

Abstract
P450 enzymes (P450s) are well known for their ability to oxidize unactivated C-H bonds with high regio- and stereoselectivity. Hence, there is emerging interest in exploiting P450s as potential biocatalysts. Although bacterial P450s typically show higher activity than their mammalian counterparts, they tend to be more substrate selective. Most drug-metabolizing P450s on the other hand, display remarkable substrate promiscuity, yet product prediction remains challenging. Protein engineering is one established strategy to overcome these issues. A less explored, yet promising alternative involves substrate engineering. This review discusses the use of small molecules for controlling the substrate specificity and product selectivity of P450s. The focus is on two approaches, one taking advantage of non-covalent decoy molecules, and the other involving covalent substrate modifications.2009 Elsevier Ltd. All rights reserved.
Keywords: Anchoring molecule, Biocatalysis, Chemical auxiliary, Decoy molecule, Directing group, Fatty acid, Molecularly imprinted polymer, Perfluorinated carboxylic acid
Graphical Abstract
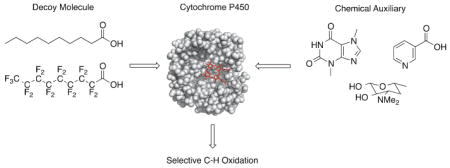
1. Introduction
P450 enzymes (P450s), are heme-dependent monooxygenases involved in processes such as biosynthesis of steroids and natural products, as well as xenobiotic metabolism.1,2 Of particular interest is their remarkable ability to hydroxylate unactivated C-H bonds with good-to-excellent regio- and stereoselectivity.1–4 Success has been achieved in the engineering of P450s for expansion of substrate scope, alteration of regioselectivity or for production of small molecules, drugs or drug metabolites.5–8 The large majority of mechanistic and engineering studies have focused on bacterial P450s due to their soluble nature and higher activity in vitro.1,9 Using mutagenesis, directed and random evolution P450BM3 for instance has been extensively engineered to oxidize unnatural substrates such as alkanes10,11, indole to indigo and indirubin12, terpenes13, steroids and alkaloids14. More recently, incorporation of unnatural amino acids into P450BM3 also allowed for enhancement of activity and alteration of regioselectivity.15 Effort has also been invested into using computational modelling16,17 and fingerprinting17,18,19 of P450s to guide rational engineering of the enzymes to obtain novel regio- and stereoselectivities. Mammalian P450s, on the other hand, have generated great interest mostly because of their important role in drug metabolism. The pharmaceutical industry has contributed to reveal the exceptional ability of some human P450s to oxidize a large variety of substrates while distinguishing between multiple sterically and electronically similar C-H bonds, yet without generating over-oxidation products.2,3,20,21 Whereas the high substrate promiscuity of P450s can be an attractive quality for a biocatalyst, our current inability to predict the site(s) of oxidation creates a paradox. Here we review some strategies aiming to manipulate substrate specificity and predict products of P450 transformations without the use of mutagenesis. These approaches use small molecules to affect substrate binding either via non-covalent or covalent interactions.
2. Non-covalent interactions between P450s and small molecules as a way to modify substrate specificity and regioselectivity
This section describes research where inert dummy molecules, also referred to as decoys, are used to control the site of P450 oxidation by blocking a part of the active site and thus constraining the substrate into a specific binding orientation.22 The decoys are often structurally similar to the natural substrate but are not turned over by the enzyme22 Two types of decoy molecules have been explored for their P450-directing capabilities: (1) short-chain fatty acids, and (2) perfluorinated fatty acids.
2.1. Effect of short-chain fatty acid decoy molecules on P450BSβ catalysis
P450BSβ is a soluble and self-sufficient P450 enzyme isolated from Bacillus subtilis. It typically catalyzes the α- and β-hydroxylation of long-chain fatty acid substrates, such as myristic acid, with good regioselectivity.23 In 2007, Watanabe and coworkers took advantage of short-chain fatty acids to expand the substrate scope of P450BSβ and enable the transformation of the unnatural substrates guaiacol, styrene, ethylbenzene and thioanisole (Figure 1).22,,24 The applied short-chain fatty acids were found to act as ligands of P450BSβ mimicking part of the substrate and leaving a small portion of the substrate binding pocket available near the heme iron for the unnatural substrate to occupy. Guaiacol oxidation by P450BSβ was optimized with the addition of carboxylic acid decoy molecules varying in length from 2 to 10 carbons.22 Although many of the acids promoted guaiacol oxidation, heptanoic acid was the most successful with a turnover rate 10-fold higher than that of myristic acid hydroxylation. The rate enhancement was attributed to the decoy molecule keeping the catalytic cycle “on”, which long-chain fatty acid substrates normally turn on when they bind and “off” when the product dissociates.22 As expected, in the presence of myristic acid, no oxidation of guaiacol occurred. Activation of P450BSβ by carboxylic acid decoy molecules was also observed for styrene epoxidation and ethylbenzene hydroxylation.22 Styrene epoxidation proceeded with both regioselectivity and enantioselectivity greater than 80%, while ethylbenzene hydroxylation reached 68% ee (R) at best. Altogether, these results demonstrate that P450BSβ can catalyze the oxidation of non-fatty acid substrates when tricked into misrecognizing short-chain fatty acids instead of the natural long chain fatty acids. Crystal structure analysis suggested that fatty acids shorter than 10 carbon long might be too short to extend into the hydrophobic substrate access channel, resulting in loose fixation of the hydrophobic tail.25 This may prevent hydroxylation of the decoy molecules while allowing entry of the small molecule.20
Figure 1.

