

Abstract
The effective delivery of therapeutics and imaging agents to a selected group of cells has been at the forefront of biomedical research. Unfortunately, the identification of the unique cell surface targets for cell selection remains a major challenge, particularly if cells within the selected group are not identical. Here we demonstrate a novel approach to cell section relying on a thermo-responsive peptide-based nanocarrier. The hybrid peptide containing cell-penetrating peptide (CPP) and collagen (COLL) domains is designed to undergo coil-to-helix transition (folding) below physiological temperature. Since only helical form undergoes effective internalization by the cells, this approach allows effective temperature-discriminate cellular uptake. The cells selected for uptake are locally cooled down thus enabling the carrier to fold and subsequently internalize. Our approach demonstrates a generic method as selected cells could differ from the adjacent cells or could belong to the same cell population. The method is fast (< 15 min) and selective; over 99.6% of cells in vitro internalized the peptide carrier at low temperatures (15°C), while less than 0.2% internalized at 37°C. In vivo results confirm the high selectivity of the method. The potential clinical applications in mixed cell differentiation carcinoma, most frequently encountered in breast and ovarian cancer, are envisioned.
The idea of targeting specific cells for drug delivery and imaging was proposed over 40 years ago.1 While multiple potential targeting moieties have been discovered (antibodies, aptamers, small protein and peptide scaffolds, and small molecule ligands), the development of the viable, unique targets for cancer cells has been a major challenge.2–7 Molecular targeting approaches for cancer treatment are of limited utility in selecting for cancerous populations because the targets are, invariably, also expressed by healthy host cells.8–14 For example HER2, one of the best currently known targetable cell surface receptor in breast cancer, is overexpressed only twice in cancer cells with comparison to healthy cells.8,15 The variability of cancer cells patient-to-patient and between different stages of the disease, and the need for sufficient number of receptors to be present on the cell surface in order to facilitate receptor-mediated endocytosis of therapeutic amount of cytotoxic agents further complicates the molecular targeting startegies.3,16,17 Moreover, the histology of the cells within a single tumor can vary significantly as observed, for example, in cases of mixed cell differentiation carcinoma in breast and ovarian cancers.18,19
In an effort to overcome difficulties in identifying the specific cell surface receptors for drug delivery, the physicochemical approaches were also explored. The pH and, to smaller extent, temperature stimuli were both employed in the past in cell selection.20 Some tumors generate lower pH in their surrounding (6.5 versus 7.4). This can be used as a trigger in carrier disintegration at lower pH resulting in cargo delivery.20–22 Hyperthermia, locally induced increase in temperature (40–45°C), was also used to initiate the release of cargo from liposomes and polymer-based carriers at tumor site.23–25 Unfortunately neither of these methods can be considered generic, and both methods suffer from low selectivity.
There are several generic methods exploiting the increased metabolic activity of malignant cells versus healthy cells. These methods avoid problems associated with identification of specific receptor targets. Tumor imaging with 2-deoxy-2-(18F)fluoro-D-glucose in positron emission tomography (PET) is current clinical standard in tumor detection.26 Recently a new generic method employing optical sensing via smart plasmonic glucose nanosensors was demonstrated in vitro.27 However many malignancies that belong to the family of mucous neoplasm can’t be targeted via glucose metabolism, because their metabolism is only slightly elevated in comparison with healthy cells.28
Here, we describe a new generic method of cell selection. This method does not rely on molecular targets. Instead, by employing a simple thermoresponsive delivery system, the cell selection process is controlled by externally-induced temperature difference between the targeted cells and the adjacent cells. The selected cells could differ from the adjacent cells or belong to the same cell population. The proposed nanocarrier is a short peptide adopting inactive coil conformation (unfolded) at HIGH (physiological) temperature and a helix conformation (folded) at LOW temperature. The peptide in a helix conformation is able to penetrate cell membranes and deliver a cargo (fluorescent tag).
