of the void areas (28). The intensity fluctuations within the identified cytoplasmic regions were not affected by the cloning procedure, because only the void areas were filled. The epidermal layers displayed different mitochondrial dynamics (Fig. 1B), consistent with our NADH intensity results. Mitochondrial clustering fluctuated minimally from baseline in the upper epidermal layer throughout the entire experiment, matching the NADH intensities for this layer. Conversely, clustering in the lower layer rose significantly during occlusion. Unlike NADH intensity though, which recovered immediately upon termination of the arterial occlusion (Fig. 1A), mitochondrial clustering remained elevated during vascular reperfusion (Fig. 1B). These findings reveal that mitochondrial biochemistry differs temporally from mitochondrial dynamics.
2D cell cultures validate in vivo response to hypoxia-induced mitochondrial changes
To confirm that variation in NADH intensity and clustering was a result of mitochondria- and hypoxia-related changes, we used a controlled 2D cell culture of human primary keratinocytes, in which precise measurements of oxygen conditions could be performed. Similar hypoxic perturbations have been conducted using perfusion chambers (13). However, to avoid shear flow effects that did not mimic the epidermally sensed in vivo hypoxia and to further enable simultaneous dynamic oxygen measurements while imaging the same optical field, we designed and built a custom setup in-house that allowed fast temporal control of the medium’s oxygen saturation (fig. S2A). The 2D cultures further offered the advantage of stable metabolic substrate availability, in contrast to the in vivo case where arterial occlusion also diminished the supply of other substrates. Last, narrow band imaging parameters were set to achieve increased selectivity in collecting NADH information.
The experiment was divided into four phases. The first phase established a baseline before hypoxia induction (0 to 5 min), the second was the hypoxic phase (5 to 16 min), and the third and fourth phases corresponded to early (16 to 23 min) and late (23 to 30 min) reperfusion time points, respectively. The control group had no oxygen manipulation. The dynamic oxygen saturation measurements, acquired from each imaged well plate for the hypoxic group (fig. S2B), verified that an oxygen saturation of less than 1% was achieved and preserved during the hypoxic phase.
As with the in vivo images, the relevant cytoplasmic space in human primary keratinocytes was isolated and evaluated (Fig. 2A). NADH intensities were stable throughout all experimental phases for the normoxic group (Fig. 2B). In contrast, the NADH intensity of the hypoxic group rapidly increased during the hypoxic phase and quickly recovered toward baseline levels when normoxia was reinstated (Fig. 2, A and B). These findings are in accordance with the NADH intensity changes observed in vivo (Fig. 1). To further investigate the possible interference of other fluorophores, we also performed fluorescence lifetime microscopy (FLIM) measurements in primary keratinocyte cultures under normoxic and hypoxic conditions. Phasor analysis of the corresponding FLIM images (32) demonstrated that the main contributors to the observed fluorescence, namely, the bound and unbound forms of NADH, were identical; however, the relative levels of bound to unbound NADH decreased during hypoxia (Fig. 2C).
Fig. 2. Cultured keratinocytes exhibit dynamic changes in NADH TPEF intensity upon exposure to hypoxia.
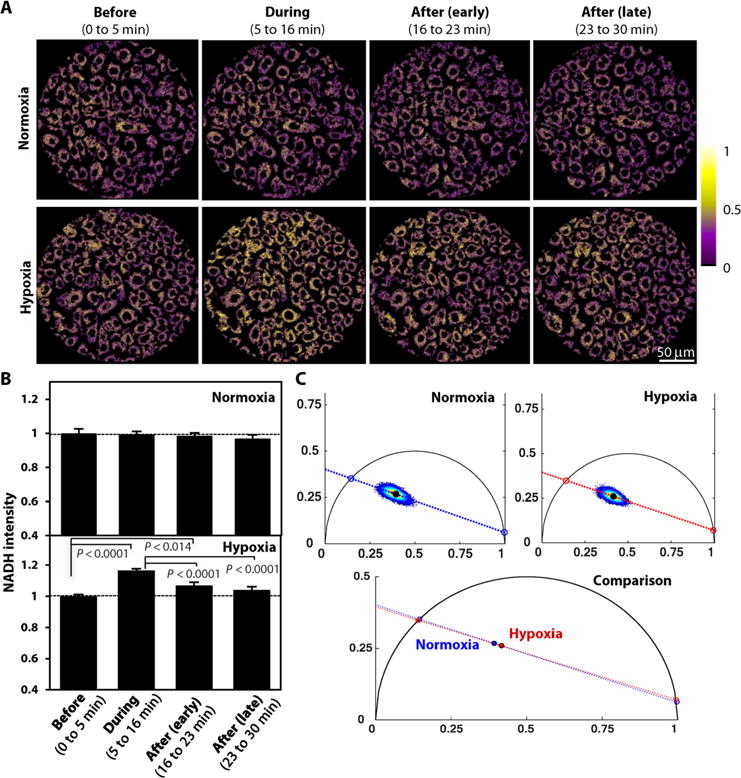
(A) Representative scaled NADH TPEF intensity images from primary neonatal human epidermal keratinocytes (NHEKs) exposed to either normoxia or before, during, and after (early and late) hypoxia induction. Image heat map reflects varying TPEF intensities. (B) Comparisons of means ± SEM NADH intensity values for the NHEKs under normoxia or hypoxia. The presented values are acquired from five independent normoxia and four independent hypoxia experiments. At least three images were captured within each experimental phase during each experiment. The mean value in each phase is normalized for each group by its respective “before” intensity. P values indicated in the figure were calculated by a mixed-model ANOVA with post hoc Bonferroni correction. (C) Phasor distributions of the fluorescence lifetime distributions acquired from NHEKs under normoxia or hypoxia (n = 3 independent wells for each treatment). Red dashed lines depict linear fits to the phasor distributions, with respective centroids indicated by black filled circles.
