

Abstract
Glioblastoma (GBM) is a lethal brain tumor composed of heterogeneous cellular populations including glioma stem cells (GSCs) and differentiated non-stem glioma cells (NSGCs). While GSCs are involved in tumor initiation and propagation, NSGCs’ role remains elusive. Here, we demonstrate that NSGCs undergo senescence and secrete pro-angiogenic proteins, boosting the GSC-derived tumor formation in vivo. We used a GSC model that maintains stemness in neurospheres, but loses the stemness and differentiates into NSGCs upon serum stimulation. These NSGCs downregulated telomerase, shortened telomeres, and eventually became senescent. The senescent NSGCs released pro-angiogenic proteins, including vascular endothelial growth factors and senescence-associated interleukins, such as IL-6 and IL-8. Conditioned medium from senescent NSGCs promoted proliferation of brain microvascular endothelial cells, and mixed implantation of GSCs and senescent NSGCs into mice enhanced the tumorigenic potential of GSCs. The senescent NSGCs seem to be clinically relevant, because both clinical samples and xenografts of GBM contained tumor cells that expressed the senescence markers. Our data suggest that senescent NSGCs promote malignant progression of GBM in part via paracrine effects of the secreted proteins.
Keywords: Glioblastoma, Cancer stem cell, Senescence, Angiogenesis, Paracrine effect
1. Introduction
Glioblastoma (GBM) is the most frequent malignant brain tumor in adults. The median survival of GBM patients is less than 2 years, because of the limited efficacy of the current therapies. One of the hallmarks of this devastating disease is hyper-vascularity; thus, targeting this feature may provide some promising results against GBM [1,2].
GBM is composed of heterogeneous cell populations with glioma stem cells (GSCs) at the apex of the cellular hierarchy [3]. GSCs can self-renew, form tumors, and exhibit resistance to radio- and chemotherapies [3–5]. Unlike GSCs, non-stem glioma cells (NSGCs), which are derived from GSCs, have no tumorigenic potential [3]. However, the roles of NSGCs in GBM propagation and therapy response remain elusive.
Recent studies in non-brain cancers have revealed unexpected roles of non-stem cancer cells in the disease malignancy [6–9]. In ovarian cancer, for example, non-stem cancer cells undergo an epithelial-mesenchymal transition-like process, accompanied by enhanced metastatic potential [7]. In prostate cancer, non-stem cancer cells behave as a critical intercellular mediator of paracrine effects on metastasis of cancer stem cells [8,9]. These observations suggest that similarly to other cancer types, NSGCs may play a crucial role in GBM development. Here, we demonstrate that NSGCs undergo senescence and secrete angiogenesis-related proteins, thereby promoting the growth of GSC-derived tumors in mouse xenografts.
2. Materials and methods
2.1. Cell culture
GSC lines, GBM146 and GBM157, were maintained under serum-free, sphere conditions as described [10,11]. Their differentiation into NSGCs was induced as described in Supplementary Materials and Methods. Sphere-forming assay was performed as described [10,11].
2.2. Flow cytometry
Flow cytometry was performed as described [10] with allophycocyanin-conjugated anti-CD133 antibody (293C3; Miltenyi Biotec, San Diego, CA, USA). DNA content was measured by propidium iodide staining [12].
2.3. Western blot analysis
Cell lysates were prepared, and western blot analysis was performed as described [12] with the antibodies listed in Supplementary Materials and Methods.
2.4. RT-qPCR
Total RNA was extracted from cell cultures using the RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and quantitated with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Gene transcripts were detected by RT-qPCR as described [13].
2.5. Mouse xenograft
The animal experiments were approved by Japanese Foundation for Cancer Research, Institutional Animal Care and Use Committee, and conducted in accordance with the institutional guidelines. GSCs were suspended in Matrigel (1:1) and subcutaneously injected into 6-week-old NOD/SCID mice. Three months after injection, tumors were collected for subsequent experiments. For the co-injection assays, 5 × 106 GSCs were mixed with 1 × 107 differentiated NSGCs (at day 15 and 10 after serum addition for GBM146 and GBM157, respectively) or senescent NSGCs (at day 30 after serum addition), and mixed with Matrigel (1:1). These cell suspensions were subcutaneously injected into 6-week-old NOD/SCID mice. After three month, the length (L) and width (W) of the tumor mass were measured, and the tumor volume (TV) was calculated using the equation: TV = (L × W2)/2.