The key to this method is a nanocarrier adapting two structurally distinctive states (ON/OFF) depending on temperature. It is known that collagen peptides fold reversibly into triple helix and the folding temperature depends on the sequence. Recently, we demonstrated that hybrid peptides containing cell-penetrating peptides (multiple Arginines; CPP) and sequences characteristic for collagen ((Proline-Hydroxyproline-Glycine)n; COLL) can penetrate the cellular membrane when folded into a triple helical conformation. The randomly coiled conformation results in little or no penetration.29 The collagen domain of the peptide controls folding, while the CPP domain affords internalization.29,30 The family of hybrid COLL/CPP peptides have many characteristics of efficient drug carriers:they are composed of non-cytotoxic peptide monomers, have ideal shape and size (rigid-rods nanoparticles, 1.4 nm diameter and 7.6 nm length when folded), and can be easily modified via peptide chemistry.29 In previous studies, we also found that the hybrid COLL/CPP peptides are resistant to enzymatic serum digestion.29 In this report we describe new COLL/CPP hybrid peptides designed for efficient temperature-discriminate cellular uptake. We also demonstrate in-vitro and in-vivo target-free temperature-based cell selection and delivery of molecular cargo (FITC Tag).
In view of future applications, a temperature-based method seems to be well suited for generic cell selection. Although, for the whole body, even small deviations from physiological temperature (36.6 °C) can cause irreversible changes or death, local temperature changes are well tolerated31–33 Local temperature control has been successfully employed in medical treatments. For example locally-induced hypothermia is routinely used to decrees swelling and to lower clotting threshold in treatment of physical body trauma.34–37
EXPERIMENTAL SECTION
Peptides
All peptides were purchased from the Tufts University Core Facility (solid support synthesis and HPLC purification) modified with fluorescein isothiocyanate (FITC; excitation wavelength (EX) = 494 nm, emission wavelength (EM) = 521 nm) at the N-terminus via a β-alanine–glycine–glycine linker.
Circular Dichroism Spectroscopy
Circular dichroism (CD) measurements were performed using a JASCO J-810 spectropolarimeter (JASCO Inc., Easton, MD, USA) equipped with a Peltier temperature control system containing a quartz cell (path length 0.2 cm). Prior to each measurement, the peptides were thermally annealed: peptide solutions in water (2 × 10−7 M) were pre-heated at 80 °C for 5 min and slowly cooled to 4 °C, then incubated for 24 h. The peptide solutions were transferred to a CD cell and equilibrated for 30 min at 37 °C. A scan speed of 50 nm/min was used, and four scans per sample were acquired. A reference spectrum containing deionized water was subtracted from the final peptide spectrum.
Thermal unfolding curves were obtained by monitoring the decrease in ellipticity in a 25–80 °C temperature range (dependent on the peptide) at a wavelength where the CD spectra show a positive maximum (224 nm) at a heating rate of 10 °C/h. The derivative of the plotted unfolding curve was calculated using the JASCO Spectra Manager II software (JASCO Inc.). The minimum of the derivative indicates the steepest slope of the unfolding process and determines the helix-to-coil transition temperature (Tm) under the described conditions. All experiments were performed in duplicate or triplicate.
Cell Culture and Cellular Uptake
NIH-3T3 Swiss mice fibroblasts were purchased from ATCC and cultured according to provided protocols. Briefly, the cells were cultured in complete media: Dulbecco’s modified Eagle’s medium (Mediatech Inc.), 10% fetal bovine serum (Cellgrow), 100-U⁄mL penicillin (Fisher Scientific); and 0.1-mg⁄ml streptomycin (Fisher Scientific). Passages 3–10 were used for all experiments.
Peptide uptake studies were performed by dissolving the given peptide in phosphate-buffered saline (PBS; pH = 7.4; Fisher Scientific) and incubating cells (90% confluence) with the peptide for variable times (5–120 min) and temperatures (4, 15, 37 °C). Subsequently, cells were washed three times and examined with microscopy or flow cytometry.