Having established that the observed outcomes were specific to the mitochondrial changes induced by the hypoxic conditions and that the in vivo and in vitro biochemical patterns were in agreement, we determined whether a similar relationship existed for mitochondrial dynamics. The mitochondrial organization of the cultured keratinocytes was evaluated using the same analytical approach we employed for the in vivo data, with the isolated cytoplasmic intensity fluctuations being cloned into the noncytoplasmic image voids (Fig. 3A). The normoxic group displayed stable overall mitochondrial dynamics through all experimental phases (Fig. 3B), consistent with the unaltered NADH intensity patterns of that group (Fig. 3C). Conversely, the mitochondrial clustering of the hypoxic group increased significantly during the hypoxic phase and remained elevated immediately after the oxygen reperfusion, although full normoxia had been reinstated (Fig. 3, B and C, and fig. S2B). Mitochondrial clustering recovered to baseline levels only during the late reperfusion phase (Fig. 3, B and C). Overall, these outcomes corroborate the temporal trends observed in vivo, where the mitochondrial dynamics changed significantly faster upon hypoxic induction than during normoxic recovery.
Fig. 3. Hypoxia-induced mitochondrial dynamics of cultured keratinocytes are similar to those observed in vivo.
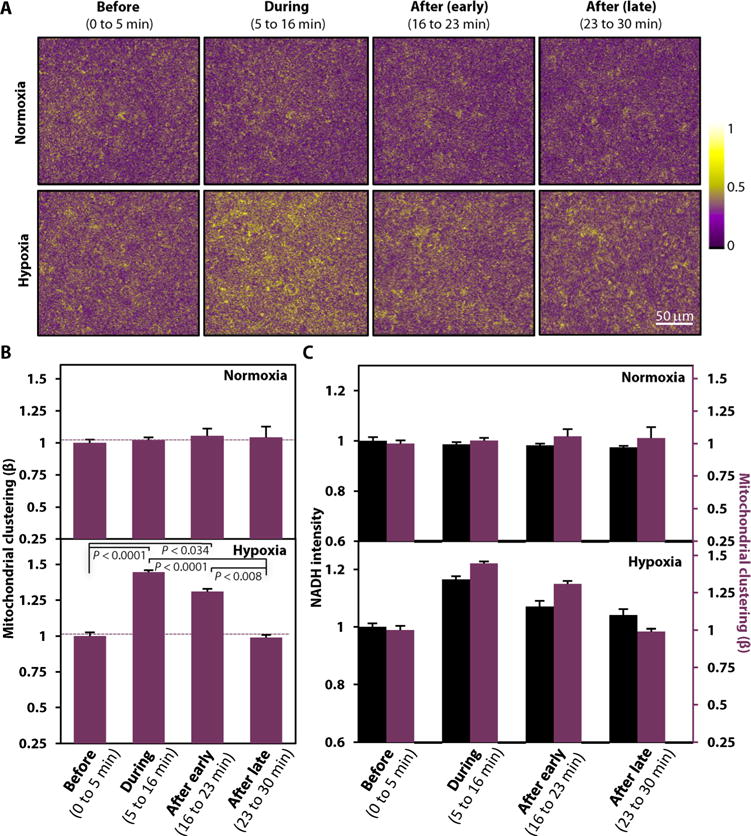
(A) Corresponding processed cloned NADH intensity images from the normoxic and hypoxic keratinocyte groups acquired before, during, and after (early and late) hypoxia induction as in Fig. 2A. Image heat map reflects varying TPEF intensities. (B) Comparisons of means ± SEM mitochondrial clustering values computed from the cloned images from all experiments performed with NHEKs under normoxic and hypoxic conditions (five independent normoxia and four independent hypoxia experiments; at least three images captured within each experimental phase during each experiment). The mean value in each phase is normalized for each group by its respective “before” baseline clustering. P values indicated in the figure were calculated by a mixed-model ANOVA with post hoc Bonferroni correction. (C) Direct comparison of NADH intensities and respective mitochondrial clustering values [as shown in (B) and Fig. 2B] for each experimental phase for the normoxia- and hypoxia-treated NHEKs. Left: NADH intensity axis. Right: Mitochondrial clustering axis.
Healthy epidermis displays depth-dependent morphological and functional patterns
We next assessed MPM imaging and mitochondrial organization as a label-free diagnostic indicator of skin cancer. For this reason, we examined full-thickness healthy and diseased skin epithelia. 3D tissue stacks were acquired in vivo from healthy human skin and diseased human skin, corresponding to BCCs and melanomas (both invasive and in situ). Fourteen participants with a total of 29 tissue stacks [12 healthy tissue stacks (n = 6 participants; 4 healthy volunteers and 2 of the patients) and 17 pathological (n = 10)] were included in our study. The 17 diseased tissues comprised 2 in situ melanoma stacks (n = 2), 7 invasive melanoma stacks (n = 3), and 8 BCC stacks (n = 5).
Owing to the wide spectral characteristics of the clinical tomograph emission filter, fluorescence was collected from NADH as well as from keratin, melanin, and collagen/elastin (Fig. 4A, colored arrows). We minimized the non-NADH signals by using Shanbhag’s entropy filtering (33), as shown explicitly in fig. S3, which includes representative images of the total TPEF collected signal, along with segmented regions that are associated with non-NADH signals. The separation accuracy was assessed by visual correlation with the natural histological localization of the chromophores. For example, keratin fluorescence was only detected in the most apical keratinized layer with minor contribution in layers below, when epidermal tissue folds were present (Fig. 4A, cyan arrows). On the other hand, melanin fluorescence was detected deeper in the normal epithelium. Its highest contribution was localized in the transition from the parabasal to the basal layer of healthy tissues (Fig. 4A, open-head red arrows), because melanin granules naturally cap the top surface of the nuclei of the proliferative cell pool and serve as photoprotectors (34). Melan A staining, when available, provided additional support to the fact that our thresholding technique for chromophore isolation yielded reliable outcomes (fig. S3).