Substrate scope for the oxidation of unnatural substrates by P450BSβ in the presence of the decoy molecules, heptanoic or hexanoic acid. Substrates shown are (a) myristic acid, (b) guaiacol, (c) styrene, and (d) ethylbenzene, along with the major product of their oxidation by P450BSβ.22
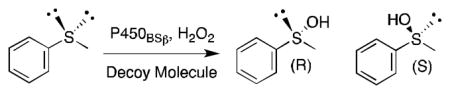
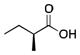
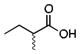
To investigate the effect of variations in the structure of the decoy molecule on the stereochemical outcome of the reaction, Watanabe and colleagues examined sulfoxidation of thioanisole (Table 1).24 Most of the decoy molecules examined favored the product with the R configuration at the sulfur with modest ee. In fact the turnover was correlated (R = 0.8187) with the % ee for the product with R configuration. Branched carboxylic acids with and without a chiral center were also tested with the aim of obtaining the S-isomer but did not induce the desired stereochemical inversion. It was noted that over-oxidation to the sulfone product was not observed.
Table 1.
Sulfoxidation of thioanisole catalyzed by P450BSβ in the presence of carboxylic acid decoy molecules.24
| ||
|---|---|---|
| Decoy Molecule | Turnover ratea (min−1) | % ee (R or S) |
| None | 24 ± 2 | 4 ± 0 (R) |
|
|
251 ± 20 | 21 ± 4 (R) |
|
|
514 ± 72 | 29 ± 1 (R) |
|
|
390 ± 53 | 21 ± 2 (R) |
|
|
274 ± 48 | 16 ± 1 (R) |
|
|
261 ± 75 | 18 ± 0 (R) |
|
|
270 ± 21 | 20 ± 1 (R) |
|
214 ± 27 | 19 ± 1 (R) |
|
146 ± 22 | 13 ± 1 (R) |
|
|
370 ± 39 | 19 ± 0 (R) |
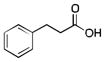
|
462 ± 37 | 21 ± 0 (R) |
|
(PAA) |
71 ± 10 | 14 ± 0 (R) |
|
(o-MPAA) |
34 ± 9 | 9 ± 1 (R) |
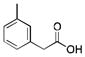 (m-MPAA) |
32 ± 8 | 12 ± 2 (R) |
|
(p-MPAA) |
89 ± 14 | 11 ± 2 (S) |
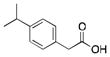
|
55 ± 2 | 10 ± 1 (S) |
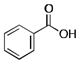
|
41 ± 5 | 12 ± 1 (R) |
The units for catalytic activity are nmol product min−1 nmol P450−1.
Interestingly, addition of p-methylphenylacetic acid (p-MPAA) or p-isopropylphenylacetic acid to the enzymatic reaction mixture was successful to favor the S enantiomeric product.24 Molecular modeling simulations with phenylacetic acid (PAA) and p-MPAA showed that for p-MPAA, the methyl group is positioned over the porphyrin ring creating steric bulk near thioanisole, which may favor positioning of the pro-S lone pair closer to the heme (Figure 2). This is an interesting example of a non-mutagenic method to reverse the stereoselectivity of a P450-catalyzed reaction.
Figure 2.