Animal Material
All in vivo experiments were performed with Pristina leidyi (Carolina biological Supply sold as Stylaria) maintained as previously described in artificial spring water.38,39 Before each experiment animals were collected from growing cultures placed in the separate container in freshly prepared spring water (1% artificial sea water) and anesthetized with 8% ethanol. Animals were incubated with peptide carriers for 15 min at 19 °C or 37 °C, washed several times with PBS and imaged with the microscope (confocal or fluorescence). Once ethanol is removed from the media animals regain mobility within 30 min. The animals were observed for additional 36 h to ensure lack of peptide toxicity. When P. leidyi was subjected to temperature gradient, it was first immobilized in 4% agar gel in spring water (about 3 mm thickness). The gels were cut and inverted to expose the specimen to peptide solution; the remaining of the protocol was the same as outlined above.
Microscopy
Cells were imaged using a confocal microscope (Olympus FluoView) and an inverted fluorescent microscope (Nikon TE2000). 3T3 Swiss mice fibroblasts were seeded at a density of 50,000 cells/mL in complete media in 35-mm Petri dishes with No. 1.5 coverslip as a bottom (MatTek Corporation, Ashland, MA, USA) and incubated overnight. Cells were incubated with peptides in PBS (0.1–100 μM) for up to 120 min. After incubation, cells were washed three times with fresh PBS. The cells were imaged immediately following treatment.
Flow Cytometry
NIH-3T3 Swiss mice fibroblast cells were used for flow cytometry assays. Cells were grown to 90% confluency and incubated with peptides for 15 min at 15 or 37 °C. After the incubation period, cells were washed three times with cold PBS, trypsinized, and then resuspended in PBS containing 0.1% Bovine Serum Albumin. Cells (10–80k/sample) were analyzed using flow cytometry (Cell Lab Quanta SC-MPL, Beckman Coulter, Inc., Brea, CA, USA) to quantify the cellular uptake of the peptides.
Temperature gradient device
The 2D temperature gradient was generated across the flat area constructed by inserting polyethylene window into two aluminum (Spellbinders, 30 gauge) compartments (Figure S1). The size of the device is 2×4.5 cm, large enough to place the cell culture on top. The cooling/heating mechanism is based on pumping cold and hot water into the two compartments connected with tubing (Tygon R-3603) to two water pumps (Aquascape XS 70 GPH). The temperature of circulating water was 0°C and 45°C. The spatial temperature distribution (Figure S2) on the top of the device was measured with infrared thermometer gun (Visual IR Thermometer, Fluke VT02).
RESULTS AND DISCUSSION
We have previously demonstrated that only folded forms of COLL/CPP peptides are able to penetrate the cell membrane.29 In this report we describe a new generic cell selection method based on induced temperature difference between the selected cells and adjacent cells. The emphasis of the current work was placed on the modification of the peptide sequence allowing highly efficient temperature-discriminate cellular uptake, and therefore cell selection.
We first report Circular Dichroism studies of hybrid peptide nanocarriers yielding temperature dependent folding characteristics, we then discusses temperature dependent uptake studies in vitro (NIH 3T3 Swiss mouse fibroblasts) and in vivo using fresh water worm as animal model (Pristina Leidyi).
Thermo-responsive peptides
The sequence of FL8V1, a hybrid peptide nanocarrier studied previously,29 was modified to lower the folding temperature below physiological temperature (37°C). Table 1 shows sequences of peptides designed to adopt an inactive, random coil conformation in physiological conditions while undergoing coil-to-helix transition at lower temperatures. The coil-to-helix transition results in triple helix conformation leading to formation of nanoparticle carrier able to penetrate the cell membrane (Scheme 1). In this design the peptide nanocarrier can be internalized by the cell (ON switch) in triple helical form while it is not internalized (OFF switch) in the random coil form. In the proof-of-concept experiment, the peptide nanocarriers were modified with a fluorescein (FITC) tag that serves as an example of molecular cargo and at the same time enables monitoring of cellular uptake.