2.6. Immunofluorescence staining
Cells were fixed with 2 or 4% paraformaldehyde and permeabilized with 0.5% Nonidet P-40. Xenograft tumors were embedded in the Optimal Cutting Temperature compound and frozen in liquid nitrogen. The tumor section was sliced and fixed with cold methanol/acetone. Immunofluorescence staining was performed as described [12] with the antibodies listed in Supplementary Materials and Methods.
2.7. Immunohistochemistry
Xenograft tumors were fixed in Mildform 10N (133–10311; Wako Pure Chemical Industries, Ltd., Osaka, Japan), embedded in paraffin, and sliced at 4–5 μm. Formalin-fixed, paraffin-embedded GBM tissue microarrays were obtained from ISU Abxis (A221V; Seoul, Korea) and US Biomax (GL806c; Rockville, MD, USA). Immunohistochemistry was performed as described [14] with the antibodies listed in Supplementary Materials and Methods.
2.8. Telomere and senescence assays
Population doubling assay, senescence-associated β-galactosidase (SA-β-gal) staining, telomere Southern blot analysis, and telomeric repeat amplification protocol (TRAP) assay were performed as described [12].
2.9. GeneChip microarrays
RNA was extracted, and GeneChip microarray analysis was performed as described [13]. The microarray data were deposited in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo) under accession number GSE74304.
2.10. ELISA
Cells were cultured for 5 days, and the culture supernatant was assayed with Human Quantikine ELISA kits for IL-6, CXCL8/IL-8, VEGF and VEGF-C (R&D Systems, Minneapolis, MN, USA).
2.11. HBMEC growth assay
HBMECs (ACBRI376; Cell Systems, Kirkland, WA, USA) were seeded in collagen-coated plates and cultured for 5 days either with the control or conditioned medium of senescent NSGCs, which had been cultured with 10% FBS-containing medium for 30 days. The cell number was counted at day 5 of the culture. Statistical evaluations were performed using the Welch t test. **, P < 0.001.
3. Results
3.1. GSCs lose their stemness and differentiate into NSGCs with telomerase repression upon serum exposure
Neurosphere cultures of GSC lines, GBM146 and GBM157, maintain the clonogenicity and possess tumor-propagating potential in vivo [11]. Consistent with a previous report [15], serum stimulation dispersed GSC neurospheres, rendering the cells adherent to the dish (Supplementary Fig. S1). These cells almost completely lost their stemness markers, such as CD133, SOX2, Nestin, and Olig2, and the sphere-forming potential by day 20 whereas GSC neurospheres maintained the properties (Supplementary Figs. S2–3). These observations confirm that GSCs lose their stemness, giving rise to NSGCs upon serum exposure.
NSGCs proliferated extensively for a week, but their growth rates decreased over passages, and the cells eventually ceased to proliferate (Fig. 1A). Most cancer cells become immortal by telomerase activation to maintain telomere length [16,17]. In GBM146 cells, telomerase activity was detected until day 3 after serum addition, but disappeared at day 10 (Fig. 1B). Consistent with this observation, the telomeres gradually shortened over passages for 35 days (Fig. 1C). The rate of shortening was about 70 bp/population doubling, which is equivalent to that in telomerase-negative cells [18]. These observations suggest that the end replication problem induces eventual growth arrest of NSGCs.
Fig. 1.