Suggested transition states for stereochemical inversion during the sulfoxidation of thioanisole by P450BSβ with (a) PAA and (b) p-MPAA as illustrated by Fujishiro et al.24
2.2. Effect of perfluorinated fatty acid decoy molecules on P450BM3 catalysis
Over two decades ago, Baker and coworkers reported that the activity of microsomal P450s could be enhanced with the addition of fluoroethane derivatives (fluorinated, chlorinated and/or brominated).26–29 More recently, perfluorinated fatty acids have proven to be useful decoy molecules to divert P450BM3 into accepting non-natural substrates, especially small alkanes.30,31 P450BM3 is another self-sufficient enzyme encoded by Bacillus megaterium. It catalyzes the hydroxylation of long-chain fatty acids such as myristic and palmitic acid at the ω–1, ω–2, and/or ω–3 positions. Although P450BM3 has a large active site which can accommodate multiple substrates at once, molecules that are not fatty acids are typically not transformed by the enzyme.32,33 The carboxylate group in particular is required for catalytic activity.32,33 The large size of the active site poses an additional challenge for biocatalytic applications; several binding orientations are accessible, potentially leading to multiple products.30 Two studies reported by Reetz30 and Watanabe32 independently demonstrated that perfluorocarboxylic acids (PFCs) can be used as decoy molecules to favor the transformation of unnatural substrates by P450BM3 without resorting to mutagenesis. In the context of oxidations, PFCs are an inert alternative to fatty acids due to the greater bond-dissociation energy of C-F bonds (116 kcal mol−1) compared to that of C-H bonds (95–99 kcal mol−1).31 Although PFCs and fatty acids have similar hydrophobicity, PFCs are considerably larger than fatty acids.34
Using PFCs with 8 to 14 carbon lengths, Reetz and Watanabe independently confirmed that PFCs bind P450BM3 and create significant steric bulk in the active site. Moreover, the PFCs activate the enzyme by displacing the distal water molecule and inducing a low-to-high spin state shift of the heme iron.31 The PFCs were assessed for their ability to direct the hydroxylation of small substrates ranging from methane to octane, including some constitutional isomers of hexane (Table 2).30,31 The results point to longer PFCs as better decoys to favor the desired transformations. In general, PFCs enhanced enzymatic turnover but had little effect on regio- and stereoselectivity. Indeed, multiple products were generated and this was attributed to inconsistent orientation of the alkanes due to the lack of functional groups.30,31 While the conversion of propane to propanol in the presence of PFC10 proceeded successfully,30 the original conclusion that methane was oxidized to methanol was later refuted.35 In the presence of decanoic acid P450BM3 did not convert propane to propanol, suggesting that the fluorine atoms in the decoy molecule are crucial for coercing the enzyme to accept small alkane substrates.31
Table 2.
Conversion rates of gaseous molecules by P450BM3 in the presence of PFCs.
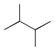
| Substrate | PFCa | Rateb (min−1) | Regioselectivity | Refs. |
|---|---|---|---|---|
|
|
C11 | 1021 | 30 | |
|
|
C10 | 67 | 31 | |
|
|
C7 | 3632 | 30 | |
|
|
C10 | 113 | 31 | |
|
|
C11 | 525 | 2-/3-hexanol = 77:23 | 30 |
|
|
C9 | 1184 | 2-/3-/4-octanol = 10:42:48 | 30 |
|
|
C9 | 110 | 31 | |
|
|
C9 | 476 | 2-/3-hydroxy = 88:12 | 30 |
|
|
C9 | 891 | Only 2-hydroxy | 30 |
|
C11 | 3241 | Only 2-hydroxy | 30 |
Optimal perfluorinated carboxylic acid reported.
Observed turnover rates.
More recently, Watanabe, Shoji and colleagues reported successful hydroxylation of aromatic rings such as those of benzene, toluene, anisole, chlorobenzene, nitrobenzene and acetophenone using P450BM3 and various PFCs (Table 3).36 The transformations proceeded with exclusive formation of phenol derivatives but the turnover rate diminished with the presence of electron-withdrawing groups on the ring. The major hydroxylation site was always ortho to the existing aromatic substituent, even with meta directing groups. Thus it was assumed that the regioselectivity is controlled by the substrate binding pocket.36
Table 3.
Hydroxylation of aromatic rings by P450BM3 in the presence of PFCs.36
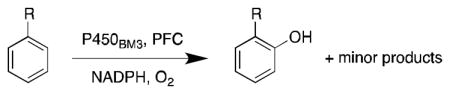
| |||
|---|---|---|---|
| Substrate | PFCa | Rate (min−1) b | Major Product |
|
|
C9 | 120 ± 9 | phenol |
|
|
C9 | 220 ± 7 | ortho |
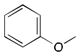
|
C9 | 260 ± 4 | ortho |
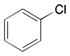
|
C9 | 120 ± 4 | ortho |
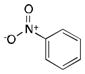
|
C9 | 0.9 ± 0.05 | ortho |
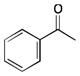
|
C9 | 2.9 ± 0.1 | ortho |
The chain length of PFC that elicited the greatest effect.
Rates are given in units of min−1 per P450. Uncertainty is given as the standard deviation from at least three measurements.
Overall, decoy molecules provide a simple and quick way for increasing the substrate scope of P450s without the need for protein mutagenesis. The molecules can also be used to influence the regio- and stereoselectivity of P450-catalyzed reactions. This elegant strategy remains to be expanded to P450s other than P450BSβ and P450BM3, and especially to drug-metabolizing P450s, which often show cooperative behavior resulting from the simultaneous binding of multiple substrates.
3. Covalent modifications of substrates as a way to control substrate specificity, regioselectivity and stereoselectivity of P450s
The second non-mutagenic strategy reviewed here applies substrate engineering to control oxidation by P450s. Early studies with monooxygenases suggested that the presence of specific functional groups can improve substrate acceptance by biocatalysts.37–44 Substrate engineering may involve the covalent addition of a specific recognition motifs, the chemical auxiliary or directing group, to an unnatural substrate with the aim of facilitating recognition by the enzyme and favoring a specific productive binding orientation (Figure 3).43,45
Figure 3.