Fig. 4. Morphological and functional differences are detected in in vivo endogenous TPEF images of normal and cancerous human epithelia.
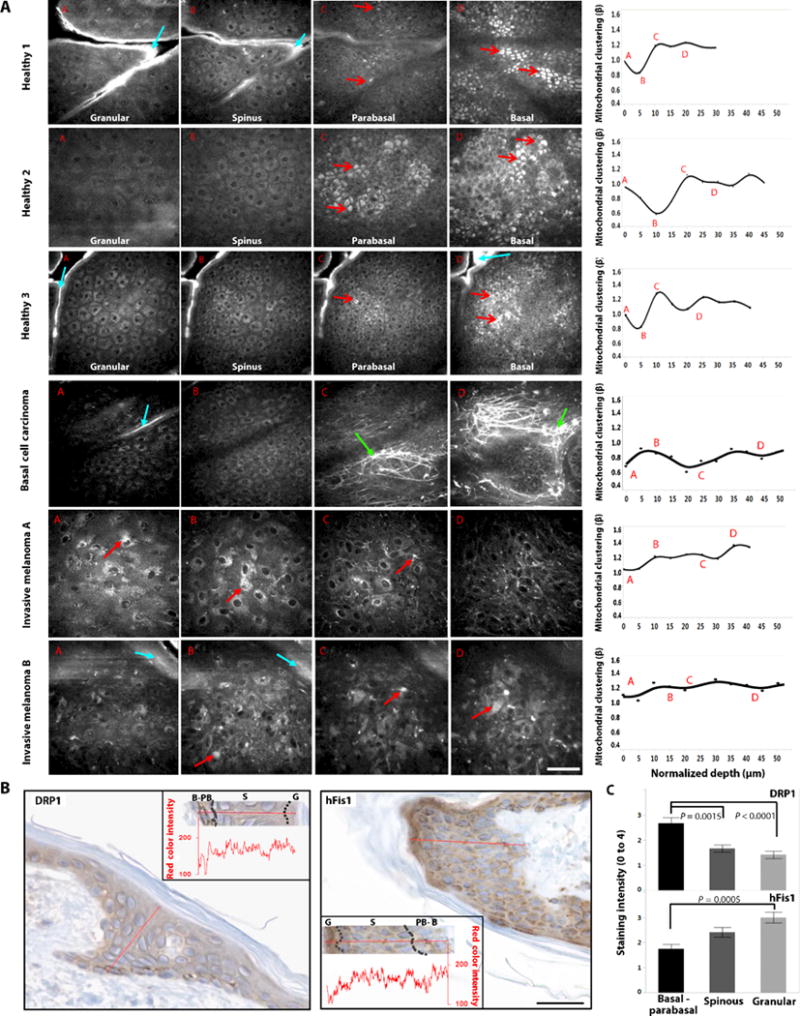
(A) Representative raw TPEF images acquired at distinct depths from healthy and diseased epithelial tissues. The corresponding mitochondrial clustering values as a function of depth extracted from the tissue stacks are presented at the rightmost column as dots. The solid lines represent penalized spline fits with λ = 0.005. The labels A, B, C, and D within the mitochondrial clustering panels represent the mitochondrial clustering values extracted from the panel’s respective labeled images. Open-head red arrows designate the melanin caps, which intensify above and regress below the nuclei of the basal cells. Closed-head red arrows designate melanin identified in melanoma tissues. Cyan and green arrows designate keratin and collagen/elastin contributions, respectively. Scale bar, 50 μm. (B) Immunohistochemical staining of DRP1 and hFis1 proteins in healthy human skin. Insets in both cases show quantitative intensity measurements as a function of depth for each respective panel. Decreased red color intensity corresponds to darker brown hues and, therefore, increased staining intensity. The borders between the basal-parabasal (B-PB), spinous (S), and granular (G) layers are identified with dashed lines. Scale bar, 25 μm. (C) Quantitative comparisons of the means ± SEM immunohistochemical staining intensity as a function of epithelial layer according to standard practice using a grading range from 0 to 4 (n = 3 healthy human samples). Statistical significance between epithelial layers (P values are indicated in the figure) was determined by nested-model ANOVAs with post hoc Tukey test.
We found that healthy epithelia displayed uniform cellular morphology within each optical section and smooth epidermal stratification, with cellular size decreasing and nuclear-to-cytoplasmic ratio increasing in a depth-dependent manner (fig. S4), as expected. The consistent morphological features of the healthy epithelia were accompanied by consistent depth-dependent mitochondrial clustering trends (Fig. 4A and fig. S5A). The basal and parabasal layers displayed high and stable values of the clustering metric (fragmented state). As epithelial cell differentiation progressed from the basal to the higher epidermal layers, clustering values declined, reaching their minima within the spinous layer (tubular mitochondria). Finally, toward the most terminally differentiated layer, as the granular keratinocytes enter an apoptotic state to create the stratum corneum, mitochondrial clustering values started to recover again, signifying a return to a more fissioned phenotype.
This interpretation of the depth-dependent mitochondrial organization changes in healthy tissues was consistent with the mitochondrial organization states identified in 2D primary keratinocytes stained with mitochondrial specific dyes, which were either maintained at an undifferentiated/basal state or induced to differentiate and then apoptose, using established Ca2+ and staurosporine treatment protocols (see the Supplementary Materials and Methods and fig. S6) (35, 36). Although little is known about the exact mechanisms of keratinization induction in vivo, keratinization is accepted to be a specialized form of apoptosis (37).
To further validate the mitochondrial organization patterns we observed in vivo, we performed immunohistochemical analysis of healthy human skin samples stained for dynamin-related protein 1 (DRP1) (38) and the human fission protein 1 (hFis1) (39), two proteins related to mitochondrial fragmentation in mammalian cells.
