

Summary
Although poly (ADP-ribose) polymerase (PARP) inhibitors are active in homologous recombination (HR)-deficient cancers, their utility is limited by acquired resistance following restoration of HR. Here, we report that dinaciclib, an inhibitor of cyclin-dependent kinases (CDKs) 1, 2, 5 and 9, additionally has potent activity against CDK12, a transcriptional regulator of HR. In BRCA-mutated triple-negative breast cancer (TNBC) cells and patient-derived xenografts (PDXs), dinaciclib ablates restored HR and reverses PARP inhibitor resistance. Additionally, we show that de novo resistance to PARP inhibition in BRCA1-mutated cell lines and a PDX derived from a PARP inhibitor-naïve BRCA1 carrier is mediated by residual HR and is reversed by CDK12 inhibition. Finally, dinaciclib augments the degree of response in a PARP inhibitor-sensitive model, converting tumor growth inhibition to durable regression. These results highlight the significance of HR disruption as a therapeutic strategy and support the broad use of combined CDK12 and PARP inhibition in TNBC.
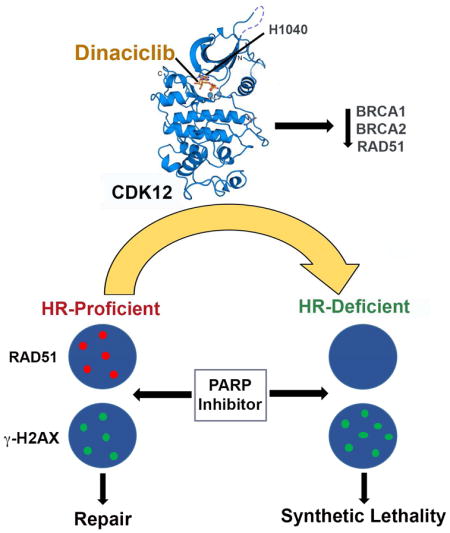
Graphical abstract
Introduction
Poly ADP(ribose) polymerase (PARP) inhibition has emerged as a compelling strategy for BRCA- or otherwise homologous recombination (HR) repair-deficient cancers (Scott et al., 2015). However, the broad utility of these drugs has been limited by their lack of activity in HR-proficient cancers, as well as acquired resistance of initially responding tumors, often mediated by restoration of HR (Bouwman and Jonkers, 2014). Additionally, a proportion of BRCA-mutated cancers display de novo (primary) resistance, potentially mediated by hypomorphic isoforms of BRCA1 (Hill et al., 2014), tumor heterozygosity (King et al., 2007), or preexisting alterations in the DNA damage response that may confer residual HR activity (Bouwman et al., 2010).
These challenges have prompted interest in combining PARP inhibitors with agents capable of disrupting HR in cancer cells as an approach to sensitize BRCA-wild type cancers to PARP inhibition, and also to overcome de novo and acquired resistance in BRCA-mutated cancers. Because complex mechanisms of HR restoration confer resistance to PARP inhibitors in BRCA-mutated cells, simultaneous suppression of multiple HR genes together with PARP inhibition may be a preferred strategy for resensitizing resistant cells to these agents. In this regard, cyclin-dependent kinase (CDK)12, an RNA polymerase II C-terminal domain (CTD) kinase, has recently been identified as an essential regulator for the transcription of various DNA damage response (DDR) and DNA repair genes, particularly those involved in the HR and Fanconi anemia (FA) pathways (Bartkowiak et al., 2010; Blazek et al., 2011; Liang et al., 2015). Somatic inactivating mutations in CDK12 have been observed in a subset of epithelial ovarian carcinomas, resulting in compromised HR (Joshi et al., 2014). Furthermore, shRNA-mediated depletion of CDK12, or its cyclin K binding partner, from BRCA- and CDK12-wild type ovarian cancer or other transformed cell lines has been shown to suppress HR gene expression and sensitize cells to cisplatin-induced interstrand cross-links and PARP inhibition (Bajrami et al., 2014; Blazek et al., 2011; Joshi et al., 2014).
These observations have led to interest in the development of pharmacological inhibitors of CDK12 to act as sensitizers to PARP inhibitors, as well as to standard DNA damaging agents. Here, we show that dinaciclib, a known inhibitor of CDKs 1, 2, 5 and 9 (Parry et al., 2010) that has produced documented responses in breast cancer (Mita et al., 2014) has previously unreported potent activity against CDK12. We studied dinaciclib as a CDK12 inhibitor in models of triple-negative breast cancer (TNBC), an aggressive breast cancer subset associated with poor outcome and absence of defined molecular targets. Dinaciclib reduces HR gene expression in BRCA-wild type TNBC cells and sensitizes these cells to PARP inhibition. We have further investigated the activity of dinaciclib in concert with PARP inhibition in BRCA-mutated TNBC cell lines and patient-derived xenograft (PDX) models and demonstrate reversal of de novo and acquired PARP inhibitor resistance. Finally, in a BRCA-mutated model in which long-term tumor growth control is achieved by PARP inhibitor monotherapy, the addition of dinaciclib converts the outcome to deep and prolonged tumor regression. Collectively, these data support the combination of dinaciclib with PARP inhibition in both BRCA-wild type and -mutant TNBCs.
Results
Dinaciclib inhibits CDK12 with greater potency than other known transcriptional CDK inhibitors
To identify potential inhibitors of CDK12, we made use of its recently elucidated crystal structure. Although the kinase domain of CDK12 shares significant primary sequence homology with CDK9, a panel of small-molecule CDK9 inhibitors was previously shown to have substantially reduced potency against CDK12 in in vitro biochemical assays (Bosken et al., 2014). To further interrogate this result, we aligned the CDK12 crystal structure 4NST with the CDK9 crystal structure 3BLQ (Baumli et al., 2008). While the two kinases share extensive tertiary structural homology (RMSD= 0.83 Å; Figure 1A), inspection of secondary structure elements demonstrated a variance in the C-terminal portion of each kinase domain (Figures 1B and S1A). CDKs that regulate transcriptional elongation have a unique extension helix that lies C-terminal to the canonical CDK kinase domain. In CDK12, this extension helix interacts with the ATP binding site and is initiated by a DCHEL motif beginning at amino acid 1038. The interaction of the C-terminal extension helix with the nucleotide binding site of CDK12 is mediated by the H1040 and E1041 residues, and loss of the helix severely disrupts activity of the kinase (Bosken et al., 2014). CDK9 shares a similar C-terminal extension helix, but does not share the initiating 1038DCHEL motif (Figure 1B). Since this structural variation occurs in close proximity to the site of binding for small molecule inhibitors of CDK9, we hypothesized that it may be responsible for the lack of shared specificity with CDK12. In silico modeling of flavopiridol, a well-described potent CDK9 inhibitor, into the ATP-binding site of CDK12 revealed a significant steric clash between the benzene ring of bound flavopiridol and the H1040 residue of the DCHEL motif of CDK12. To determine if this occlusion was a shared feature of other compounds that tightly bind CDK9, we modeled dinaciclib, a CDK9 inhibitor that had not been tested against CDK12, into the CDK12 ATP-binding site. In contrast to flavopiridol, there does not appear to be steric hindrance between the CDK12 H1040 aromatic ring and the pyridine-N-oxide ring of dinaciclib (Figure 1B).