The use of chemical auxiliaries to control the transformation of unnatural substrates by P450s. Typical steps include (1) covalent attachment of the auxiliary to the substrate of interest, (2) transformation of the auxiliary-substrate by the enzyme, (3) product release, and (4) chemical cleavage of the auxiliary.
3.1. Covalent modifications to impose unnatural substrates to bacterial P450s
One of the earlier groups to apply substrate engineering with P450s was that of Munzer for the expansion of the substrate scope of P450BM3.46 Protein mutagenesis was combined with the use of 2-aminophenol as a chemical auxiliary (via formation of a benzoxazole ring, see Figure 4a) for establishing cyclopentanoic acid as a substrate of numerous P450BM3 mutants.46 The auxiliary not only ensured that cyclopentanoic acid was transformed by the enzyme but also imparted selectivity towards production of two out of four possible diastereoisomers. In a separate study with the same P450, Arnold and coworkers manipulated protecting-group moieties and the stereochemistry at the anomeric carbon of globally-protected monosaccharides to control the regioselectivity of P450BM3-catalyzed deprotection further away on the sugar (Figure 4b).47 Interestingly inverting the stereochemistry from α to β and benzylation of the anomeric position turned otherwise unreactive molecules into substrates that are regioselectively deprotected at O-4 by P450BM3 F87A or F87I mutants. In combination, these studies demonstrate the ability of substrate engineering to expand the substrate scope of P450BM3 mutants while imparting some selectivity.
Figure 4.

Examples of regio- and stereoselective oxidation by P450BM3 mutants via substrate engineering. a) Oxidation of cyclopentanoic acid46, b) deprotection of globally-methylated glucose (glc) and galactose (gal)47.
PikC is a P450 enzyme isolated from Streptomyces venezuelae and involved in the hydroxylation of the 12- and 14-membered ring macrolides YC-17 and narbomycin.48,49 Inspired by the structures of YC-17 and narbomycin, a desosamine glycoside moiety was designed to favor hydroxylation of carbocyclic rings by PikC.45 A self-sufficient PikC fusion mutant, PikCD50N-RhFRED50 was evaluated for the oxidation of a series of carbocyclic rings ranging in size from 12 to 15 carbons and linked to a desosamine glycoside (Table 4). In each case multiple hydroxylation products were detected with the major regioisomer typically displaying the hydroxyl group at the site most distant from the chemical auxiliary. No over-oxidation to ketone products and no hydroxylation of the auxiliary were detected. Molecules other than carbocyclic rings were poor substrates or were not transformed at all by the enzyme. Crystal structure analysis suggested that rigidity of the substrates is a major determinant for the regioselectivity of PikCD50N-RhFRED. The desosamine moiety can also adopt two significantly different binding modes, causing flipping of the substrate. This work has recently been extended by the same group to examine other auxiliaries containing the tertiary amine moiety found in desosamine.51 The natural substrate YC-17 is hydroxylated by PikC at C-10 and C-12 in a 1:1 ratio. The regioselectivity of the monohydroxylation of YC-17 analogues by PikC was investigated with various tertiary amines. The authors showed that the regioselectivity of YC-17 hydroxylation depends on the presence, length, rigidity and stereochemistry of the tertiary amine.51 By varying the chemical auxiliary from 3-(dimethylamino)propanoic acid to m-(dimethylamino)benzoic acid they were able to tune the selectivity of oxidation to favor either C12 (3 fold), or C10 (20 fold) (Figure 5). Interestingly, replacement of the tertiary amine group with an isopropyl group did not yield turnover of the analogues by the enzyme. By removing the majority of the desosamine structure and keeping only the tertiary amine moiety, the authors demonstrated the importance of preserving the active site salt bridge formed between the tertiary amine moiety of an auxiliary and an active site glutamic acid.
Table 4.
The scope and turnover of desosamine-linked unnatural substrates by PikCD50N-RhFRED45
| Substrate | Kd (μM) | Yield (%) | No. of Products |
|---|---|---|---|
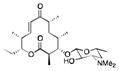
|
19 | >99 | 2 |
|
|
309 | 47 | 7 |
|
|
218 | 65 | 6 |
|
|
289 | 63 | 6 |
|
|
243 | 35 | 9 |
|
|
NBa | 0 | 0 |
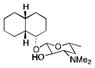
|
NB | 0 | 0 |
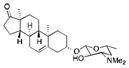
|
NB | <1 | 0 |
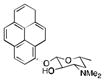
|
>5000 | 4 | 1 |
|
|
NB | 0 | 0 |
|
|
2900 | 8 | 5 |
|
|
2300 | 14 | 7 |
NB, binding below detection limit
Figure 5.