Table 1.
Peptide nanocarrier sequences. “O” stands for hydroxyproline. FF is peptide folded fraction calculated from CD.41
| Peptide | Sequence | Tm (°C) | FF @ 37 °C | Conformation@ 37 °C |
|---|---|---|---|---|
| FL8V1 | FITC-AβGG-(POG)8-RRGRRG | 48.8 | 0.90 | Triple helix |
| FL7V1 | FITC-AβGG-(POG)7-RRGRRG | 17.2 | — | Random coil |
| FL6V1 | FITC-AβGG-(POG)6-RRGRRG | 15.3 | — | Random coil |
Scheme 1.
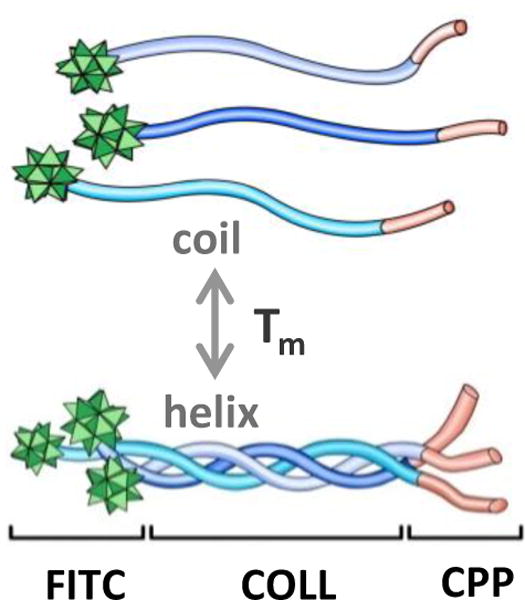
Scheme of hybrid peptide domains and reversible thermal folding. FITC (fluorescein tag), COLL (collagen folding domain), CPP (cell penetrating peptide domain).
To accomplish the scheme outlined above the sequence of previously studied hybrid peptide FL8V1 was truncated to form new peptide nanocarriers FL7V1 and FL6V1. All studied nanocarriers: FL8V1 (control), FL7V1, and FL6V1 (new nanocarriers) were synthesized, purified and their sequence confirmed (MS, HPLC) by Tufts University Core Facility. To monitor the helix-to-coil transition (unfolding), the decrease in molar ellipticity at 224 nm, characteristic of a triple helix conformation (positive nπ* transition), was measured with Circular Dichroism Spectroscopy (Figure 1).40 The transition temperature (Tm) was calculated as a minimum of the first derivative of molar ellipticity (Table 1). The Tm of FL8V1 nanocarrier was determined to be 48.8°C. Thus, the peptide persists in solution in the folded conformation at 37°C; FF@37=0.90, where FF is the folded fraction of peptide defined as:41
| (1) |
and CDx represents molar ellipticity measured at 224 nm. We used Brodsky model of sequence-dependent thermal stability to design the peptide sequence in order to achieve efficient temperature-discriminate cellular uptake based on temperature dependent folding into nanoparticle. Based on this model, we expected the Tm to fall to about 25 °C for FL7V1 and to about 18 °C for FL6V1.42,43 Truncation of the COLL domain of the FL8V1 nanocarrier sequence by one (FL7V1) and two (FL6V1) POG repeat units, results in the peptide Tm drop to 17.2 °C and 15.3 °C, respectively. Therefore both FL7V1 and FL6V1 nanocarriers persist in a random coil conformation at physiological conditions.
Figure 1.