Figure 1. Dinaciclib is a potent inhibitor of CDK12 in addition to CDK9.

(A) Tertiary structural alignment of CDK9 and CDK12. (B) Sequence corresponding to the variance in the C-terminal extension helix of the kinase domains of CDK12 and CDK9 (see also Figure S1), as well as structural modeling of the orientations of flavopiridol and dinaciclib in relation to H1040 and E1041 of the CDK12 ATP binding site. The benzene ring of flavopiridol shows a steric clash with H1040 of Cdk12, whereas the pyridine-N-oxide ring of dinaciclib overlaps the aromatic H1040 side chain, resulting in a possible stacking of the aromatic ring systems that stabilizes the interaction and contributes to binding specificity. (C) In vitro kinase assays using pS7-CTD[3] as substrate and 0.2 μM cyclin T-CDK9 and cyclin K-CDK12 holoenzyme complexes alone or with and 10x or 1000x dinaciclib. (D) Concentration series of dinaciclib and flavopiridol for cyclin T1-CDK9 and cyclin K-CDK12 at 0.2 μM kinase concentration. The IC50 values against Cdk9 and CDK12 are comparable for dinaciclib but disparate for flavopiridol. Introduction of the indicated mutations sensitizes CDK12 to flavopiridol. All data are reported as the mean ± SD from three independent experiments.
We predicted that this favorable interaction would afford potent CDK12 inhibitory activity to dinaciclib. The addition of 10x or 1000x concentration of dinaciclib to 0.2 μM cyclin K-CDK12 or cyclin T-CDK9 holoenzyme complexes reduced CDK12 activity by approximately 20-fold and CDK9 activity by 12–25-fold (Figure 1C). Compared to previously reported results of similar assays using other CDK9 inhibitors (Bosken et al., 2014), dinaciclib demonstrates strong inhibition of CDK12 kinase activity. Concentration series were then performed to determine IC50 values against CDK12 and other CDK family members (Figures 1D and S1B). While flavopiridol had only modest activity against CDK12 with potency compared to CDK9 reduced by more than 10-fold (Bosken et al., 2014), dinaciclib demonstrated robust inhibitory activity against both kinases, with IC50’s in the 40–60 nM range, making it the most potent known inhibitor of CDK12. Furthermore, mutation of the H1040 site to glycine, or mutation of either the DCHEL motif or the adjacent polybasic region to alanine, conferred sensitivity of CDK12 to flavopiridol, consistent with the predictions of structural modeling. In contrast, these three CDK12 mutations had no effect on the IC50 of dinaciclib (Figure 1D).
Dinaciclib displays hallmarks of CDK12 inhibition in BRCA wild type TNBC cells
We next characterized the transcriptional effects of dinaciclib treatment on TNBC cells. Eukaryotic gene transcription is regulated by a coordinated sequence of phosphorylation events along the CTD of RNA Polymerase II. CDK9 is recruited to the 5’ ends of gene bodies where it primarily phosphorylates CTD-Ser5, releasing the assembled transcription complex from promoter-proximal pausing and initiating transcription (Eick and Geyer, 2013; Ghamari et al., 2013). CDK12 is predominantly associated with the 3’ ends of genes, where it has been shown to coordinate transcript elongation and processing largely by phosphorylation of CTD-Ser2 (Bartkowiak et al., 2010; Blazek et al., 2011; Eick and Geyer, 2013). Treatment of MDA-MB-231 cells with low nanomolar concentrations of dinaciclib for 6 hours resulted in concentration-dependent reduction in phospho-CTD levels, with greater effects on Ser2 compared to Ser5 phosphorylation (Figure 2A).
Figure 2. Dinaciclib is a transcriptional CDK inhibitor that reduces expression of genes in DNA damage response and DNA repair pathways.

(A) MDA-MB-231 cells were treated with the indicated concentrations of dinaciclib for 6 hrs, demonstrating reduced phosphorylation at the Ser2 and Ser5 sites of the CTD of RNA pol II. (B) Cells were collected before and after treatment with 10 nM dinaciclib for 12 hrs and changes in transcription were measured using the Affymetrix HG-U133A2 arrays. Analyses were performed in triplicate. 21% of genes were significantly downregulated in dinaciclib-treated vs. untreated samples (P < 0.05). (C) Genes statistically significantly downregulated in response to dinaciclib were analyzed by Ingenuity Pathway Analysis (IPA) software, demonstrating downregulation of DNA damage response and DNA repair pathways. (D) Expression of genes in the “Role of BRCA1 in DNA Damage Response” pathway. (E) Downregulation of expression of BRCA1 and RAD51 mRNAs in cells treated with the indicated concentrations of dinaciclib was confirmed utilizing RT-PCR. (F) Concentration-dependent reduction in expression of BRCA1, BRCA2, RAD51 and FANCD2 in cells treated with dinaciclib for 24 hrs. (G) Time-dependent reduction in expression of BRCA1 and RAD51 in response to dinaciclib. (H) Cell cycle patterns following dinaciclib exposure demonstrate the absence of G1 arrest in MDA-MB-231 cells (see also Figure S2).