Regioselective oxidation of YC-17 analogues with tertiary amine auxiliaries.51
3.2. Covalent modifications to extend the substrate promiscuity and predict the regio-and stereoselectivity of mammalian P450s
Substrate promiscuity is considered a highly attractive quality in a versatile biocatalyst, and many mammalian P450s are unequalled in this aspect.2,52–54 Human P450 3A4 for example accounts for the metabolism of approximately 50% of all xenobiotics.55 Due to its large and flexible active site P450 3A4 is inherently highly promiscuous and unpredictable in terms of substrate specificity and product selectivity. Through substrate engineering the Auclair group has demonstrated that the regio- and stereoselectivity of P450 3A4 and P450 2E1 can be controlled in a predictable manner.56,57
The use of theobromine as a chemical auxiliary was reported to not only favor the transformation of unnatural substrates by P450 3A4 but also to allow prediction of the enzyme regio- and stereoselectivity.56 The theobromine auxiliary was modeled after a known P450 3A4 substrate, lisofylline, which is hydroxylated in part at the fourth carbon from the theobromine moiety (Figure 6).58 With a series of small alkanes and alkenes covalently attached to theobromine it was shown that theobromine successfully directs hydroxylation by P450 3A4 at the fourth atom away from the auxiliary for all the compounds that had a fourth methylene carbon (Figure 7). Double bonds at the third and fourth, or fourth and fifth atoms from the auxiliary led to epoxide formation. Moreover, there was a preference for pro-R facial selectivity at the fourth carbon for both hydroxylation and epoxidation. The presence of double bonds and tertiary C-H bonds elsewhere in the molecule did not affect the regioselectivity, in spite of their higher susceptibility towards oxidation. Additionally, no over-oxidation to the ketone and no oxidation of the auxiliary were observed. With its heterocycle, compound 8 had two different carbons positioned four atoms away from the auxiliary, and as expected the fourth position closest to the heteroatom was preferred due to electronics. This was the first example of a chemical auxiliary that can predictably direct the site of oxidation based on the distance from the auxiliary.
Figure 6.

Oxidation of lisofylline and theobromine derivatives by P450 3A4.
Figure 7.

The scope of functionalized theobromine substrates and their conversion products by P450 3A4; Tb, theobromine.56
The authors investigated another potential role for the theobromine auxiliary: facilitating product recovery from whole- cell reaction mixtures.59 Product isolation from fermentation reactions is an expensive and time-consuming process that generates large amounts of organic solvent waste. Molecularly imprinted polymers (MIPs) are polymers assembled in the presence of a template.60–68 After removal of the template, MIPs tend to show selective affinity for molecules containing the template moiety. This occurs mainly through non-covalent interactions such as complementarity in size, shape, electrostatics and hydrogen bonding. With the aid of MIPs, the theobromine auxiliary turned out to be an asset to facilitate isolation of the starting materials and products from complex matrices typically used in biotransformations.59 Overall, the MIPs captured >85% of the theobromine derivatives from complex mixtures and showed high reusability. It also yielded an isolated product of >90% purity. These results suggest that chemical auxiliaries can find use not only in controlling the selectivity of P450 transformations but also in facilitating product recovery.
In a subsequent study by the same group, type II P450 ligands proved successful as chemical auxiliaries to direct unnatural substrates to the P450 2E1 active site.57 Type II ligands are molecules which coordinate to the heme iron of P450s and are typically viewed as P450 inhibitors.57,69,70 Usually, type II ligands have a higher affinity for the active site than similar substrates lacking nitrogen.71–73 However, more recent studies have shown that some type II ligands can be metabolized by P450s.71,74–78 To design a system with complementary selectivity to that observed with the theobromine auxiliary and P450 3A4, the authors investigated a variety of type II ligands as directing groups for the transformation of various substrates by P450 2E1.57 Modeled after methyl nicotinate, a known type II ligand of P450 2E172, and nicotine-derived nitrosamine ketone, a naturally occurring P450 2E1 substrate, nicotinate proved to be the most useful chemical auxiliary among those tested (Figure 8). Using a series of nicotinate esters the authors confirmed the preference of P450 2E1 to oxidize aliphatic CH2 or alkenyl CH groups furthest from the auxiliary (ω–1), as previously observed for the hydroxylation of fatty acids such as lauric, myristic, palmitic and stearic acids.53,79 In the absence of the auxiliary, however, the enzyme did not significantly transform any of the substrates, confirming the role of the auxiliary in forcing biocatalysis in a predictable manner.
Figure 8.

Oxidation of nicotinate containing unnatural substrates by P450 2E1.57
4. Conclusion
We discussed here two simple and quick strategies reported to increase the substrate scope of P450 biocatalysts without the need for protein mutagenesis. Both decoy molecules and substrate engineering have the ability to influence the substrate specificity as well as the regio- and stereoselectivity of P450-catalyzed reactions. Although these strategies require extra molecules or synthetic steps, this is often offset by a number of advantages, the key one being that the desired products can otherwise require several synthetic steps to be produced. Substrate engineering is largely unexplored in the area of P450 biocatalysis. As well, there is room for further investigation of decoy molecules and other cooperative modulators of P450 activity. The successful stories summarized here justify additional efforts in this area.