Although both proteins were present in all epidermal layers, DRP1 was primarily expressed within the basal-parabasal cells. Overexpression of DRP1 is an independent cause of increased mitochondrial fragmentation (40). On the other hand, hFis1 expression was significantly increased within the granular layer, which consists of the most terminally differentiated keratinocytes before cornification (Fig. 4, B and C). This is consistent with studies showing that transient overexpression of hFis1 induces excessive DRP1-mediated mitochondrial fragmentation followed by cytochrome c release and, ultimately, apoptosis (39). Therefore, the observed immunohistochemistry findings of increased mitochondrial fission proteins in the basal and granular layers agree with the 3D optical trends and the 2D differentiation-apoptosis studies. These findings suggest a potential interplay between mitochondrial dynamics and the keratinization process, with mitochondria being key participants in the apoptotic cascade, which transforms granular keratinocytes into the stratum corneum.
Diseased epidermis lacks depth-dependent morphological and functional patterns
Several features were present within the optical stacks obtained from cancerous lesions, suggestive of underlying histopathology. In BCC, cellular nests resided in the dermis surrounded by excessive collagen/elastin stromal deposition (Fig. 4A, green arrows) (24), findings representing typical histological BCC presentation. The cellular morphological features were more highly variable among the melanomas than among the BCCs, with dendritic cells (melanocytes) appearing within the upper epidermal layers, haphazard cellular morphology, or cells invading the dermis (Fig. 4A). These abnormal morphological features of the diseased tissues were accompanied by functional deviations. Specifically, the depth-dependent mitochondrial clustering behavior of the lesion-derived optical stacks did not display any stratification-related correlation (Fig. 4A).
We did not detect any consistent depth-dependent mitochondrial clustering patterns when comparing BCCs and melanomas, nor did we detect any such patterns among lesions of the same gross histological category. In part, this result was expected, because the lesions originated from different cell types and displayed varying degrees of intraepithelial and/or intradermal invasion. In some of the lesions characterized by tumor cells invading the dermis, the mitochondrial organization was stabilized over the invaded dermal depth, suggesting a locally uniform metabolic lesion profile (fig. S5B).
Thus, in contrast to the consistent, depth-dependent variations in the morphological (nuclear-to-cytoplasmic ratio) and functional (mitochondrial clustering) features of the healthy epithelia that relate to histological stratification and indicate an interdependence of structure and function, the cancer-containing tissues exhibited depth-independent morphological and functional patterns. Consequently, metrics quantifying depth-dependent variations in functional features such as mitochondrial organization are candidates for useful tissue diagnostics.
Depth-dependent mitochondrial organization distinguishes diseased from healthy human skin tissues
To test the potential of the mitochondrial organization assessment by MPM imaging as a quantitative diagnostic biomarker, we considered three uncorrelated tissue metrics that evaluated both the metabolic and morphological features of human skin and could together differentiate healthy and diseased epithelia. The first captured the degree of depth-dependent stratification-related variations in mitochondrial clustering, absent in diseased epithelia. The second was the median mitochondrial clustering value of the tissue. The third was the depth-dependent variation of the nuclear-to-cytoplasmic ratio, which is related to a standard histopathological morphological marker of healthy, differentiated squamous epithelia. Large variations in this ratio correlate with higher values of the PSD depth-dependent variance over scales that correspond to the size of nuclei and cell borders (29). Accordingly, the value of this third metric decreased with the loss of epithelial cell differentiation in cancerous lesions (fig. S4C).
The values of the three metrics were separated between healthy and diseased tissues (Fig. 5A). To quantitatively gauge the efficacy of our classification system, we used linear discriminant analysis to differentiate healthy and diseased tissue stacks. Significant mean differences at α = 0.05 were observed for all three predictor variables (Fig. 5A). The log determinants were similar, and Box’s M indicated that the assumption of equality of covariance matrices was not violated (Fig. 5A). The discriminate function calculated with the entire data set revealed a significant association between groups and all predictors, accounting for 72.4% of between-group variability, and resulting in 100% specificity and 94.1% sensitivity (Fig. 5B). Because the data set was relatively small, we also classified the sites using leave-one-out cross-validation, which resulted in correct classification of 89.7% of the cases, with a specificity of 100% and a sensitivity of 82.4% (Fig. 5B).
Fig. 5. Mitochondrial depth-dependent assessments provide accurate tissue classification.