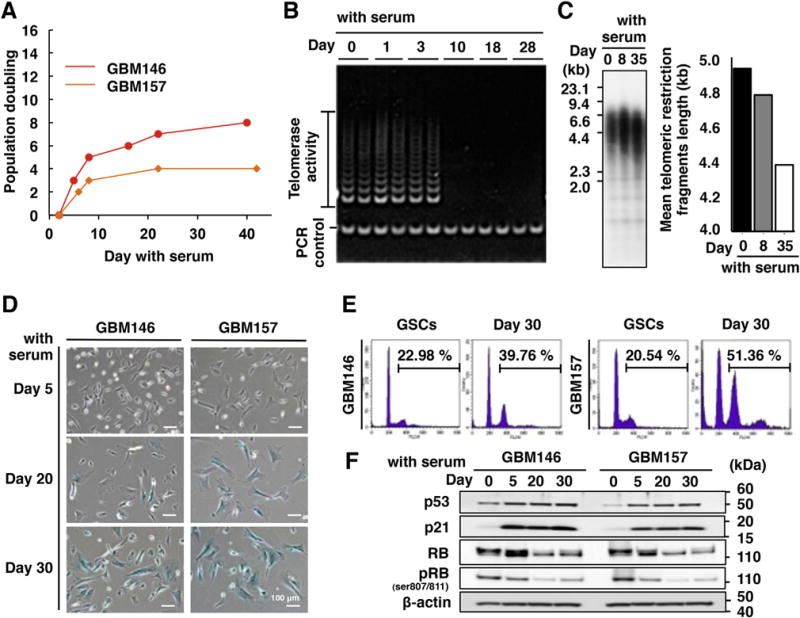
Non-stem glioma cells (NSGCs) undergo senescence with telomerase downregulation. (A) Growth curves of NSGCs. The cumulative number of population doublings in serum-containing medium was measured. (B) Telomerase activity in GBM146 cells. (C) Telomere Southern blot analysis of GBM146 cells. The signal indicates the telomere restriction fragment, which contains both telomeric and subtelomeric sequences. Thus, the graph on the right shows the sum of telomere and subtelomere (several kb) lengths. (D) SA-β-gal staining. Blue-colored cells are SA-β-gal-positive. (E) Propidium iodide staining. Value indicates the percentage of cells at ploidy larger than 4N. (F) Western blot analysis of cell cycle-inhibitory proteins. RB gave double bands of phosphorylated (upper, inactive) and dephosphorylated (lower, active) proteins. pRB means inactivated RB that is phosphorylated at Ser 807 and 811. β-actin was used as an internal control.
3.2. NSGCs undergo cellular senescence
We found that the arrested NSGCs appeared enlarged and flattened (Fig. 1D and Supplementary Fig. S1). These morphological changes are reminiscent of senescent cells [19]. Fig. 1D shows that GBM146 and GBM157 cells were negative for SA-β-gal, a senescence marker, at day 5 after serum addition. However, frequency of the stain-positive cells and staining intensity gradually increased, depending on the time of serum exposure. By day 30, most cells were positive for SA-β-gal, and the tetraploid cell fractions were increased, which is another biomarker of cellular senescence (Fig. 1E).
The cell cycle inhibitory pathways are essential for senescence induction [19]. p53 and its downstream effector, p21, were upregulated at day 5 after serum stimulation, which was maintained at later time points (Fig. 1F and Supplementary Fig. S4). Moreover, RB dephosphorylation, which activates its cell cycle inhibitory function, was detected at day 5, and further enhanced at day 20 and 30 (Fig. 1F). Another senescence biomarker, p16 [19], was not detected (Supplementary Fig. S5).
To exclude the possibility that senescence of NSGCs was caused by an artificial “culture shock” in the serum-containing medium, we exposed GBM157 GSCs to bone morphogenetic protein 4 (BMP4), which is often used for GSC differentiation [20]. BMP4 almost completely abolished the CD133-positive cells, and significantly reduced the levels of SOX2 and Olig2 (Supplementary Fig. S6A and B). Under these conditions, the cells eventually stopped dividing and became enlarged, flattened and positive for SA-β-gal (Supplementary Fig. S6C and D). Thus, NSGCs become senescent, and this phenomenon is an intrinsic property of NSGCs, not caused by acute culture shock.
3.3. Senescence markers in clinical GBM samples and mouse xenografts
We next investigated whether NSGCs in a senescent state reside in vivo. First, human GBM tissue microarrays consisting of patient-derived primary tumor sections were subjected to immunohistochemistry. Glial fibrillary acidic protein (GFAP), a differentiation marker of GSCs [3], was expressed in wide areas of the tumors of all GBM patients (Fig. 2A). Within those GFAP-positive areas, DNA damage response/senescence biomarkers, including formation of 53BP1 foci (Fig. 2A, inset), and accumulation of p53 and p21 proteins, were observed in 77.0% (23/30), 81.0% (17/21), and 71.4% (15/21) of the patients, respectively (Fig. 2A).
Fig. 2.