Acknowledgments
Writing of this article and research in the area of P450 enzymes in the Auclair group have been funded by the National Science and Engineering Research Council of Canada (NSERC), the Center in Green Chemistry and Catalysis, Merck Frosst Canada Ltée, Boehringer Ingelheim Canada, and AstraZeneca Canada. V.P. was supported by scholarships from the Dr. Richard H. Tomlinson Foundation, Walter C. Sumner Foundation, and the Centre in Green Chemistry and Catalysis.
Biographies

Karine Auclair was born in 1972 in Jonquiere, Canada. As a graduate student (1994–1999) in the lab of Prof. J. C. Vederas at the University of Alberta (Edmonton, Canada), she studied the biosynthesis of the cholesterol-lowering drug lovastatin (Mevacor). Amongst her thesis achievements, she reported the first purified natural Diels-Alderase. Next, she moved to the University of California at San Francisco (USA) to pursue post-doctoral studies with Prof. P. R. Ortiz de Montellano (1999–2001). Her research results contributed to the mechanistic understanding of heme oxygenases and P450 enzymes. In 2002, she started her independent career as an Assistant Professor of Chemistry at McGill University (Montreal, Canada). She was promoted Associate Professor of Chemistry with tenure in 2006. She was a Visiting Scientist at Boehringer Ingelheim (Laval, Canada) in 2010. Her current research at McGill covers the areas of antibiotic resistance and P450 enzymes. Some of her key contributions include aminoglycoside resistance inhibitors active in cells, new antibacterial agents, new insights into enzyme allostery, as well as mechanistic and biocatalytic studies of P450 enzymes.

Vanja Polic was born in 1989 in Zenica, Bosnia and Herzegovina. She graduated from the University of Waterloo (Waterloo, Canada) with an Honors B.Sc. in Biochemistry. Her undergraduate research, funded by the National Science and Engineering Council of Canada focused on the calcium-binding domain of calmodulin. Vanja is a graduate student in chemistry at McGill University (Montreal, Canada) since 2011. Her current interests focus on the chemical and mutagenic modification of P450s for mechanistic studies.
References
- 1.Bernhardt R. J Biotechnol. 2006;124:128. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Publishers; New York: 2005. p. 377. [Google Scholar]
- 3.Gillam EMJ. Clin Exp Pharmacol Physiol. 2005;32:147. [Google Scholar]
- 4.Guengerich FP. Chem Res Toxicol. 2001;14:611. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 5.Urlacher VB, Girhard M. Trends Biotechnol. 2011;30:26. doi: 10.1016/j.tibtech.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Jung ST, Lauchli R, Arnold FH. Curr Opin Biotechnol. 2011;22:809. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munro AW, Girvan HM, Mason AE, Dunford AJ, McLean KJ. Trends Biochem Sci. 2013;38:140. doi: 10.1016/j.tibs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Grogan G. Curr Opin Chem Biol. 2011;15:241. doi: 10.1016/j.cbpa.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Kumar S. Expert Opin Drug Metab Toxicol. 2011;6:115. doi: 10.1517/17425250903431040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meinhold P, Peters MW, Hartwick A, Hernandez AR, Arnold FH. Adv Synth Catal. 2006;348:763. [Google Scholar]
- 11.Fasan R, Chen MM, Crook NC, Arnold FH. Angew Chem Int Ed Engl. 2007;46:8414. doi: 10.1002/anie.200702616. [DOI] [PubMed] [Google Scholar]
- 12.Li QS, Schwaneberg U, Fischer P, Schmid RD. Chem Eur J. 2000;6:1531. doi: 10.1002/(sici)1521-3765(20000502)6:9<1531::aid-chem1531>3.3.co;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Seifert A, Vomund S, Frohmann K, Kriening S, Urlacher VB, Laschat S, Pleiss J. Chem Bio Chem. 2009;10:853. doi: 10.1002/cbic.200800799. [DOI] [PubMed] [Google Scholar]
- 14.Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong CH, Arnold FH. Chem Bio Chem. 2010;11:2502. doi: 10.1002/cbic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolev JN, Zaengle JM, Ravikumar R, Fasan R. Chem Bio Chem. 2014;15:1001. doi: 10.1002/cbic.201400060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fasan R, Meharenna YT, Snow CD, Poulos TL, Arnold FH. J Mol Biol. 2008;383:1069. doi: 10.1016/j.jmb.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang K, El Damaty S, Fasan R. J Am Chem Soc. 2011;133:3242. doi: 10.1021/ja109590h. [DOI] [PubMed] [Google Scholar]