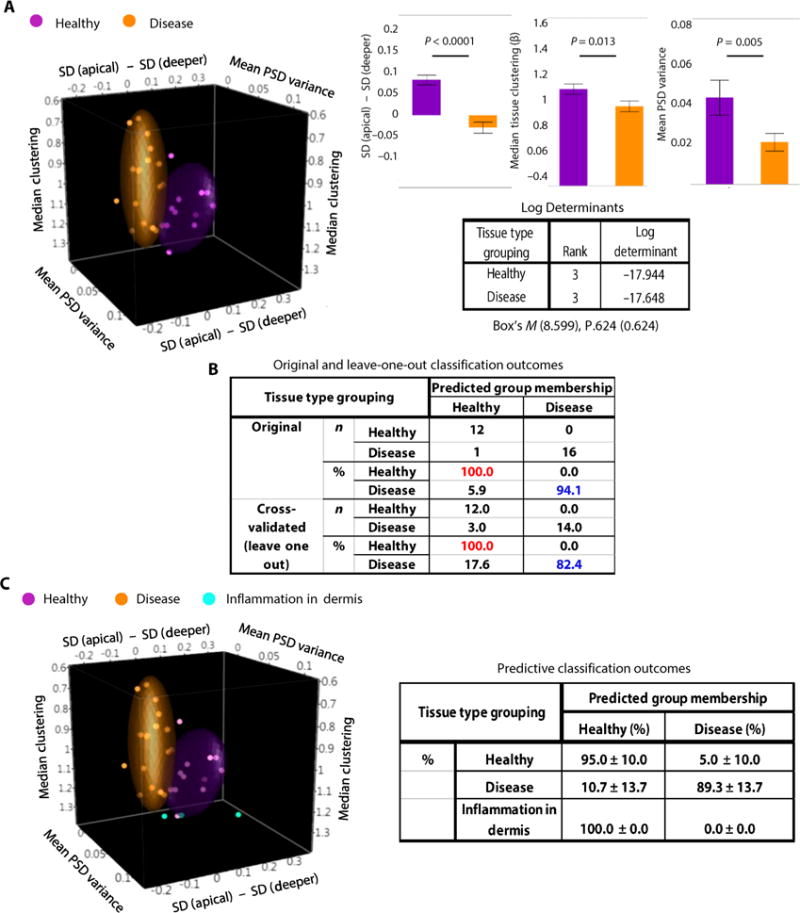
(A) 3D scatterplot (left) showing in space the tissue separation of the healthy (purple; n = 12) and diseased (orange; n = 17) tissue stacks, on the basis of three uncorrelated tissue metrics extracted from TPEF image analysis. Each point represents one tissue stack. Colored ellipsoids represent 70% of data coverage. Comparisons of the means ± SEM values for each of the metrics from all data acquired from healthy and diseased tissue stacks. Significant mean differences at α = 0.05 were observed by univariate ANOVAs for all three predictor variables (P values are indicated in the figure). The log determinants, Box’s M, and P values supported the assumption of equality of covariance matrices. (B) Original and cross-validated classification outcomes based on the comparison of the three-metric model predictions with corresponding histopathological evaluations. (C) 3D scatterplot (left) showing the separation of the healthy (purple), diseased (orange), and dermal inflammation (cyan; n = 3) tissue stacks. Each point represents one tissue stack. Colored ellipsoids represent 70% of data coverage. Classification outcomes (right) from the predictive discriminant analyses using randomized 70%/30% training–test data separation, respectively. The tissue stacks representing the areas of dermal inflammation were also evaluated by the predictive functions. Means and SDs are reported from five independent randomized runs.
We then tested our classification approach in a predictive model, using randomized 70%/30% training–test data separation, with the separation performed at the participant level. The predictive classification on the test data displayed 91.9% mean accuracy, 95% mean specificity, and 89.3% mean sensitivity (Fig. 5C). As a preliminary but potential future application, we applied the predictive classification prospectively to a small sample (n = 3) of TPEF tissue stacks acquired from one of the in situ melanoma patients, from locations that appeared macroscopically suspicious (erythematous with loss of normal pigmentation) but histologically exhibited solely dermal inflammation (fig. S7A). These optical stacks, in agreement with the histological findings, displayed no intraepithelial morphological abnormalities (fig. S7B). The stacks representing areas of dermal inflammation (fig. S7B) were classified as healthy tissues 100% of the time (Fig. 5C).
DISCUSSION
Here, we established the ability to evaluate mitochondrial organization noninvasively in vivo within 3D, intact human tissues using solely endogenous TPEF images. Using fast monitoring under hypoxia-reperfusion conditions, we validated the sensitivity and specificity of our analytical technique to real physiological mitochondrial organization changes occurring in vivo and in vitro. These studies also illustrated the capability of this method to quantify dynamic changes occurring over several seconds. To demonstrate the translational diagnostic potential of assessing mitochondrial organization in vivo, we used this analytical approach to differentiate healthy skin epithelia from cancerous skin, as one example among a broader range of pathologies in which mitochondrial organization dysfunctions are implicated.
The mitochondrial responses to arterial occlusion and reperfusion in the upper epithelial layers are distinct from those in the lower layers, with the deeper layers having a stronger dependence on vascular oxygenation. This finding adds to existing studies (30) that point to the importance of vascular oxygen to the oxygen supply of the human epidermis. In addition to validating the sensitivity of our approach to established mitochondrial organization changes, the oxygen-reperfusion measurements reveal an interesting distinction in the dynamics of biochemical and organizational mitochondrial changes during reperfusion. Specifically, whereas NADH TPEF intensity recovers almost immediately, mitochondrial fusion, which reinstates the networks that enable exchange of the mitochondrial matrix content, is delayed. Such a delay is not observed during the onset of hypoxia, when an increase in both NADH TPEF intensity and mitochondrial fissioning occurs within seconds. Our finding indicates that mitochondrial biochemistry can differ temporally from mitochondrial dynamics. The mitochondrial organization changes that we have observed noninvasively are similar to the fission-fusion dynamics of mitochondria observed after hypoxia-reperfusion that until now have been studied in vitro with exogenous dyes (13, 14, 38). Mitochondrial fusion requires high levels of control and coordination (15), because it is a multistep process involving several main and accessory proteins (41). Furthermore, the protein complexes rely on the mitochondrial membrane potential (ΔΨm) and adenosine 5′-triphosphate (ATP) availability (42). During acute hypoxia, although carbon flux into the tricarboxylic acid cycle is not altered, the mitochondrial membrane potential is abolished by blockage of the electron transport chain due to the lack of oxygen. Ample recovery of the ΔΨm after reoxygenation is not instantaneous (43), which, in turn, can further hinder ATP production, because the electrochemical gradient is the main force driving ATP production. Therefore, disturbance of any of these finely balanced processes can delay fusion.
On the other hand, mitochondrial fission requires the recruitment of a single protein, DRP1 (38). Although several proteins (39) are associated with recruiting DRP1 to the mitochondria, fission can happen within seconds (44), even outside a cellular environment. Mitochondrial depolarization by hypoxia is well known to induce DRP1-dependent fission (38). Hence, the fast hypoxia-driven fissioning response that we detected contrasts with the delayed fusion dynamics and both are consistent with the biochemistry of the mitochondrial fission-fusion protein machinery.