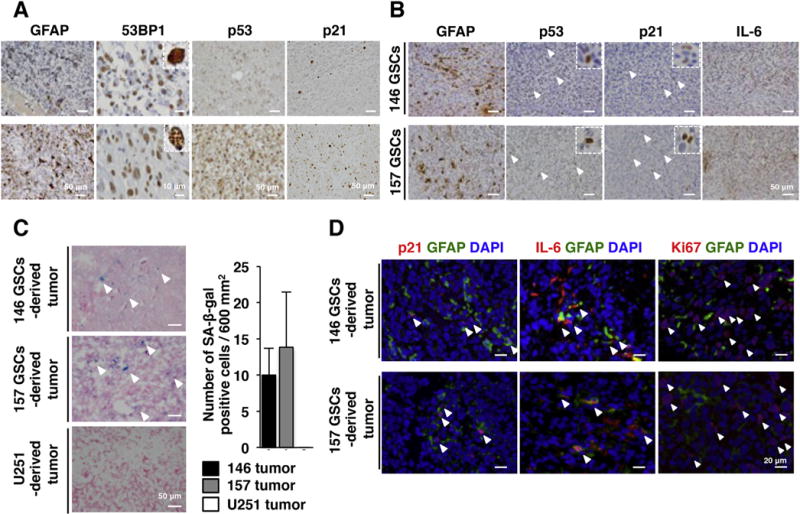
Senescent NSGCs in clinical and xenograft GBM in vivo. (A) Immunohistochemistry of human GBM tissue microarray (21–30 patient-derived tissue sections on each slide). Upper and lower panels represent the tissue sections with low and high frequencies of positive cells, respectively. In the case of 53BP1, presence of multiple nuclear foci (inset in the lower panel of 53BP1) was judged as marker-positive. (B) Immunohistochemistry of GSC-derived xenograft tumors. Each panel shows the same area derived from multiple slices of a tumor. Arrowheads indicate the antigen-positive cells. 146 GSCs and 157 GSCs mean xenograft tumors derived from GBM146 and GBM157 GSCs, respectively. (C) SA-β-gal staining of xenograft tumors. Arrowheads indicate SA-β-gal positive cells. (D) Immunofluorescence staining of GSC-derived tumors. Arrowheads indicate the p21+/GFAP+, IL-6+/GFAP+, and Ki67+/GFAP− cells, respectively. DNA was counterstained with DAPI (blue). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Xenograft tumors derived from GBM146 and GBM157 GSCs also contained GFAP, p53 and p21-positive cells (Fig. 2B). Furthermore, the inflammatory cytokine, interleukin-6 (IL-6), which is a major secreted factor in senescent cells [19], was detected in the tumors (Fig. 2B). These tumors contained SA-β-gal-positive cells, suggesting that accumulation of p53 and p21 proteins is not simply due to DNA damage response or quiescence (Fig. 2C). Meanwhile, xenograft tumors derived from U251, an established p53-deficient GBM cell line that had no hierarchy of stem/non-stem cells, did not contain SA-β-gal-positive cells. The senescence markers p21 and IL-6, but not a proliferation marker Ki67, were localized closely around the GFAP-expressing cells (Fig. 2D), implying that some NSGCs were senescent. These observations support that GBM-derived senescent cells exist in vivo.
3.4. Senescent NSGCs show an angiogenesis-related response at the transcriptional level
We next monitored the gene expression profile of senescent NSGCs. By using GeneChip microarrays, we identified 303 and 401 upregulated probes in the senescent NSGCs derived from GBM146 and GBM157 compared with GSCs and differentiated/pre-senescent NSGCs, respectively (Fig. 3A). Since senescent cells play an important role in cancer via secreting humoral factors [21], we focused on secretory proteins. Fig. 3B shows that vascular endothelial growth factor-C (VEGF-C), which controls angiogenesis and lymphangiogenesis, was significantly upregulated in the senescent NSGCs. Moreover, gene ontology analysis revealed that angiogenesis-related categories were enriched in the senescent NSGCs (Fig. 3C).
Fig. 3.