- 18.Zhang K, Shafer BM, Demars MD, Stern HA, Fasan R. J Am Chem Soc. 2012;134:18695. doi: 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolev JN, O’Dwyer KM, Jordan CT, Fasan R. ACS Chem Biol. 2014;9:164. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guengerich FP. Chem Res Toxicol. 2008;21:70. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 21.Guengerich FP. Annu Rev Pharmacol Toxicol. 1999;39:1. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Shoji O, Fujishiro T, Nakajima H, Kim M, Nagano S, Shiro Y, Watanabe Y. Angew Chem Int Ed Engl. 2007;46:3656. doi: 10.1002/anie.200700068. [DOI] [PubMed] [Google Scholar]
- 23.Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, Adachi S-I, Park S-Y, Shiro Y. J Biol Chem. 2003;278:9761. doi: 10.1074/jbc.M211575200. [DOI] [PubMed] [Google Scholar]
- 24.Fujishiro T, Shoji O, Watanabe Y. Tetrahedron Lett. 2011;52:395. [Google Scholar]
- 25.Shoji O, Fujishiro T, Nagano S, Tanaka S, Hirose T, Shiro Y, Watanabe Y. J Biol Inorg Chem. 2010;15:1331. doi: 10.1007/s00775-010-0692-4. [DOI] [PubMed] [Google Scholar]
- 26.Baker MT, Bates JN, Leff SV. Anesth Analg. 1987;66:1141. [PubMed] [Google Scholar]
- 27.Baker MT, Ronnenberg WC., Jr Toxicol Appl Pharmacol. 1992;114:25. doi: 10.1016/0041-008x(92)90092-7. [DOI] [PubMed] [Google Scholar]
- 28.Want Y, Baker MT. Drug Metab Dispos. 1993;21:299. [PubMed] [Google Scholar]
- 29.Wang Y, Olson MJ, Baker MT. Biochem Pharmacol. 1993;46:87. [PubMed] [Google Scholar]
- 30.Zilly FE, Acevedo JP, Augustyniak W, Deege A, Häusig UW, Reetz MT. Angew Chem. 2011;123:2772. doi: 10.1002/anie.201006587. [DOI] [PubMed] [Google Scholar]
- 31.Kawakami N, Shoji O, Watanabe Y. Angew Chem Int Ed Engl. 2011;50:5315. doi: 10.1002/anie.201007975. [DOI] [PubMed] [Google Scholar]
- 32.Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Biochemistry. 1997;36:370. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- 33.Atkins WM. Annu Rev Pharmacol Toxicol. 2005;45:291. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- 34.Leroux F. Chem Bio Chem. 2004;5:644. doi: 10.1002/cbic.200300906. [DOI] [PubMed] [Google Scholar]
- 35.Zilly FE, Acevedo JP, Augustyniak W, Deege A, Häusig UW, Reetz MT. Angew Chem Int Ed Engl. 2011;52:13483. doi: 10.1002/anie.201006587. [DOI] [PubMed] [Google Scholar]
- 36.Shoji O, Kunimatsu T, Kawakami N, Watanabe Y. Angew Chem Int Ed Engl. 2013;52:1. doi: 10.1002/anie.201300282. [DOI] [PubMed] [Google Scholar]
- 37.Pietz S, Wolker D, Haufe G. Tetrahedron. 1997;4020:17067. [Google Scholar]
- 38.Dawson MJ, Lawrence GC, Mayall J, Noble D, Roberts SM, Turner MK, Wall WF. Tetrahedron Lett. 1986;27:1089. [Google Scholar]
- 39.Holland HL, Brown FM, Larsen BG, Zabic M. Tetrahedron: Asymmetry. 1995;6:1569. [Google Scholar]
- 40.Sundby E, Azerad R, Anthonsen T. Biotechnol Lett. 1998;20:337. [Google Scholar]
- 41.Virne B, Archelas A, Fustoss R. Tetrahedron. 1991;47:1447. [Google Scholar]
- 42.Holland HL, Morris TA, Nava PJ, Zabic M. Tetrahedron. 1999;55:7441. [Google Scholar]
- 43.Braunegg G, de Raadt A, Feichtenhofer S, Griengl H, Kopper I, Lehmann A, Weber H. Angew Chem Int Ed Engl. 1999;38:2763. [PubMed] [Google Scholar]
- 44.De Raadt A, Griengl H, Weber H. Chemistry. 2001;7:27. doi: 10.1002/1521-3765(20010105)7:1<27::aid-chem27>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 45.Li S, Chaulagain MR, Knauff AR, Podust LM, Montgomery J, Sherman DH. Proc Natl Acad Sci USA. 2009;106:18463. doi: 10.1073/pnas.0907203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Münzer DF, Meinhold P, Peters MW, Feichtenhofer S, Griengl H, Arnold FH, Glieder A, De Raadt A. Chem Commun. 2005:2597. doi: 10.1039/b501527h. [DOI] [PubMed] [Google Scholar]
- 47.Lewis JC, Bastian S, Bennett CS, Fu Y, Mitsuda Y, Chen MM, Greenberg WA, Wong C, Arnold FH. Proc Natl Acad Sci USA. 2009;106:16550. doi: 10.1073/pnas.0908954106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue Y, Zhao L, Liu HW, Sherman DH. Proc Natl Acad Sci USA. 1998;95:12111. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xue Y, Wilson D, Zhao L, Sherman DH. Chem Biol. 1998;5:661. doi: 10.1016/s1074-5521(98)90293-9. [DOI] [PubMed] [Google Scholar]