Changes in the relative expression of the major molecular machinery components of mitochondrial fission may also be, at least in part, related to the consistent depth-dependent changes we observe within healthy keratinizing squamous epithelia. Specifically, the increased mitochondrial clustering seen within the basal/parabasal and granular layers are correlated with higher intraepithelial expression of DRP1 and hFis1 within those strata, respectively (Fig. 4, B and C). Furthermore, our depth-dependent results are a manifestation of the structure-function interdependence that governs the epidermal physiology. During differentiation, as cells turn from glycolysis to oxidative phosphorylation for energy production, mitochondria fuse and create more extensive networks (45), whereas during proliferation or apoptosis, mitochondria assume fragmented phenotypes (39, 46, 47), with mitochondrial fission often being the initiating step of the apoptotic pathway (39). Our in vitro studies, although performed in oversimplified models of the in vivo epidermis, conjointly support these associations. The consistent depth-dependent variations in both morphological and functional characteristics observed in healthy tissues fade away with disease, which is pathophysiologically expected as cancer cells overtake the normal parenchyma (23, 24), and these differences can be used for accurate tissue classification. In summary, our findings establish the ability to assess mitochondrial organization in humans in vivo in a quantitative manner, which obviates the need for tissue excision or exogenous contrast agents.
Although our results are promising, they are derived from a limited number of participants. The incorporation of morphofunctional evaluation in the analysis of NADH MPM images could be further developed beyond the current proof-of-principle demonstration by evaluating a larger human population. Such an extensive patient data pool could also allow a deeper understanding of the optimal combination of structure-function patterns that could predict patient prognosis, lesion metastatic potential, and treatment response/outcomes. Furthermore, the widespread adoption of MPM imaging in the clinic for diagnostic applications is still limited by the high technological costs associated with the required laser source and by the inability to image large fields of view. Moreover, current commercially available MPM systems consist of fairly bulky articulated arms that rely on traditional objectives for imaging, preventing access to epithelial tissues that can only be visualized endoscopically or laparoscopically. However, rapid advances in photonic components such as portable, cost-efficient near-infrared laser technology (48); rapid scanning mechanisms (49); and hand-held MPM-compatible probes (50) are expected to overcome these obstacles and enable broad clinical translation of MPM imaging for tissue diagnosis.
Despite the aforementioned limitations, our data display direct evidence of proof-of-concept translatability to humans. The technique described here is uniquely capable of simultaneously providing high-resolution images and functional information, a feature that could enhance standard diagnostic criteria for early cancer, which currently rely primarily on morphological features after tissue fixation and processing. One of the key advantages of mitochondrial clustering as a diagnostic biomarker is its relevance to both mitochondrial structure and cellular function. Our analysis can be automated and implemented in near real time. Thus, although there may be some training required for the operation of the microscope, image interpretation can be performed algorithmically, resulting in procedural time and cost efficiency as well as enhanced screening efficacy. Finally, mitochondrial monitoring may have broad diagnostic, therapeutic, and disease monitoring applicability, a result of the association of mitochondrial dysfunction with a variety of human diseases.
MATERIALS AND METHODS
Study design
Our overall objective was to demonstrate that characterization of mitochondrial dynamics can be performed noninvasively in vivo in human subjects by using only endogenous contrast from the metabolic coenzyme NADH. We used both in vivo and in vitro systems to meet this goal. To validate temporal sensitivity in assessing mitochondrial dynamics changes, hypoxia-reperfusion experiments were performed, because hypoxia is a well-known factor that affects mitochondrial dynamics and causes mitochondrial fragmentation. To demonstrate the potential medical application of mitochondrial organization as a diagnostic biomarker, we then proceeded to evaluate the depth-dependent mitochondrial organization of the human epidermis from healthy and cancerous skin. All in vivo measurements were conducted according to approved University of California, Irvine institutional review board protocol with written informed consent from all participants. Participants older than 18 years were included in the study, with pregnancy being the only exclusion criterion. Lesion sites were imaged before biopsy. All lesions were diagnosed by a dermatopathologist at University of California, Irvine (R.M.H.), using standard hematoxylin and eosin histology or Melan A staining when necessary. Immunohistochemical staining of fission proteins was also performed using healthy human skin discarded from cosmetic surgeries to validate the optically derived depth-dependent observations. Sample sizes were set so as to secure validation of the statistical analysis assumptions. No blinding or randomization was used, except for the predictive classification model, where a random number generator was used to assign the participants into the training and test data sets. Replication information is provided in the figure legends and in the respective section in the Supplementary Materials.
Imaging
Detailed protocols for the imaging and image analysis procedures are provided in the Supplementary Materials.
Statistical analyses
Statistical analysis was performed using IBM SPSS Statistics 22 and JMP 11 (SAS). To assess the significance of the NADH TPEF intensity or the mitochondrial clustering differences between hypoxia-reperfusion phases, mixed-model ANOVAs were performed with the post hoc Bonferroni correction for multiple comparisons. To account for the time series/temporal interdependence nature of the measurements within each experimental repeat, a nested, repeated-measures design was implemented. Because of a minor movement of the subject during the in vivo time series acquisition, the fixed focus of the epidermal layer was sometimes lost. Those images were not used in the analysis and comprised about 15% of the temporal data set. To recover those time points during the intensity and mitochondrial clustering evaluation described previously, linear interpolation was used. To evaluate the three-metric tissue separation model, multivariate and canonical linear discriminant analyses were performed. Multivariate analysis was implemented to evaluate the extent of correlation between the three metrics for each tissue group using the Pearson product-moment correlation coefficient. Before proceeding with the discriminant analysis, the mean values for each group and predictor variable were compared using univariate ANOVAs, and the test for the equality of the group covariance matrices was used to examine whether the null hypothesis of multivariate normality was not rejected. Sensitivity and specificity were calculated, with the linear discriminant function determined and applied using the entire data set as well as a leave-one-out cross-validation scheme. Similarly, linear discriminant analyses were used to evaluate the predictive performance of the three-metric model, with the tissue data set randomly grouped into training and test data using a 70%/30% ratio, respectively. The discriminant functions derived from the training set were also applied in every run to the stacks representing dermal inflammation. To avoid data overfitting, five independent randomized runs were performed. To evaluate the immunohistochemical visual grading scores between the different epithelial layers for each protein, ANOVAs were performed followed by a post hoc Tukey test for multiple comparisons. Each ANOVA model accounted for the multiple fields sampling within each individual tissue using a nested design. Significance level for all statistics was set to α = 0.05. All statistical analyses were reviewed by an independent statistician.