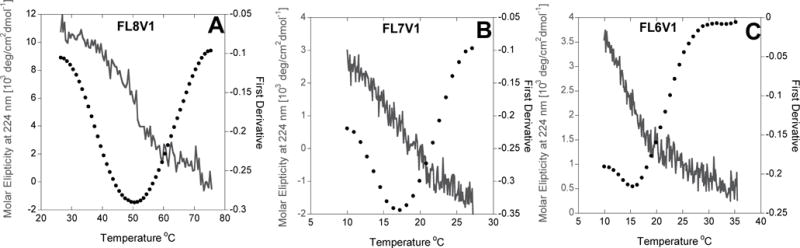
Molar ellipticity at 224 nm (solid line) of (a) FL8V1, (b) FL7V1, and (c) FL6V1, and first derivative (dotted curve). Thermal unfolding shows that the peak (224nm) is eliminated in a cooperative transition what indicates the helix-to-coil transition.
In both instances, the difference between the predicted (Brodsky model) and experimental value of Tm was unexpectedly large. It was reported that some of the experimental Tm and predicted Tm in the model vary by as much as 13 °C, particularly if the sequence contains multiple arginine and lysine residues.42,43 Clearly, the presence of multiple arginines in close proximity (CPP domain) induces strong repulsion between the folded strands. Moreover, comparing FL7V1 and FL8V1 nanocarriers, the addition of a single POG sequence has a dramatic effect in structural stabilization of peptide, while truncating FL7V1 to FL6V1 by single POG sequence has very small effect on peptide Tm. These differences indicate the presence of strong cooperative effects in folding stabilization of nanocarrier. Because the effect is based on cooperation the temperature dependence of folding/unfolding process is affected: to fully unfold the FL8V1 the temperature difference between Tm and fully unfolded state is over 20°C, while in the case of FL6V1 and FL7V1, it only takes about 10°C (Figure 1) in the same conditions. This suggests that nanocarriers FL6V1 and FL7V1 are well suited to thermally discriminate the delivery of molecular cargo to cells.
Peptide nanocarrier uptake studies in vitro
While our previous studies concluded that peptide nanocarrier is able to translocate across cellular membrane carrying molecular cargo (FITC tag) in folded conformation only, thermally controlled delivery was not considered.29 Lowering the temperature from physiological value affects not only peptide folding, but also the effectiveness of nanocarrier cellular uptake due to changes induced in the cell membrane.
When we consider the general mechanism of cellular uptake of CPP, the number of positive charges present in the sequence is a main factor allowing the CPP to cross the cell membrane. In the unfolded state (coil conformation) the four arginine residues present within the CPP domain of FL6V1 and FL7V1 peptides do not carry sufficiently positive charge to translocate across the membrane, what is consistent with other CPPs.44,45 Once folded into nanocarrier (helix conformation) the number of arginine residues is increased to twelve per peptide (Scheme 1) resulting in cellular uptake.
Internalization studies using the FITC-conjugated nanocarriers at and below physiological temperature were performed in NIH 3T3 Swiss mouse fibroblasts cells. The cells were treated across a range of peptide carrier concentrations (5–75 μM), incubation temperatures (4–37 °C), and incubation times (5–120 min) (Figure S3–S6). The images were acquired using confocal microscopy (excitation 488 nm, Figure 2). At physiological temperature (37 °C) the fluorescence is observed only in cells exposed to FL8V1 nanocarrier. The fluorescence is dim because in order to preserve viability of the cells we have used very low laser power for excitation (488nm). None of the cells exhibited auto-fluorescence (Figure 2d,f) consistent with previously published results.29 The first visible fluorescent cells are observed after about 7 min of exposure. On the other hand FL7V1 and FL6V1 peptide nanocarriers (for both peptide nanocarriers Tm<20°C) are not internalized into the cells at 37 °C for up to 2h of incubation time. At an incubation temperature of 15 °C (below Tm for all peptide nanocarriers), fluorescence was detected after exposure to all three nanocarriers. For all conditions, the fluorescence signal is observed within cell cytoplasm, as previously shown for tagged peptides that carry an RRGRRG sequence in the CPP domain.29 If the desired location of peptide carrier is nucleus, the CPP domain can be altered to R6, what causes accumulation of peptide in the nuclear bodies that contain high concentration of nucleic acids.29
Figure 2.