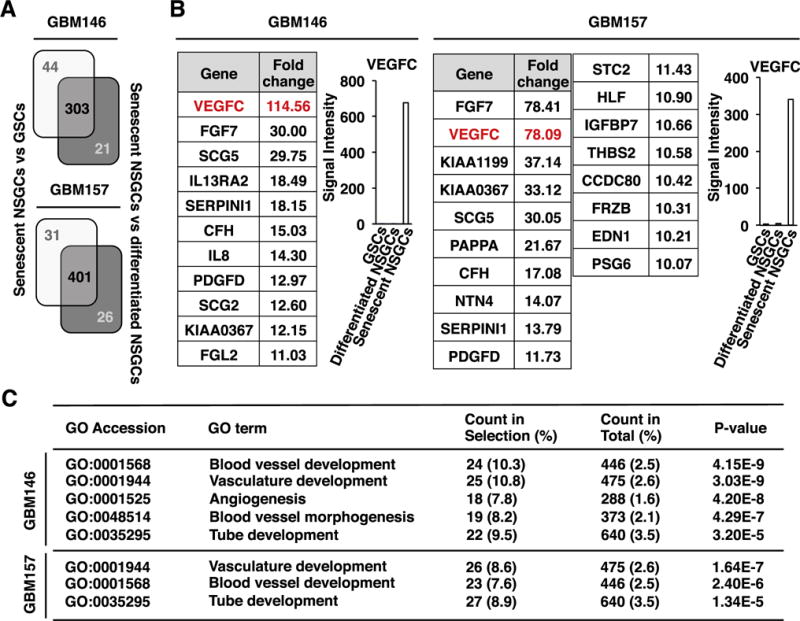
Pro-angiogenic gene expression in senescent NSGCs. (A) Gene expression profiles were compared between senescent NSGCs (day 30 after serum exposure) and GSCs, as well as between senescent NSGCs and differentiated/pre-senescent NSGCs (day 7 after serum exposure). Expression of 303 and 401 probes was upregulated >10-fold in the senescent GBM146 and GBM157 cells, respectively. (B) Signal intensities of the 10-fold-upregulated gene probes (only secretory protein-coding genes) in senescent NSGCs. Fold change indicates the ratio of signal intensity of senescent NSGCs to the average of GSCs and differentiated NSGCs. (C) Gene ontology (GO) analysis of the senescent NSGC-associated genes (P < 0.005, Fisher’s exact test).
3.5. Paracrine effects of senescent NSGCs promote vascular endothelial cell growth
While VEGF-C was undetected or barely detected in conditioned medium (CM) of serum-stimulated GSCs from day 0 and 5, its level was much higher at day 30 (Fig. 4A). The level of VEGF-A, another major contributor to angiogenesis [22], also increased (Fig. 4A).
Fig. 4.
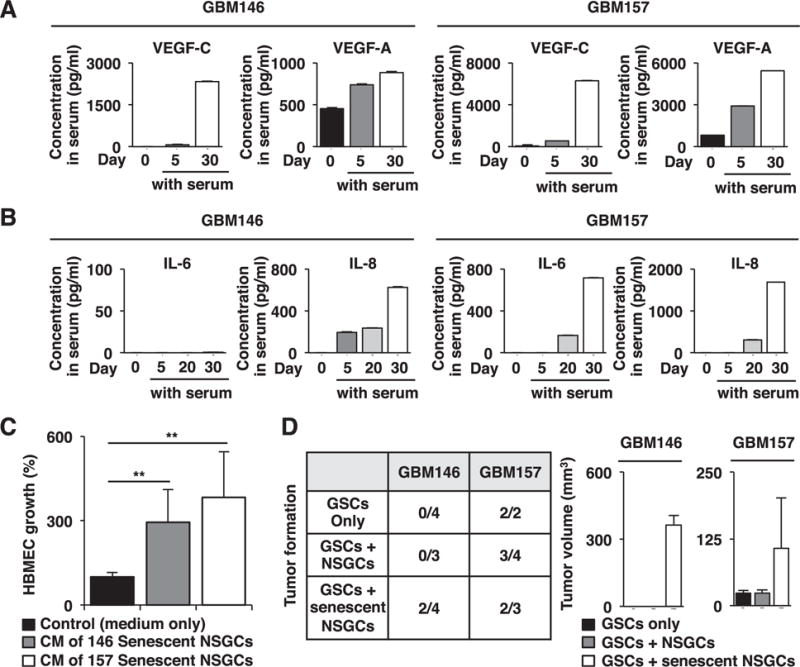
Senescent NSGCs enhance HBMEC growth and GSC-derived tumor formation. (A) ELISA of VEGFs in the conditioned medium (CM) of GBM146 and GBM157 cells precultured in serum-containing medium for the indicated time periods. For CM preparation, cells were cultured in serum-containing medium for 5 days, and the culture supernatants were collected. (B) ELISA of IL-6 and IL-8 in CM prepared as in A. (C) Promotion of HBMEC growth by the CM of senescent NSGCs (day 30 after serum exposure). Error bars, standard deviations. Statistical evaluations were performed using the Welch t test. **, P < 0.001. (D) Promotion of GSC-derived tumor formation by senescent but not differentiated NSGCs. GSCs with or without differentiated/senescent NSGCs were injected subcutaneously into NOD/SCID mice. After three months, tumor formation and its volume were evaluated.