While CDK9-mediated phosphorylation of RNA Pol II occurs globally across transcripts (Garriga and Grana, 2004), CDK12 predominantly associates with the 3’ ends of genes involved with DNA damage and repair (Blazek et al., 2011). Gene expression analysis of RNA collected from MDA-MB-231 cells after 12 hours of dinaciclib exposure showed a significant reduction in expression of only a limited number of genes, in contrast to the global transcriptional repression that has been reported with potent CDK9 inhibitors (Lam et al., 2001) (Figure 2B). Pathway analysis revealed that the differentially expressed genes were significantly enriched for those involved in HR repair and DNA damage sensing (Figures 2C and S2A), with representation from multiple genes previously reported to be repressed by disruption of CDK12 activity (Blazek et al., 2011) (Figure 2D). We confirmed these results via qPCR using primers for BRCA1 and RAD51 (Figure 2E). Consequently, the expression of multiple proteins involved in HR was decreased in dinaciclib-treated cells, demonstrated in both concentration- and time-dependent experiments, with substantial reduction of these proteins by 24 hours (Figures 2F and 2G). Importantly, the transcriptional effects of dinaciclib could not be attributed to a block in cell cycle progression, as we observed only minimal cell cycle perturbations in asynchronous or hydroxyurea-synchronized cells (Figures 2H and S2B). Taken together, these data suggest that dinaciclib acts primarily as a transcriptional CDK inhibitor in TNBC cells, and that the transcriptional consequences of dinaciclib exposure are predominantly associated with its inhibition of CDK12.
Dinaciclib compromises HR repair and sensitizes BRCA wild type TNBC cells to PARP inhibition
We reasoned that the transcriptional effects of dinaciclib we observed would severely impair HR, as reported in multiple myeloma cells (Alagpulinsa et al., 2016). To test this prediction, we assessed functional metrics of HR in BRCA wild-type TNBC cells. Irradiated MDA-MB-231 cells pretreated with dinaciclib showed significant concentration-dependent reduction in the recruitment of BRCA1 and RAD51 to sites of double-strand DNA breaks (Figure 3A and B). To directly measure HR, we utilized U2OS cells with stable integration of the DR-GFP reporter. Transfection of the I-SceI restriction enzyme resulted in 13.3% and 3.6% GFP-positive cells following vehicle or dinaciclib treatment, respectively (Figures 3C and S3A). The profound disruption of HR suggested that dinaciclib could sensitize HR-proficient cell to PARP inhibition. We found that dinaciclib treatment sensitized a panel of BRCA wild-type TNBC cell lines to the PARP inhibitor veliparib (Figures 3D and S3B). In the presence of dinaciclib, the IC50 to veliparib was reduced between 2.5 and 12.5-fold (Table S1).
Figure 3. Disruption of HR by dinaciclib and sensitization to PARP inhibition of BRCA wild type and BRCA-mutated cells with acquired PARP inhibitor resistance.

(A) Cells were treated with vehicle or 10 nM dinaciclib for 18 hrs prior to treatment with 10 Gy γ-irradiation (IR). Six hrs post-IR, cells were analyzed for BRCA1, RAD51 and γ-H2AX focus formation. (B) Quantification of cells with > 5 foci in irradiated cells pretreated with vehicle or dinaciclib at the indicated concentrations. (C) USOS DR-GFP cells were transfected I-SceI in the presence of vehicle or 15 nM dinaciclib. The percentage of GFP-positive cells is significantly reduced (P < 0.0001) in the presence of dinaciclib, consistent with direct inhibition of HR repair (See also Figure S3). (D) BRCA-proficient TNBC cell lines were treated with veliparib in the absence and presence of dinaciclib, demonstrating reduced IC50 values in the presence of dinaciclib. (E) (Left) An MDA-MB-436 PARP inhibitor-resistant derivative (MDA-MB-436-RR2) (Johnson et al., 2013) was treated with veliparib in the absence or presence of dinaciclib, demonstrating reduced IC50 in the presence of dinaciclib. (Middle) Reduced expression of the mutant BRCA1 protein and RAD51 in response to dinaciclib. (Right) MDA-MB-436-RR2 cells were treated with vehicle or dinaciclib at the indicated concentration for 18 hours, subjected to 10 Gy IR and assessed for RAD51 focus formation 6 hours later. (P < 0.0001) (F) Treatment history of the BRCA2 carrier; PDX 12-58 was procured after progression on cisplatin and olaparib (see also Figure S4A) . SD, stable disease. (G) (Left) Mice bearing 12–58 xenografts were treated with vehicle (n = 3) or cisplatin (n = 3) on days 1 and 22 (arrows), with tumor volume measured over 36 days. (Right) Mice were treated with vehicle (n = 4), veliparib (n = 8), dinaciclib (n = 8) or the combination (n = 8). Combination treatment produced significant tumor growth inhibition at day 42 compared to vehicle (P < 0.0001) or monotherapies (P < 0.0001 for both veliparib and dinaciclib). * P < 0.01, ** P < 0.001, ***P < 0.0001 for experimental value vs. control.
Effects of dinaciclib in TNBC cells are phenocopied by CDK12 knockout
To provide further evidence that the effects of dinaciclib are mediated by CDK12 inhibition, we utilized CRISPR/Cas9-mediated knockout of CDK12 in MDA-MB-468 and BT549 cells (Figure S4A). Knockout of CDK12 caused reduced expression of BRCA1, BRCA2 and RAD51, compromised RAD51 focus formation after γ-irradiation and substantial sensitization to veliparib. Importantly, treatment with dinaciclib did not further sensitize CDK12-depleted cells to veliparib. CDK9 knockout did not reduce HR gene expression (Figure S4A). Consistent with previously published results, CDK9 knockout over several days was lethal to TNBC cells (Wang et al., 2015). We therefore used low concentrations of flavopiridol (Figure S4B), which reduced phosphorylation of Ser5 but not Ser2 of the CTD, and observed no impact on HR gene expression or RAD51 focus formation after DNA damage. In contrast to dinaciclib or CDK12 knockout, flavopiridol did not sensitize TNBC cells to veliparib or olaparib-mediated PARP inhibition.
BRCA mutant TNBC cells with acquired PARP inhibitor resistance are resensitized to PARP inhibition by dinaciclib
Many mechanisms of acquired PARP inhibitor resistance have shared a common feature in that they restore RAD51-loading and rescue HR repair. We hypothesized that the multifocal disruption of HR resulting from CDK12 inhibition could potentially resensitize BRCA-mutated cells that have developed resistance to PARP inhibition. We made use of a previously generated PARP inhibitor-resistant clone of the BRCA1-mutated MDA-MB-436 cell line, in which heterozygous mutation of the TP53BP1 gene and stabilization of a hypomorphic BRCT domain-mutated BRCA1 protein results in rescue of DNA end resection, RAD51 loading and HR (Johnson et al., 2013). Dinaciclib treatment substantially reduced protein levels of both RAD51 and the hypomorphic BRCA1 mutant protein, as well as formation of RAD51 foci following irradiation and resensitized the resistant cells to PARP inhibition (Figure 3E and Table S1).