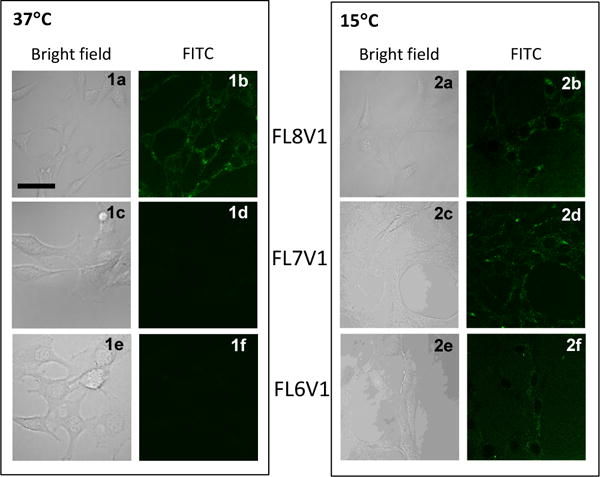
Confocal microscopy images of 3T3 mouse fibroblasts incubated for 15 min with FL6V1, FL7V1 and FL8V1 peptide nanocarrier (15μM) at 37 °C (panel 1) and 15 °C (panel 2); bar represents 20 μm.
The molecular pathways of cellular uptake of CPP-related peptides are widely discussed and there is no consensus which pathway dominates the uptake, and if the pathway is sequence specific.44,45 However, majority of available pathways are dependent on the endosome formation that requires ATP, thus all those pathways would be strongly affected by low temperature conditions. Therefore we investigated peptide nanocarrier uptake under conditions that significantly limit transport mechanisms that require ATP by incubating the cells with peptides at 4 °C.46–47 We observed cytoplasmic fluorescence in cells incubated with nanocarriers at 4 °C, however, the number of cells exhibiting fluorescence was less than at 15 °C. The reduced, but still significant, peptide internalization at 4 °C demonstrates that ATP-dependent translocation is not necessary for internalization of studied nanocarriers and direct translocation is sufficient to accomplish cellular uptake (Figure S7).
In view of possible future applications of peptide nanocarriers in vivo the bioavailability has to be considered. Many of the short peptides suffer from the fast clearing process through the renal pathway, thus limiting the peptide circulation time to 15–30 min and ultimately resulting in decreased bioavailability. The nanocarriers presented here show effective cellular internalization process within 15 min of peptide incubation with cells(most peptides from CPP family will be effectively delivered within 1–3h). This fast internalization raises hopes that the studied system could be used in the future in vivo applications.
Although the microscopy studies of hybrid peptide nanocarriers uptake at different temperatures result in carrier localization information, one can observe at most tens of cells at the time. In order for the temperature to be considered as a differentiation factor for cell selection, there has to be a well-defined difference in the nanocarrier uptake at different temperatures for the entire cell population. We analyzed the cell populations (10,000 – 80,000) with flow cytometry. The protocol of cell incubation with peptides was identical to cells imaged with microscope. As expected, there are no dramatic differences in the cellular internalization of FL8V1 nanocarrier, 69% of cells at 15 °C compared to 81% of cells at 37 °C. However, when incubated with FL6V1 nanocarrier, over 99.6% of cells at 15 °C internalized the peptide, while only 0.2% of cells at 37 °C were able to uptake the nanocarrier (Figure 3). Based on these results, we concluded that FL6V1 nanocarrier could be utilized in the molecular cargo delivery to thermally selected cells.
Figure 3.