Senescent cells release secretory proteins, termed senescence-associated secretory phenotype (SASP) factors that elicit autocrine/paracrine biological effects [21,23]. In GSCs, secretion of the major SASP components, IL-6 and IL-8 gradually increased by serum stimulation, and significant amounts were detected when the cells were fully senescent, although GBM146 did not secrete IL-6 (Fig. 4B). As SASP components, including IL-6 and IL-8, are known as angiogenesis inducers [21], senescent NSGCs may play a role in angiogenesis. In fact, growth of human brain microvascular endothelial cells (HBMEC) was increased 4–5 times by the CM of senescent NSGCs compared with the control (Fig. 4C).
3.6. Senescent NSGCs support the tumorigenic potential of GSCs
Senescent NSGCs lost tumorigenic potential when 2 × 107 GBM146 cells were subcutaneously injected into NOD/SCID mice (our unpublished observation). To examine the significance of NSGCs in vivo, we subcutaneously implanted GSCs into NOD/SCID mice with or without matched differentiated or senescent NSGCs (Fig. 4D). In the case of GBM146, GSCs alone or in combination with differentiated NSGCs did not form tumors under the present experimental conditions. By contrast, GSCs co-injected with senescent NSGCs formed tumors in two out of four mice. In the case of GBM157, GSCs alone or in combination with differentiated or senescent NSGCs formed tumors at comparable frequencies. Importantly, when co-injected with senescent NSGCs, the tumors grew much larger than those derived from GSCs alone or co-injected with differentiated NSGCs, suggesting that senescent NSGCs promote tumor formation of GSCs.
4. Discussion
We found that the serum-induced NSGCs eventually became senescent at least in part by telomerase downregulation and the resulting telomere shortening. In general, senescence occurs through the p16/RB and/or p53/p21 pathways [24,25]. Senescent NSGCs described here accumulated p53 and p21, but not p16, indicating that this senescence was triggered by the p53/p21 pathway and subsequent RB dephosphorylation. Because telomere dysfunction-induced replicative senescence is induced primarily by activation of the p53/p21 pathway [26,27], it is not surprising that the senescent NSGCs did not induce p16. Of note, loss or mutation of CDKN2A, which encodes p16, is detected in nearly 90% of GBM cases, whereas loss or mutation of TP53, which encodes p53, is only observed in 30% of cases [28–30]. This may reflect the larger dependence of NSGC senescence on p53 rather than p16. Our immunohistochemical analysis also indicated that more than 70% of GBM tumors exhibited p53 and p21 accumulation. Because p53 and p21 rather promptly accumulated upon serum stimulation (Fig. 1F), this pathway would also be involved in GSC differentiation.
Originally, cellular senescence is recognized as an important mechanism for tumor suppression [31–33]. However, recent studies have demonstrated that senescent cells can also accelerate tumor initiation and its malignant progression through a non-cell-autonomous effect of SASP [21,23]. In GBM, the most prominent SASP factors, IL-6 and IL-8, play an important role in tumor propagation by supporting GSC properties [34–36]. Consistently, IL-6 and IL-8 are detected in GBM primary tissues and are poor prognostic markers [37,38]. We found that GSC-derived tumors express IL-6, and the IL-6-expressing cells were colocalized with the differentiation marker, GFAP-positive cells in the tumors. Furthermore, the senescent NSGCs released IL-6 and IL-8 in vitro. Established GBM cell lines sometimes produce IL-6 and IL-8, but they have not been acknowledged as SASP factors. We propose a new model in which these cytokines are produced as SASP factors from senescent NSGCs and contribute to GBM malignancy.