To test the ability of dinaciclib to reverse acquired PARP inhibitor resistance in vivo, we generated a PDX model derived from a TNBC patient carrying a germline S1970* BRCA2 mutation. This heavily pretreated patient achieved stable disease for approximately 10 months on combined cisplatin/olaparib followed by olaparib alone (Balmana et al., 2014), prior to disease progression. After brief intervening chemotherapy, a biopsy was performed when new hepatic metastases developed, used for establishment of the PDX 12–58 model (Figure 3F) (Tao et al., 2014). Although targeted sequencing did not demonstrate evidence of a BRCA2 reversion mutation (Figure S5A), the model was refractory to cisplatin as well as veliparib (Figure 3G and H), requiring animal euthanasia at approximately 40 days for progressive tumor growth, suggesting alternative mechanisms governing resistance. However, the combination of dinaciclib and veliparib resulted in tumor growth inhibition lasting at least 60 days (Figure 3H). End-of-experiment histology revealed no abnormalities in lung, liver, GI tract and bone marrow of combination-treated mice, with similar appearance of organs harvested from vehicle-treated mice and only modest staining for γ-H2AX in marrow (Figure S5B).
To further study the selectivity of combination treatment for transformed cells, human mammary epithelial cells (HMECs) were exposed to olaparib in the absence or presence of dinaciclib (Figure S5C). In contrast to transformed cells, dinaciclib improved the viability of HMECs treated with olaparib; this protective effect was likely due to the much greater degree of G2 arrest observed, which should preclude PARP inhibitor-mediated cytotoxicity that typically occurs in S phase.
Characterization of BRCA1 mutated TNBC cells with de novo (primary) PARP inhibitor resistance
In addition to acquired PARP inhibitor resistance, there is a high rate of de novo resistance to PARP inhibition in BRCA-mutated tumors. To address the utility of CDK12 inhibition in this setting, we first identified and characterized BRCA-mutated cell lines with primary PARP inhibitor resistance. Relative PARP inhibitor sensitivity was determined for a panel of BRCA1-mutated TNBC cell lines using hormone receptor-positive and non-transformed breast cell lines as a reference standard for insensitivity to PARP inhibition. While MDA-MB-436 and HCC1395 displayed exquisite sensitivity to PARP inhibition, SUM149 and HCC1937 were relatively insensitive to PARP inhibitor treatment, either with veliparib or olaparib (Figures 4A and S6A). To determine whether the variability in sensitivity to PARP inhibition was due to differences in susceptibility to apoptosis, we performed mitochondrial BH3 profiling on the BRCA-mutated cell lines. No significant differences were observed (Figure S7), suggesting that resistance to PARP inhibition in SUM149PT and HCC1937 was not due to an anti-apoptotic phenotype.
Figure 4. Characterization of BRCA1-mutated TNBC cell lines.

(A) Panel of indicated cell lines was treated with the indicated concentrations of veliparib or cisplatin and viability assessed after 7 days of treatment. (B) Cells were treated with vehicle, veliparib or mitomycin C and metaphase spreads were prepared; radials quantified in vehicle and drug-treated cells. (C) Cells were treated with vehicle, veliparib or cisplatin and analyzed by immunofluorescence for RAD51 and FANCD2 foci. Graphs show quantification of cells with > 5 foci in vehicle and drug-treated cells.
In addition to PARP inhibition, HR-deficient tumors are sensitive to the accumulation of DNA interstrand crosslinks (ICLs). To our surprise, all four BRCA1-mutated cell lines displayed marked sensitivity to cisplatin, regardless of PARP inhibitor sensitivity (Figure 4A). Furthermore, we observed that treatment with another DNA crosslinking agent, mitomycin C, resulted in accumulation of chromosomal aberrations in both PARP inhibitor-resistant and -sensitive BRCA1-mutated lines, whereas veliparib only produced chromosomal aberrations in MDA-MB-436 and HCC1395 cells (Figure 4B).
While sensitivity to PARP inhibition is associated with defects in HR, the repair of ICLs requires the activity of multiple DNA repair pathways, including nucleotide excision repair (NER) and the Fanconi Anemia (FA) pathway, in addition to HR. BRCA1 function is essential for both HR and the FA pathway, and BRCA1-deficient tumors have been observed to also carry NER defects. We hypothesized that the BRCA1-mutated cell lines SUM149PT and HCC1937 may have selectively retained functional HR while maintaining a defect in NER, as described for a subset of BRCA1-mutated ovarian carcinomas (Ceccaldi et al., 2015), or the FA pathway, as in Brca1−/− 53BP1−/− mouse embryonic fibroblasts (MEFs) that display resistance to PARP inhibition but sensitivity to crosslinking agents (Bunting et al., 2012).
We first assessed NER-proficiency in SUM149 and HCC1937and failed to detect an NER defect in either cell line (Figure S6B). We next determined HR and FA pathway proficiency by monitoring the recruitment of repair factors immediately downstream of BRCA1 in both pathways. Recent work has demonstrated that in addition to its role in RAD51 loading following end resection in HR, BRCA1 is required for the removal of stalled replication machinery and subsequent recruitment of the FA complex to the site of crosslinks (Schlacher et al., 2012). We therefore measured RAD51 and FANCD2 foci formation as surrogate markers for repair activity downstream of the role of BRCA1 in the HR and FA pathways, respectively (Figure 4D). Following PARP inhibitor treatment, a significant increase in RAD51 foci was observed in SUM149PT and HCC1937, suggesting the presence of functional HR. Strikingly, none of the BRCA1-mutated lines displayed recruitment of FANCD2 foci following cisplatin treatment. These data suggest that similar to the phenotype observed in MEF genetic studies (Bunting et al., 2012), human pathogenic mutations in BRCA1 may affect separate DNA repair pathways to varying extents.