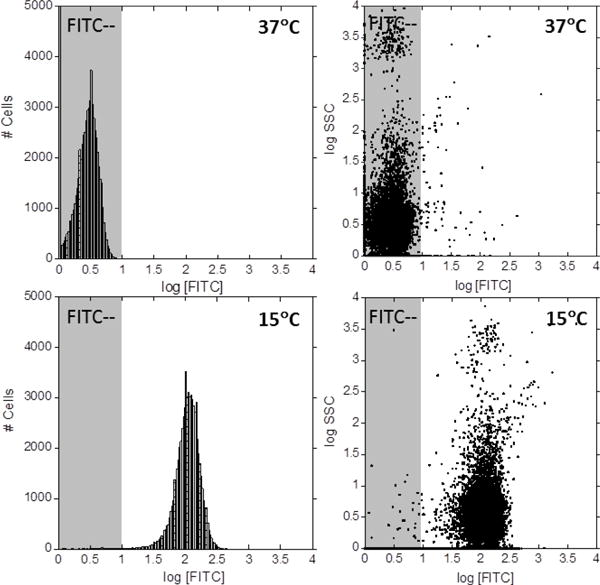
Flow cytometry of NIH 3T3 Swiss mouse fibroblasts incubated for 15 min with FL6V1 at 15 °C and 37 °C.
To test the limits of the switch-like (ON/OFF) uptake of the FL6V1 nanocarrier we thermally selected a small number of cells from a larger population of the same cells. To technically accomplish the selection, we designed a simple device to vary the temperature within a small area, and monitored the population of cells exposed to the temperature gradient (4–40 °C). The details of the device design are described in supplemental materials (Figure S1, S2). The spatial distribution of temperatures was achieved within 2 cm and measured with infrared thermal gun. 3T3 fibroblast cells were seeded on the cover slip and subjected to temperature gradient. The device was placed in the incubator and the peptide nanocarrier was delivered in the fully unfolded state (annealed directly before addition to cell culture). After 15 min the cells were washed and imaged with inverted fluorescent microscope (Figure 4). At temperatures below 20 °C, fluorescence is observed, and it appears that the internalization of FL6V1 is very effective. Although the Tm of FL6V1 was determined to be 15.3 °C, there is still visible, but small, uptake of nanocarrier at 24 °C (FF@24=0.08), though almost no uptake at 28 °C (FF@28=0.02). The cells located within areas with temperatures above 30 °C do not appear fluorescent, and are concluded not to uptake the nanocarrier. By spatially controlling the temperature across the device, we effectively direct the delivery of FITC tag with hybrid peptide nanocarrier to selected cells. Because there is a soft transition in ON/OFF behavior of FL6V1 temperature dependent uptake (between 20 °C and 30 °C), the spatial cell selection for uptake can be improved by generating a sharper temperature gradient. Our device generates a temperature gradient of 24 °C/cm, thus the objects (i.e. clusters of cells) larger than ~600 μm can be thermally selected for uptake.
Figure 4.
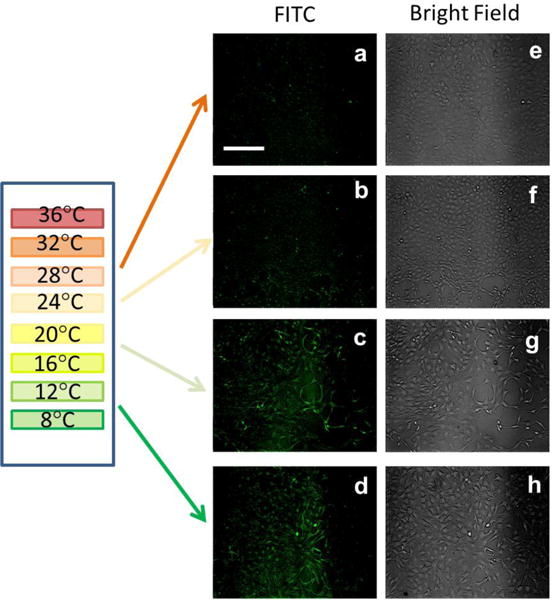
Fluorescence (a–d) and bright-field (e–h) images of NIH 3T3 fibroblasts subjected to temperature gradient and incubated with FL6V1 for 15 min; bar represents 50 μm.