We found that senescent NSGCs also secrete VEGF-A and –C. In GBM, VEGF-A is necessary for angiogenesis, but it also induces GSC differentiation to endothelial cells that can form tumor vasculatures [39,40]. Furthermore, VEGF-A promotes GSC viability and tumor growth, because the agonistic VEGF receptor 2 is preferentially expressed on GSCs [41,42]. VEGF-C is also expressed in GBM primary tumors [43,44]. Moreover, the VEGF-C receptor, VEGFR3, which is mainly expressed in lymphatic vessels, has been observed in blood endovascular cells [43] and can promote tumor angiogenesis [45,46]. Thus, it is likely that VEGF-C released from senescent NSGCs plays an important role in GBM angiogenesis. In clinical settings, bevacizumab, an anti-VEGF-A antibody, has not displayed effects on GBM as expected. One possibility is that VEGF-C compromises the effect of VEGF-A blockade. In fact, bevacizumab induces not only VEGF-C expression in GBM cells, but also VEGF-C reactivity of tumor-associated endothelial cells [47]. Moreover the presence of lymphatic vessel in central nerve systems of brain has been recently identified [48], suggesting that VEGF-C may promote both angiogenesis and lymphangiogenesis in GBM. For these reasons, we consider that both VEGF-A and –C released from senescent NSGCs contribute to GBM malignancy.
Recent studies have demonstrated that NSGCs can be reprogrammed to resume the cancer stemness [49]. Furthermore, as shown in this study, NSGCs undergo senescence and have a paracrine effect on the GSC-derived tumor formation. We propose that NSGCs hold pathological significance in GBM malignancy in a bimodal manner, one by replenishing the tumor-propagating stem cells and the other by eliciting a senescence-mediated perturbation of the tumor microenvironment. This study sheds light on the biological significance of NSGCs in GBM and may help develop a therapeutic intervention against this fatal disease.
Supplementary Material
Acknowledgments
We thank Haruka Yoshida for technical assistance with immunohistochemistry, preparation of tumor sections, and care of mice. We also thank Tetsuo Mashima and laboratory members for critical comments and valuable discussions. R.O. is Research Fellow (DC2) of the Japan Society for the Promotion of Science (JSPS). This work was supported in part by KAKENHI from JSPS to R.O. (14J08396) and H.S. (25290060), and by R01NS083767, R01NS087913 and P01CA163205 to I.N.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.bbrc.2016.01.071.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Transparency document
Transparency document related to this article can be found online at http://dx.doi.org/10.1016/j.bbrc.2016.01.071.
References
- 1.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, et al. Dirks, identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 4.Cheng L, Bao S, Rich JN. Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol. 2010;80:654–665. doi: 10.1016/j.bcp.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaffer CL, Brueckmann I, Scheel C, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long H, Xiang T, Qi W, et al. CD133þ ovarian cancer stem-like cells promote non-stem cancer cell metastasis via CCL5 induced epithelial-mesenchymal transition. Oncotarget. 2015;6:5846–5859. doi: 10.18632/oncotarget.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celia-Terrassa T, Meca-Cortes O, Mateo F, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122:1849–1868. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateo F, Meca-Cortes O, Celia-Terrassa T, et al. SPARC mediates metastatic cooperation between CSC and non-CSC prostate cancer cell subpopulations. Mol Cancer. 2014;13:237. doi: 10.1186/1476-4598-13-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyazaki T, Pan Y, Joshi K, et al. Telomestatin impairs glioma stem cell survival and growth through the disruption of telomeric G-quadruplex and inhibition of the proto-oncogene, c-Myb. Clin Cancer Res. 2012;18:1268–1280. doi: 10.1158/1078-0432.CCR-11-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakano I, Joshi K, Visnyei K, et al. Siomycin A targets brain tumor stem cells partially through a MELK-mediated pathway. Neuro Oncol. 2011;13:622–634. doi: 10.1093/neuonc/nor023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seimiya H, Muramatsu Y, Ohishi T, et al. Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell. 2005;7:25–37. doi: 10.1016/j.ccr.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 13.Hirashima K, Seimiya H. Telomeric repeat-containing RNA/G-quadruplex-forming sequences cause genome-wide alteration of gene expression in human cancer cells in vivo. Nucleic Acids Res. 2015;43:2022–2032. doi: 10.1093/nar/gkv063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirashima K, Migita T, Sato S, et al. Telomere length influences cancer cell differentiation in vivo. Mol Cell Biol. 2013;33:2988–2995. doi: 10.1128/MCB.