SUM149PT and HCC1937 cells require BRCA proteins for HR, which can be depleted by dinaciclib
We next sought to determine pathway components necessary for the residual HR function of SUM149PT and HCC1937 cells and if removal of these factors would result in sensitization to PARP inhibition. SUM149PT cells carry a 2288delT mutation in exon 11 of BRCA1, resulting in loss of the full-length p220 isoform of BRCA1 but detectable levels of a truncated BRCA1Δ672-4095 isoform, BRCA1Δ11b, produced from an in-frame splicing event that removes exon 11 (Hill et al., 2014). We hypothesized that the BRCA1Δ11b isoform, which retains the C-terminal BRCT domains necessary for RAD51 loading, may facilitate HR and confer PARP inhibitor resistance. siRNA-mediated depletion of BRCA1 Δ11b resulted in sensitization of SUM149PT to PARP inhibition (Figure 5A). Additionally, we observed that siRNA targeting of either BRCA2 or PALB2 also sensitized SUM149PT to PARP inhibition, suggesting that the 11 isoform functions in place of p220 in the BRCA1-PALB2-BRCA2 axis. While the 5382insC BRCA1 mutation in HCC1937 cells ablates expression of both p220 and Δ11b BRCA1 isoforms, we observed a similar sensitization to PARP inhibition following siRNA-mediated depletion of both BRCA2 and PALB2 (Figure 5B).
Figure 5. BRCA1-mutated SUM149PT and HCC1937 cells are sensitized to PARP inhibition by siRNA- or dinaciclib-mediated depletion of the BRCA1-PALB2-BRCA2 axis and RAD51.

(A) SUM149PT cells were transfected with the indicated siRNAs targeting BRCA1, BRCA2 or PALB2, followed by veliparib treatment at the indicated concentrations. Colony formation was assessed over a 14-day period. (B) Similar experiments were performed with HCC1937 cells using siRNAs targeting BRCA2 and PALB2. (C) Cells were treated with vehicle (0 nM) or the indicated concentrations of dinaciclib for 24 hrs and nuclear lysates subjected to Western blotting with the indicated antibodies. (D) Cells were pretreated with vehicle or 10 nM dinaciclib for 18 hours followed by 10 Gy IR. RAD51 and γH2AX focus formation was assessed by immunofluorescence 6 hrs after IR. (E) Quantification of RAD51 focus formation 6 hrs after IR in cells pretreated with vehicle (0 nM) or the indicated concentrations of dinaciclib (* P < 0.0001 for dinaciclib vs. vehicle). (F) Cells were treated with the indicated concentrations of veliparib in the absence or presence of dinaciclib, demonstrating reduced IC50 values in the presence of dinaciclib.
Based on these observations, we hypothesized that CDK12 inhibition could additionally sensitize PARP inhibitor-naïve BRCA1-mutated cells to PARP inhibition. Dinaciclib treatment resulted in a concentration-dependent reduction of BRCA2, RAD51, and BRCA1 Δ11b protein levels (Figure 5C), as well as a reduction in RAD51 foci following IR (Figures 5D and 5E). Additionally, dinaciclib treatment sensitized both SUM149PT and HCC1937 cells to PARP inhibition (Figure 5F and Table S1).
A 185delAG BRCA1-mutated PDX model demonstrates cisplatin sensitivity and primary PARP inhibitor resistance with residual HR activity that is ablated by dinaciclib
The significance of residual HR function as a mechanism of primary resistance to PARP inhibition has not been clarified due to the limited availability of clinical samples. To address this issue, we established a xenograft model (PDX127) from a PARP inhibitor-naïve germline 185delAG BRCA1 carrier presenting with metastatic TNBC (Figure 6A). The patient received cisplatin/olaparib in combination. After experiencing a partial response (PR; ~60% tumor regression) at a dose of 60 mg/m2 cisplatin and 50 mg twice-daily olaparib, her course was complicated by cisplatin-associated peripheral neuropathy, prompting a transition to 400 mg twice-daily olaparib monotherapy, on which she experienced rapid disease progression. Based on the observation that the patient’s PR had occurred on a suboptimal dose of olaparib, we hypothesized that her disease may have possessed a platinum-sensitive/PARP inhibitor-resistant phenotype similar to SUM149PT and HCC1937. As the biopsy utilized for generation of the PDX model was procured prior to receiving cisplatin/olaparib in combination, we were able to address this question in vivo. Cisplatin treatment of PDX127 resulted in tumor regression (Figure 6B), whereas olaparib monotherapy demonstrated minimal tumor growth inhibition (Figure 6C). Whole exome sequencing of the PDX127 model ruled out BRCA1 reversion mutation as a cause of PARP inhibitor resistance (Figure S5A). Tumor samples taken from vehicle- and olaparib-treated PDX127 bearing mice demonstrated the formation of RAD51 foci following olaparib treatment, supporting our hypothesis that preservation of HR-function had resulted in PARP inhibitor resistance (Figure 6D).
Figure 6. Generation and treatment of the PDX127 model from a 185delAG BRCA1 carrier.

(A) Treatment history of the BRCA1 carrier; the model was procured prior to exposure to cisplatin/olaparib or olaparib monotherapy. PR, partial response; PD, progressive disease. (B) Mice bearing xenografts were treated with vehicle (n = 4) or cisplatin (n = 6) on the days 1 and 34 (arrows) demonstrating tumor regression in response to platinum-based treatment. (C) Mice bearing xenografts were treated with vehicle (n = 8), olaparib (n = 7), dinaciclib (n = 3) or the combination (n = 7). Combination treatment produced significant tumor growth inhibition compared to vehicle or monotherapies. At day 35, P = 0.018 (*) for combination vs. dinaciclib and P < 0.0001 (**) for combination vs. olaparib. (D) Mice bearing xenografts were treated with vehicle, olaparib, dinaciclib or the combination (n = 6/group). (Left) After 15 days, mice were sacrificed and tumors subjected to immunofluroescence for RAD51 and γ-H2AX foci. (Right) Quantification of cells with > 5 γ-H2AX foci, as well as γ-H2AX-positive cells with > 5 RAD51 foci. For γ-H2AX foci, P values for control vs. dinaciclib, olaparib or the combination are 0.103, 0.013 (*) and 0.005, respectively. P values for dinaciclib or olaparib vs. the combination are < 0.0001 and 0.0076 (**), respectively. For RAD51 quantification in γ-H2AX-positive cells, P values for control vs. dinaciclib, olaparib or the combination are 0.69, 0.04 and 0.74 (NS), respectively. P values for dinaciclib or olaparib vs. the combination are 0.158 and 0.0035 (**), respectively. (E) Tumor RNA from mice in D treated with vehicle or dinaciclib (n = 3/group) was subjected to RT-PCR for BRCA1 and RAD51. P values for vehicle vs. dinaciclib are 0.000068 (**) in both cases. (F) Tumor lysates from mice in D treated with vehicle or dinaciclib were subjected to Western blotting with the indicated antibodies.