Supplementary Material
Fig. S1. Automated image analysis steps before quantitative extraction of cellular-related biomarkers.
Fig. S2. Setup for dynamic media oxygen saturation and live oxygen saturation measurements.
Fig. S3. Automated digital filter–based identification of endogenous TPEF contributions from non-NADH chromophores.
Fig. S4. Detailed mitochondrial clustering profiles.
Fig. S5. The depth-dependent mean PSD variance is correlated with the corresponding N/C ratio (nuclear-to-cytoplasmic ratio) variation.
Fig. S6. Organization patterns in stained mitochondria images of 2D cultures of undifferentiated, differentiated, and apoptotic cells.
Fig. S7. Mitochondrial organization distinguishes benign from malignant macroscopically suspicious sites.
Acknowledgments
We thank JenLab GmbH for loaning the MPTflex clinical tomograph.
Funding: We acknowledge support from the American Cancer Society (RSG-09-174-01-CCE), the NIH National Institute of Biomedical Imaging and Bioengineering Laser Microbeam and Medical Program (P41-EB015890), the Alexander S. Onassis Public Benefit Foundation, and the Arnold and Mabel Beckman Foundation.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/8/367/367ra169/DC1)
Materials and Methods
Author contributions: D.P., M.B., B.J.T., and I.G. were responsible for conceiving and designing the study; K.M.K., M.B., and B.J.T. were involved in patient recruitment and acquisition of the human in vivo data; D.P. performed the data analysis and the cellular experiments; C.A.A., Z.L., and K.P.Q. contributed to the development of data analysis algorithms; F.R.-D. consulted on the study design and statistical analysis of the data; R.M.H. evaluated all histopathological samples; and B.J.T. and I.G. supervised all aspects of data acquisition and analysis, respectively.
Competing interests: Provisional patents related to this work have been filed as follows: (i) provisional application no. 62/400430, titled “Methods and systems for mitochondrial imaging,” filed 27 September 2016; (ii) provisional application no. 62/400402, titled “Multifunctional devices for dynamic control of cell culturing conditions,” filed 27 September 2016. D.P., K.P.Q., and I.G. are listed as inventors on the patents. All other authors declare that they have no competing interests.
Data and materials availability: All materials used in this study are commercially available. All data are available upon request as mandated by NIH guidelines.
REFERENCES AND NOTES
- 1.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 2.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y-i, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 3.Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peinado JR, Diaz-Ruiz A, Fruhbeck G, Malagon MM. Mitochondria in metabolic disease: Getting clues from proteomic studies. Proteomics. 2014;14:452–466. doi: 10.1002/pmic.201300376. [DOI] [PubMed] [Google Scholar]
- 6.Sebastián D, Hernández-Alvarez MI, Segalés J, Sorianello E, Muñoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Orešič M, Pich S, Burcelin R, Palacín M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharp WW, Fang Y Hu, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, Yu T, Lee H, O’Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. 2015;106:272–283. doi: 10.1093/cvr/cvv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–4824. doi: 10.1038/onc.2012.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 12.Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem. 2010;285:11740–11752. doi: 10.1074/jbc.M109.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giedt RJ, Yang C, Zweier JL, Matzavinos A, Alevriadou BR. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: Role of nitric oxide and reactive oxygen species. Free Radical Biol Med. 2012;52:348–356. doi: 10.1016/j.freeradbiomed.2011.10.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Hajnóczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia–reoxygenation stress. Cell Death Differ. 2011;18:1561–1572. doi: 10.1038/cdd.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malka F, Guillery O, Cifuentes-Diaz C, Guillou E, Belenguer P, Lombès A, Rojo M. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005;6:853–859. doi: 10.1038/sj.embor.7400488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 17.Georgakoudi I, Quinn KP. Optical imaging using endogenous contrast to assess metabolic state. Annu Rev Biomed Eng. 2012;14:351–367. doi: 10.1146/annurev-bioeng-071811-150108. [DOI] [PubMed] [Google Scholar]
- 18.Blinova K, Levine RL, Boja ES, Griffiths GL, Shi ZD, Ruddy B, Balaban RS. Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry. 2008;47:9636–9645. doi: 10.1021/bi800307y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K, Georgakoudi I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci Rep. 2013;3:3432. doi: 10.1038/srep03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivera DR, Brown CM, Ouzounov DG, Pavlova I, Kobat D, Webb WW, Xu C. Compact and flexible raster scanning multiphoton endoscope capable of imaging unstained tissue. Proc Natl Acad Sci USA. 2011;108:17598–17603. doi: 10.1073/pnas.1114746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.König K. Clinical multiphoton tomography. J Biophotonics. 2008;1:13–23. doi: 10.1002/jbio.200710022. [DOI] [PubMed] [Google Scholar]
- 22.Dimitrow E, Ziemer M, Johannes Koehler M, Norgauer J, König K, Elsner P, Kaatz M. Sensitivity and specificity of multiphoton laser tomography for in vivo and ex vivo diagnosis of malignant melanoma. J Invest Dermatol. 2009;129:1752–1758. doi: 10.1038/jid.2008.439. [DOI] [PubMed] [Google Scholar]