- 50.Li S, Podust LM, Sherman DH. J Am Chem Soc. 2007;129:12940. doi: 10.1021/ja075842d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Negretti S, Narayan ARH, Chiou KC, Kells PM, Stachowski JL, Hansen DA, Podust LM, Montgomery J, Sherman DH. J Am Chem Soc. 2014;136:4901. doi: 10.1021/ja5016052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Julsing MK, Cornelissen S, Bühler B, Schmid A. Curr Opin Chem Biol. 2008;12:177. doi: 10.1016/j.cbpa.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 53.Chefson A, Zhao J, Auclair K. Chem Bio Chem. 2006;7:916. doi: 10.1002/cbic.200600006. [DOI] [PubMed] [Google Scholar]
- 54.Podust LM, Sherman DH. Nat Prod Rep. 2012;29:125. doi: 10.1039/c2np20020a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Redlich G, Zanger UM, Riedmaier S, Bache N, Giessing ABM, Eisenacher M, Stephan C, Meyer HE, Jensen ON, Marcus K. J Proteome Res. 2008;7:4678. doi: 10.1021/pr800231w. [DOI] [PubMed] [Google Scholar]
- 56.Larsen AT, May EM, Auclair K. J Am Chem Soc. 2011;133:7853. doi: 10.1021/ja200551y. [DOI] [PubMed] [Google Scholar]
- 57.Menard A, Fabra C, Huang Y, Auclair K. Chem Bio Chem. 2012;13:2527. doi: 10.1002/cbic.201200524. [DOI] [PubMed] [Google Scholar]
- 58.Shin HS, Slattery JT. J Pharm Sci. 1998;87:390. doi: 10.1021/js970382f. [DOI] [PubMed] [Google Scholar]
- 59.Larsen AT, Lai T, Polic V, Auclair K. Green Chem. 2012;14:2206. [Google Scholar]
- 60.Cormack PAG, Elorza AZ. J Chromatog B. 2004;804:173. doi: 10.1016/j.jchromb.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 61.He C, Long Y, Pan J, Li K, Liu F. J Biochem Biophys Meth. 2007;70:133. doi: 10.1016/j.jbbm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 62.Kist TBL, Mandaji M. Electrophoresis. 2004;25:3492. doi: 10.1002/elps.200406114. [DOI] [PubMed] [Google Scholar]
- 63.Qiao F, Sun H, Yan H, Row KH. Chromatographia. 2006;64:625. [Google Scholar]
- 64.Turiel E, Martín-Esteban A. Anal Chim Acta. 2010;668:87. doi: 10.1016/j.aca.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 65.Jin Y, Row KH. Polymer. 2007;28:276. [Google Scholar]
- 66.Theodoridis G, Manesiotis P. J Chromatog A. 2002;948:163. doi: 10.1016/s0021-9673(01)01457-1. [DOI] [PubMed] [Google Scholar]
- 67.Tse Sum Bui B, Haupt K. Anal and Bioanal Chem. 2010;398:2481. doi: 10.1007/s00216-010-4158-x. [DOI] [PubMed] [Google Scholar]
- 68.Cirillo G, Curcio M, Parisi OI, Puoci F, Iemma F, Spizzirri UG, Restuccia D, Picci N. Food Chem. 2011;125:1058. [Google Scholar]
- 69.Vasaitis TS, Bruno RD, Njar VCO. J Steroid Biochem Mol Biol. 2011;125:23. doi: 10.1016/j.jsbmb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mercer EI. Lipids. 1991;26:584. doi: 10.1007/BF02536422. [DOI] [PubMed] [Google Scholar]
- 71.Peng CC, Pearson JT, Rock DA, Joswig-Jones CA, Jones JP. Arch Biochem Biophys. 2010;497:68. doi: 10.1016/j.abb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones JP, Joswig-Jones CA, Hebner M’, Chu Y’, Koop DR. Chem Biol Interact. 2011;193:50. doi: 10.1016/j.cbi.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng CC, Cape JL, Rushmore T, Crouch GJ, Jones JP. J Med Chem. 2008;51:8000. doi: 10.1021/jm8011257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pearson JT, Hill JJ, Swank J, Isoherranen N, Kunze KL, Atkins WM. Biochemistry. 2006;45:6341. doi: 10.1021/bi0600042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dahal UP, Joswig-Jones CA, Jones JP. J Med Chem. 2012;55:280. doi: 10.1021/jm201207h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pearson J, Dahal UP, Rock DA, Peng C-C, Schenk JO, Joswig-Jones CA, Jones JP. Arch Biochem Biophys. 2011;511:69. doi: 10.1016/j.abb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chiba M. Drug Metab Dispos. 2001;29:1. [PubMed] [Google Scholar]
- 78.Hutzler JM, Melton RJ, Rumsey JM, Schnute ME, Locuson CW, Wienkers LC. Chem Res Toxicol. 2006;19:1650. doi: 10.1021/tx060198m. [DOI] [PubMed] [Google Scholar]
- 79.Adas F, Salaün JP, Berthou F, Picart D, Simon B, Amet Y. J Lipid Res. 1999;40:1990. [PubMed] [Google Scholar]