We next sought to determine if the combination of dinaciclib and olaparib could be extended to this model of primary PARP inhibitor resistance. Treatment with dinaciclib resulted in reduced HR gene mRNA and protein expression (Figures 6E and 6F) in the absence of evidence of cycle arrest (Figure S5D); as a result, the formation of RAD51 foci in response to olaparib treatment was significantly suppressed in the combination treated mice. Tumors treated with the combination also displayed significantly greater induction of γ-H2AX foci compared to those treated with olaparib or dinaciclib alone (Figure 6D). These effects translated to prolonged disease stabilization in mice treated with the combination (Figure 6C). These results provide evidence for the role of residual HR as a mechanism of resistance in PARP inhibitor-naïve BRCA1 mutant human tumors and suggest that combining a CDK12 inhibitor with a PARP inhibitor may be an effective treatment strategy in this setting.
Activity of combined CDK12 and PARP inhibition in a model with initial PARP inhibitor sensitivity
Although many BRCA1/2 mutated tumors display initial sensitivity to PARP inhibition, treatment in the metastatic setting is not curative and clinical benefit is manifested by transient and incomplete tumor regressions or sustained stable disease. We established an additional PDX model (11–26) from a patient with early stage TNBC harboring a somatic BRCA1 R1443* mutation. Consistent with HR deficiency, although the PDX model expressed RAD51, foci were not observed after short-term exposure to PARP inhibition (Figures 7A and B). As expected, dinaciclib reduced RAD51 expression (Figure 7B) not attributable to cell cycle arrest (Figure S5D).
Figure 7. Treatment of the 11–26 PDX model harboring somatic BRCA1 R1443* mutation.

(A) Mice bearing xenografts were treated with vehicle or veliparib (n = 5/group). (Left) After 15 days, mice were sacrificed and tumors subjected to immunofluorescence for RAD51 and γ-H2AX foci. (Right) Quantification of γ-H2AX-positive cells with > 5 RAD51 foci. P, non-significant. (B) Mice bearing xenografts were treated with vehicle or dinaciclib for 2 doses over 5 days (n = 3/group), after which mice were sacrificed and tumors stained for RAD51. P = 0.059. Bar, 100 μm. (C) Mice bearing xenografts were treated with vehicle (n = 3), dinaciclib (n = 7), veliparib (n = 4) or the combination (n = 10), demonstrating long-term growth control with veliparib and sustained tumor regressions with combination treatment. After 2 months of treatment (day 61), P < 0.001 (*) for combination treatment vs. either monotherapy. (D) Waterfall plot demonstrating % change in tumor volume at the time of sacrifice for individual mice in the four treatment groups. (E) Representative end-of-experiment histology (H & E) and γ-H2AX staining of tumors isolated from mice in the 4 treatment groups. Bar, 100 μm. (F) Quantification of % nuclei staining positively for γ-H2AX at end-of-experiment (* P < 0.05 for combination vs. control treatment).
PARP inhibitor monotherapy produced stable disease over a prolonged 156-day time course, although palpable primary tumors were detectable for the duration of the experiment. The addition of dinaciclib to veliparib resulted in substantial and durable tumor regression (Figures 7C and 7D).
Immunohistochemistry performed on tumors harvested at the end of the experiment demonstrated viable cell populations in vehicle and monotherapy-treated tumors (Figure 7E). In contrast, bland fibrous tissue predominated in combination-treated tumors; small nests of tumor cells were present, which displayed expression of γ-H2AX, consistent with the induction of persistent DNA damage (Figures 7E and F). Finally, histologic analyses of liver, lung, GI tract and bone marrow harvested at the end-of-experiment from combination-treated mice revealed no abnormalities, with only minimal γ-H2AX staining, indicating that the combination of dinaciclib and veliparib was tolerable to normal tissues over a prolonged treatment course (Figure S5B). These data suggest that the addition of dinaciclib to PARP inhibition can augment the quality and degree of response, even in a tumor initially susceptible to PARP inhibitor monotherapy.
Discussion
The development of PARP inhibitor resistance in BRCA-mutated cancers is a pressing clinical problem. Restoration of HR plays a major role in acquired resistance and may occur by varied and complex mechanisms, highlighting the need for a therapeutic strategy that can be broadly applied across patients with tumors resistant to PARP inhibitors. Here, we demonstrate that the combination of CDK12 and PARP inhibition represents a viable approach for reversing such resistance.
In MDA-MB-436 BRCT domain BRCA1 mutant derivatives with acquired PARP inhibitor resistance, RAD51 loading is facilitated by an HSP90-stabilized splice variant mutant BRCA1 (Johnson et al., 2013). As transcription of the BRCA1 splice variant is driven by the intact BRCA1 promoter, inhibition of CDK12 is expected to reduce its expression. Dinaciclib ablated expression of the mutant protein, inhibited HR and re-established PARP inhibitor sensitivity.
The multiple components of the HR pathway that are transcriptionally downregulated by CDK12 inhibition suggest that this strategy may be effective even when the precise mechanism of HR restoration is unknown. In the PDX model of acquired PARP inhibitor resistance (12–58), a BRCA2 reversion was not detected, and the events underlying PARP inhibitor and platinum resistance are under further investigation. Nonetheless, tumor growth inhibition was imposed by combined dinaciclib and PARP inhibition.
Hypomorphic BRCA1 proteins may account for some instances of de novo PARP inhibitor resistance, illustrated by mutations arising in exon 11 of the BRCA1 reading frame. Consequently, BRCA1 Δ11b-expressing SUM149PT cells exhibit primary PARP inhibitor resistance that may be reversed by siRNA- or CDK12 inhibitor-mediated depletion of the hypomorphic protein or other components of the BRCA1-PALB2-BRCA2 axis. In a second BRCA1-mutated cell line, HCC1937 (BRCA1 5382insC), PARP inhibitor resistance is BRCA1-independent, possibly related to a compensatory role for RAD52 (Lok et al., 2013). HCC1937 cells continue to be dependent on remaining components of the BRCA1-PALB2-BRCA2 axis, allowing them to be similarly sensitized to PARP inhibition by dinaciclib treatment. Additionally, the existence and clinical significance of residual HR in tumors arising in PARP inhibitor-naïve BRCA carriers has remained unknown. Here, we show evidence of residual HR in the clinical course of a 185delAG BRCA1 carrier, whose tumor was resistant to olaparib, but sensitized with dinaciclib.
Despite exhibiting PARP inhibitor resistance, SUM149PT and HCC1937 cells, as well as the PDX127 model, demonstrated sensitivity to cisplatin. The retention of residual HR in these models is not adequate to confer FA pathway proficiency, required for the repair of damage induced by DNA crosslinking agents. These results further emphasize that PARP inhibitor and platinum sensitivity are not always concordant (Bunting et al., 2012), and provide additional evidence for the importance of platinum agents in BRCA-mutated TNBC.