- 23.Balu M, Kelly KM, Zachary CB, Harris RM, Krasieva TB, König K, Durkin AJ, Tromberg BJ. Distinguishing between benign and malignant melanocytic nevi by in vivo multiphoton microscopy. Cancer Res. 2014;74:2688–2697. doi: 10.1158/0008-5472.CAN-13-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balu M, Zachary CB, Harris RM, Krasieva TB, König K, Tromberg BJ, Kelly KM. In vivo multiphoton microscopy of basal cell carcinoma. JAMA Dermatol. 2015;151:1068–1074. doi: 10.1001/jamadermatol.2015.0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gambichler T, Jaedicke V, Terras S. Optical coherence tomography in dermatology: Technical and clinical aspects. Arch Dermatol Res. 2011;303:457–473. doi: 10.1007/s00403-011-1152-x. [DOI] [PubMed] [Google Scholar]
- 26.Guitera P, Menzies SW, Longo C, Cesinaro AM, Scolyer RA, Pellacani G. In vivo confocal microscopy for diagnosis of melanoma and basal cell carcinoma using a two-step method: Analysis of 710 consecutive clinically equivocal cases. J Invest Dermatol. 2012;132:2386–2394. doi: 10.1038/jid.2012.172. [DOI] [PubMed] [Google Scholar]
- 27.DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: What do metabolic outliers teach us? Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xylas J, Quinn KP, Hunter M, Georgakoudi I. Improved Fourier-based characterization of intracellular fractal features. Opt Express. 2012;20:23442–23455. doi: 10.1364/OE.20.023442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xylas J, Varone A, Quinn KP, Pouli D, McLaughlin-Drubin ME, Thieu HT, Garcia-Moliner ML, House M, Hunter M, Munger K, Georgakoudi I. Noninvasive assessment of mitochondrial organization in three-dimensional tissues reveals changes associated with cancer development. Int J Cancer. 2015;136:322–332. doi: 10.1002/ijc.28992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balu M, Mazhar A, Hayakawa CK, Mittal R, Krasieva TB, König K, Venugopalan V, Tromberg BJ. In vivo multiphoton NADH fluorescence reveals depth-dependent keratinocyte metabolism in human skin. Biophys J. 2013;104:258–267. doi: 10.1016/j.bpj.2012.11.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palero JA, Bader AN, de Bruijn HS, van den Heuvel AP, Sterenborg HJCM, Gerritsen HC. In vivo monitoring of protein-bound and free NADH during ischemia by nonlinear spectral imaging microscopy. Biomed Opt Express. 2011;2:1030–1039. doi: 10.1364/BOE.2.001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Digman MA, Caiolfa VR, Zamai M, Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008;94:L14–L16. doi: 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shanbhag AG. Utilization of information measure as a means of image thresholding. CVGIP Graph. Model Image Process. 1994;56:414–419. [Google Scholar]
- 34.Yamaguchi Y, Brenner M, Hearing VJ. The regulation of skin pigmentation. J Biol Chem. 2007;282:27557–27561. doi: 10.1074/jbc.R700026200. [DOI] [PubMed] [Google Scholar]
- 35.Belmokhtar CA, Hillion J, Ségal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- 36.Borowiec AS, Delcourt P, Dewailly E, Bidaux G. Optimal differentiation of in vitro keratinocytes requires multifactorial external control. PLOS ONE. 2013;8:e77507. doi: 10.1371/journal.pone.0077507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamanishi K, Shen CS, Mizutani H. Apoptosis in the epidermis. Methods Mol Biol. 2005;289:171–174. doi: 10.1385/1-59259-830-7:171. [DOI] [PubMed] [Google Scholar]
- 38.Ghose P, Park EC, Tabakin A, Salazar-Vasquez N, Rongo C. Anoxia-reoxygenation regulates mitochondrial dynamics through the hypoxia response pathway, SKN-1/Nrf, and stomatin-like protein STL-1/SLP-2. PLOS Genet. 2013;9:e1004063. doi: 10.1371/journal.pgen.1004063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 40.Szabadkai G, Simoni A Maria, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 41.Escobar-Henriques M, Anton F. Mechanistic perspective of mitochondrial fusion: Tubulation vs. fragmentation. Biochim Biophys Acta. 2013;1833:162–175. doi: 10.1016/j.bbamcr.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 42.Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bahar S, Fayuk D, Somjen GG, Aitken PG, Turner DA. Mitochondrial and intrinsic optical signals imaged during hypoxia and spreading depression in rat hippocampal slices. J Neurophysiol. 2000;84:311–324. doi: 10.1152/jn.2000.84.1.311. [DOI] [PubMed] [Google Scholar]
- 44.Pucadyil TJ, Schmid SL. Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell. 2008;135:1263–1275. doi: 10.1016/j.cell.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 46.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 47.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakadžić S, Demirbas U, Mempel TR, Moore A, Ruvinskaya S, Boas DA, Sennaroglu A, Kartner FX, Fujimoto JG. Multi-photon microscopy with a low-cost and highly efficient Cr:LiCAF laser. Opt Express. 2008;16:20848–20863. doi: 10.1364/oe.16.020848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lechleiter JD, Lin DT, Sieneart I. Multi-photon laser scanning microscopy using an acoustic optical deflector. Biophys J. 2002;83:2292–2299. doi: 10.1016/S0006-3495(02)73989-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rivera DR, Brown CM, Ouzounov DG, Webb WW, Xu C. Multifocal multiphoton endoscope. Opt Lett. 2012;37:1349–1351. doi: 10.1364/OL.37.001349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Automated image analysis steps before quantitative extraction of cellular-related biomarkers.
Fig. S2. Setup for dynamic media oxygen saturation and live oxygen saturation measurements.
Fig. S3. Automated digital filter–based identification of endogenous TPEF contributions from non-NADH chromophores.
Fig. S4. Detailed mitochondrial clustering profiles.
Fig. S5. The depth-dependent mean PSD variance is correlated with the corresponding N/C ratio (nuclear-to-cytoplasmic ratio) variation.
Fig. S6. Organization patterns in stained mitochondria images of 2D cultures of undifferentiated, differentiated, and apoptotic cells.
Fig. S7. Mitochondrial organization distinguishes benign from malignant macroscopically suspicious sites.