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 16.Shay JW, Wright WE. Telomerase activity in human cancer. Curr Opin Oncol. 1996;8:66–71. doi: 10.1097/00001622-199601000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Holt SE, Shay JW. Role of telomerase in cellular proliferation and cancer. J Cell Physiol. 1999;180:10–18. doi: 10.1002/(SICI)1097-4652(199907)180:1<10::AID-JCP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 18.Huffman KE, Levene SD, Tesmer VM, et al. Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J Biol Chem. 2000;275:19719–19722. doi: 10.1074/jbc.M002843200. [DOI] [PubMed] [Google Scholar]
- 19.Kuilman T, Michaloglou C, Mooi WJ, et al. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 21.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coppe JP, Patil CK, Rodier F, et al. Campisi, Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Beausejour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herbig U, Jobling WA, Chen BP, et al. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 28.Ohgaki H, Schauble B, zur Hausen A, et al. Genetic alterations associated with the evolution and progression of astrocytic brain tumours. Virchows Arch. 1995;427:113–118. doi: 10.1007/BF00196514. [DOI] [PubMed] [Google Scholar]
- 29.Bogler O, Huang HJ, Kleihues P, et al. The p53 gene and its role in human brain tumors. Glia. 1995;15:308–327. doi: 10.1002/glia.440150311. [DOI] [PubMed] [Google Scholar]
- 30.Ichimura K, Schmidt EE, Goike HM, et al. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–1072. [PubMed] [Google Scholar]
- 31.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 32.Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 33.Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11:461–469. doi: 10.1016/j.ccr.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiou GY, Chien CS, Wang ML, et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol Cell. 2013;52:693–706. doi: 10.1016/j.molcel.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Jin X, Kim SH, Jeon HM, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and Notch signalling. Brain. 2012;135:1055–1069. doi: 10.1093/brain/aws028. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Lathia JD, Wu Q, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27:2393–2404. doi: 10.1002/stem.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salmaggi A, Eoli M, Frigerio S, et al. Intracavitary VEGF, bFGF, IL-8, IL-12 levels in primary and recurrent malignant glioma. J Neurooncol. 2003;62:297–303. doi: 10.1023/a:1023367223575. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki A, Ishiuchi S, Kanda T, et al. Analysis of interleukin-6 gene expression in primary human gliomas, glioblastoma xenografts, and glioblastoma cell lines. Brain Tumor Pathol. 2001;18:13–21. doi: 10.1007/BF02478920. [DOI] [PubMed] [Google Scholar]
- 39.Ricci-Vitiani L, Pallini R, Biffoni M, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 40.Wang R, Chadalavada K, Wilshire J, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 41.Hamerlik P, Lathia JD, Rasmussen R, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209:507–520. doi: 10.1084/jem.20111424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu C, Wu X, Zhu J. VEGF promotes proliferation of human glioblastoma multiforme stem-like cells through VEGF receptor 2. ScientificWorldJournal. 2013;2013:417413. doi: 10.1155/2013/417413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jenny B, Harrison JA, Baetens D, et al. Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. J Pathol. 2006;209:34–43. doi: 10.1002/path.1943. [DOI] [PubMed] [Google Scholar]
- 44.Xu Y, Zhong Z, Yuan J, et al. Collaborative overexpression of matrix metalloproteinase-1 and vascular endothelial growth factor-C predicts adverse prognosis in patients with gliomas. Cancer Epidemiol. 2013;37:697–702. doi: 10.1016/j.canep.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Tammela T, Zarkada G, Wallgard E, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 46.Tvorogov D, Anisimov A, Zheng W, et al. Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding and receptor dimerization. Cancer Cell. 2010;18:630–640. doi: 10.1016/j.ccr.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Grau S, Thorsteinsdottir J, von Baumgarten L, et al. Bevacizumab can induce reactivity to VEGF-C and -D in human brain and tumour derived endothelial cells. J Neurooncol. 2011;104:103–112. doi: 10.1007/s11060-010-0480-6. [DOI] [PubMed] [Google Scholar]
- 48.Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flavahan WA, Wu Q, Hitomi M, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373–1382. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.