Lastly, our work suggests that combined CDK12 and PARP inhibition is applicable to BRCA-deficient breast cancers that are PARP inhibitor-susceptible. Regressions in such tumors are rarely complete, and in some cases short-lived. In the K14cre;BRCA1F/F;p53F/F genetically-engineered mouse model challenged with PARP inhibitor monotherapy, not all tumors regress, with some demonstrating stable disease (Rottenberg et al., 2008). Similarly, in sensitive BRCA mutated TNBC PDX models, outcomes frequently demonstrate stable tumor growth inhibition or minor regression, without complete tumor response (Juvekar et al., 2012). Therefore, the 11–26 PDX model derived from a patient with early-stage TNBC harboring somatic BRCA1 mutation is representative of PARP inhibitor-sensitive breast cancers, and demonstrated prolonged tumor growth inhibition with veliparib monotherapy. The addition of dinaciclib converted the outcome to sustained regression in all of the mice treated, with only minimal residual disease evident on analysis of end-of-treatment histology.
Although dinaciclib inhibits several cell cycle and transcriptional CDKs, the modest effects on cell cycle progression coupled with the transcriptional profile suggests that the phenotype we observed in tumor cells is primarily driven by inhibition of CDK12. The highly potent CDK12 inhibitory activity in biochemical assays distinguishes dinaciclib from all other CDK inhibitors tested that target CDK family members to varying degrees (Bosken et al., 2014). These results have implications for the future development of dinaciclib and suggest that combinatorial strategies including PARP inhibitors or other DNA damaging agents should be prioritized.
An important requisite of any HR targeting strategy for PARP inhibitor sensitization is selectivity for tumor cells. Dinaciclib-mediated inhibition of cell cycle CDKs appears to arrest mammary epithelial cells in G2/M to a greater degree than TNBC cells. This is expected to impede PARP inhibitor-mediated cell death that occurs in S phase, affording a favorable therapeutic index. Further work will be required to determine if concomitant cell cycle CDK inhibition will be required for the tolerability of combined CDK12 and PARP inhibition. It is also possible that cells with inherent genomic instability may be highly dependent on CDK12-directed transcription to accomplish necessary repair, and therefore particularly vulnerable to reduced CDK12 activity in concert with PARP inhibition, whereas genetically stable non-transformed cells may only require low rates of transcription of such genes and are thus able to tolerate the degree of suppression achieved by reversible kinase inhibition. Whatever the precise mechanism, prolonged exposure to combined dinaciclib and veliparib had no apparent toxicity to normal mouse organs, while achieving profound tumor regression in the 11–26 model.
In summary, dinaciclib is a potent inhibitor of CDK12 that effectively sensitizes BRCA wild type and mutated models of TNBC to PARP inhibition, overcoming primary and acquired resistance. A Phase 1 trial of dinaciclib and veliparib is currently in progress (NCT01434316). Once recommended phase 2 doses of the agents are established, the trial will enroll expansion cohorts assessing preliminary activity in both BRCA wild-type and BRCA-mutated TNBCs.
Experimental Procedures
Compounds
Dinaciclib, veliparib and olaparib were provided by the NCI Division of Cancer Treatment and Diagnosis (DCTD) or purchased from Selleck Chemicals. Flavopiridol and hydroxyurea were purchased from Enzo Life Sciences and Sigma-Aldrich, respectively.
In vitro kinase assays
Kinase reactions were carried out using recombinant full length human CDK9 (1-372) and Cyclin T1 (1-272), and human CDK12 (696-1082) and Cyclin K (1-267), as previously described (Bosken et al., 2014). Measurements were performed in triplicate.
Cell lines and cell viability assays
Cell lines were obtained from the American Type Culture Collection (Manassas, Virginia). For viability assays, cells were seeded at 500–5,000/well on 96-well plates, cultured in the presence of drugs or vehicle for 7 days, and assessed by Cell-Titer-Glo (Promega). IC50 values, determined in the absence or presence of dinaciclib, represent veliparib concentrations at which viability was reduced by 50% of vehicle-treated cells. Cells treated with control siRNAs or those targeting BRCA1, BRCA2 or PALB2 were replated in media containing vehicle or veliparib and viability determined after 7 days. Mean viability relative to vehicle-treated cells was calculated from a minimum of three experiments.
Western blotting
Western blotting was performed with antibodies recognizing BRCA1 (OP- 92, EMD Millipore), BRCA2 (OP-95, EMD Millipore), RAD51 (H-92, Santa Cruz Biotechnology), FANCD2 (NB100-182, Novus Biologicals), γ-H2AX [pS139] (JBW301, EMD Millipore), histone H3 (AB1791, Abcam), CTD [pSer2] (3E10, EMD Millipore), CTD [pSer5] (3E8, EMD Millipore), and total CTD (8WG16, Abcam).
Gene expression array analysis
MDA-MB-231 cells were treated in triplicate with either vehicle or dinaciclib for 12 hours and RNA was collected using Trizol and Qiagen RNeasy mini kit. Changes in transcription were measured using the Affymetrix HG-U133A2 array platform. Genes statistically significantly downregulated in response to dinaciclib were imported into Ingenuity Pathway Analysis (IPA) software and networks of these focused genes were built based on the Ingenuity Knowledge Base.
Establishment and Treatment of Patient-Derived Xenografts
Patient consent was obtained under IRB approved-protocols. TNBC samples were implanted into the cleared 4th mammary fat pads of NOD-SCID-IL2Rγc−/− mice (Jackson Laboratories) or subcutaneously implanted in female HsdCpb:NMRI-Foxn1nu mice (Harlan Laboratories) at 6 weeks of age. For the PDX 127 model, animals were supplemented with 1 μmol/L estradiol (Sigma) in the drinking water. After engraftment, tumor tissue was re-implanted into recipient mice, which were randomized when volumes reached 100–300 mm3 to receive vehicle, veliparib or olaparib, dinaciclib or the combination of PARP inhibitor and dinaciclib, with 5–10 mice/group. Animals were treated by oral gavage with veliparib (50 mg/kg twice-daily) (Donawho et al., 2007), olaparib (50mg/kg six days per week) (Juvekar et al., 2012) or with intraperitoneal dinaciclib (8mg/kg six days per week for PDX 127 or 30 mg/kg twice-weekly for PDXs 11–26 and 12–58) (Parry et al., 2010). Cisplatin was administered at 6 mg/kg (PDX127) or 8 mg/kg (PDX 12–58) (Rottenberg et al., 2007). Caliper measurements were used to determine tumor volumes as length x width2. Tumor volumes are plotted as mean +/− SEM.
Immunofluoresence and focal microscopy
Primary antibodies recognizing BRCA1, RAD51 and γ-H2AX [pS139] were followed by secondary antibodies conjugated to FITC or Texas Red (Jackson ImmunoResearch Laboratories). Confocal immunofluorescence images were acquired using Andor iQ software (Belfast, United Kingdom). For metaphase spreads, cells were exposed for 2 hours to colcemid, harvested and stained with Wright’s stain. Fifty metaphase spreads were scored for aberrations, captured using CytoVision software (Applied Imaging).
Histologic and immunohistochemical staining
Formalin-fixed paraffin-embedded sections of harvested xenografts were stained with hematoxylin and eosin (H&E) or antibodies against γ-H2AX [pS139] or RAD51. At least 3 xenografts, each with at least five 40X fields, were manually scored or quantified by Aperio image analysis for each treatment. For toxicology assessments, mouse organs from vehicle or combination-treated mice were harvested, formalin-fixed, H&E stained and evaluated histologically, as well as for γ-H2AX staining.
Statistical analysis
P < 0.05 was considered statistically significant in two-tailed, unpaired Student’s t tests.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants R01 CA090687 (GIS), a Research Supplement to Promote Diversity in Health-Related Research (SFJ, GIS) and P50 CA168504 [Dana-Farber Specialized Program of Research Excellence (SPORE) in Breast Cancer] (GIS, AD), as well as Susan G. Komen Investigator Initiated Research Grant IIR12223953 (GIS). EL was supported by a Dana-Farber Claudia Adams Barr Award and a National Health and Medical Research Council of Australia Fellowship. Work at Vall d’Hebron Institute of Oncology (VHIO) was supported by Instituto de Salud Carlos III PI12/02606 (JB) and the Asociación Española Contra el Cancer (CC). MG is a member of the DFG excellence cluster ImmunoSensation and is supported by a grant from the Deutsche Forschungsgemeinschaft GE 976/9-1. U2OS-pDR-GFP cells and Lenti-CRISP/Cas9 constructs were provided by Maria Jasin and Jean Zhao, respectively. We thank Donna Skinner of the DF/HCC Research Pathology Core, Pilar Antón of VHIO and Lan Hu of the DFCI Center for Cancer Computational Biology for technical assistance, as well as Bjoern Chapuy and Margaret Shipp for sharing preliminary data.
Footnotes
Accession Numbers
The data for the gene expression analysis in vehicle and dinaciclib-treated MDA-MB-231 cells have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE88822.
Author Contributions
S.F.J., C.C., A.K.G., S.D., D.G.S., D.C., B.P., S.C., A.J.B., R.C., J.B.L., B.K., H.S., C.U., L.A.M., K.A.S., S.J.R. V.S. and E.L. conducted the experiments. R.C., M.S., D.J., J.B., A.L.R., J.B., V.S. and E.L. oversaw the procurement and establishment of PDX models, with overall coordination of in vivo experiments by E.L. S.F. J, A.D.D., J.B., N.J., M.G., V.S., E.L. and G.I.S. designed the experiments. S.F.J., M.G. and G.I.S. wrote the paper, which was reviewed and approved by all authors. G.I.S. supervised the entire project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alagpulinsa DA, Ayyadevara S, Yaccoby S, Schmookler Reis RJ. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol Cancer Ther. 2016;15:241–250. doi: 10.1158/1535-7163.MCT-15-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, et al. Genome-wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014;74:287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmaña J, Tung NM, Isakoff SJ, Grana B, Ryan PD, Saura C, Lowe ES, Frewer P, Winer E, Baselga J, et al. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Annals Oncol. 2014;25:1656–1663. doi: 10.1093/annonc/mdu187. [DOI] [PubMed] [Google Scholar]
- Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, Johnson LN. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27:1907–1918. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, Peterlin BM. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, Geyer M. The structure and substrate specificity of human Cdk12/Cyclin K. Nature Commun. 2014;5:3505. doi: 10.1038/ncomms4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20:540–547. doi: 10.1158/1078-0432.CCR-13-0225. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46:125–135. doi: 10.1016/j.molcel.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, O'Connor KW, Mouw KW, Li AY, Matulonis UA, D'Andrea AD, Konstantinopoulos PA. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015;75:628–634. doi: 10.1158/0008-5472.CAN-14-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- Eick D, Geyer M. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev. 2013;113:8456–8490. doi: 10.1021/cr400071f. [DOI] [PubMed] [Google Scholar]
- Garriga J, Grana X. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene. 2004;337:15–23. doi: 10.1016/j.gene.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Ghamari A, van de Corput MP, Thongjuea S, van Cappellen WA, van Ijcken W, van Haren J, Soler E, Eick D, Lenhard B, Grosveld FG. In vivo live imaging of RNA polymerase II transcription factories in primary cells. Genes Dev. 2013;27:767–777. doi: 10.1101/gad.216200.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Clark AP, Silver DP, Livingston DM. BRCA1 pathway function in basal-like breast cancer cells. Mol Cell Biol. 2014;34:3828–3842. doi: 10.1128/MCB.01646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110:17041–17046. doi: 10.1073/pnas.1305170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J Biol Chem. 2014;289:9247–9253. doi: 10.1074/jbc.M114.551143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2:1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TA, Li W, Brogi E, Yee CJ, Gemignani ML, Olvera N, Levine DA, Norton L, Robson ME, Offit, et al. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann Surg Oncol. 2007;14:2510–2518. doi: 10.1245/s10434-007-9372-1. [DOI] [PubMed] [Google Scholar]
- Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-10-research0041. Res. 0041.0041-0041.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, Shilatifard A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. 2015;35:928–938. doi: 10.1128/MCB.01426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, Statkevich P, Zhu Y, Yao SL, Small K, et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. 2014;14:169–176. doi: 10.1016/j.clbc.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–2353. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, Liu X, de Visser KE, Gilhuijs KG, van Tellingen O, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci U S A. 2007;104:12117–12122. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol. 2015;33:1397–1406. doi: 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, Weitsman G, Barber P, Vojnovic B, Ellis H, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Science Signal. 2014;7:ra29. doi: 10.1126/scisignal.2005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163:174–186. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.