Peptide nanocarrier uptake studies in vivo
Since the in vitro results reported above were very encouraging we next performed in vivo testing using Pristina leidyi (P. leidyi) as an animal model (the anatomy is described in Zattara et al.) uptake.39 P. leidyi is a fresh water worm, typically 2–4 mm long and 0.15–0.20 mm in diameter. The body wall epidermis consists of single layer epidermal cells positioned on top of two layers of body wall muscles. The epidermal cells secrete very thin layer of cuticle that coats the skin. P. leidyi was chosen as the in vivo model because it is easy to obtain, culture, and it is transparent, affording convenient analysis by microscopy.
P.leidyi was anesthetized and incubated with the peptide nanocarrier (FL7V1, 15 μM) for 15 min at various temperatures. After the incubation, the specimen were washed with PBS several times and imaged by confocal microscopy (Figure 5). The images of samples at 19 °C and 37 °C with no peptide present during incubation (Figure 5a–c, g–i) show the auto-fluorescence localized in coelomocytes granules (inner body) and chaetae (bristles), possibly from high concentration of riboflavin. This is consistent with the literature observations.38,39 The animals exposed to FL7V1 nanocarrier at 37 °C (Figure 5d–f) show no differences when compared to the control animals. Specimens exposed to FL7V1 nanocarrier at 19 °C exhibit strong fluorescence localized within the skin of the animal (indicated by arrows in Figure 5j–l). From the observed fluorescence, we concluded that there is a significant uptake of nanocarriers by the epidermal cells. Judging by the intensity of the fluorescence the uptake of nanocarriers seems very effective. We confirmed that the fluorescence was present throughout the entire body wall by imaging P. leidyi with fluorescent microscopy (Figure 6). We confirmed that cells across the body wall undergo peptide internalization to similar extent. We also observed that the auto-fluorescence originating from coelomocytes is present in the control animal (Figure 6b) and it is not related to fluorescence from FL7V1 nanocarrier. The auto-fluorescence intensity originating from coelomocytes (Figure 6b) depends on their position with respect to the focal plane. A very bright image is observed for coelomocytes positioned in the focal plane, while reduced fluorescence intensity is seen for coelomocytes positioned in the focal view but not the focal plane. In order to compare the relative intensities of coelomocytes’ auto-fluorescence for worms exposed to peptide and the control animal, confocal images must be used. For those images (Figure 5h, k) the intensity of fluorescence from coelomocytes is similar for the sample and the control, and the skin exhibits very bright fluorescence after exposure to peptide carriers.
Figure 5.
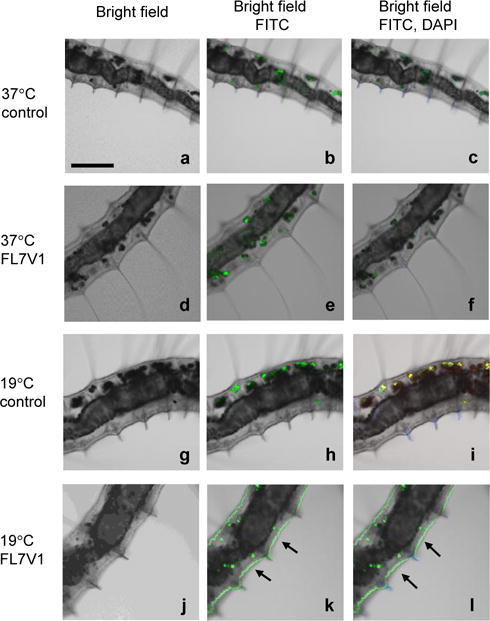
Confocal microscopy images of Pristina leidyi incubated for 15 min with FL7V1 peptide at 37°C (a–f) and 19 °C (g–l); (k, l) arrows indicate FL7V1internalization; bar represents 100 μm; control experiments were performed with no peptide present.
Figure 6.
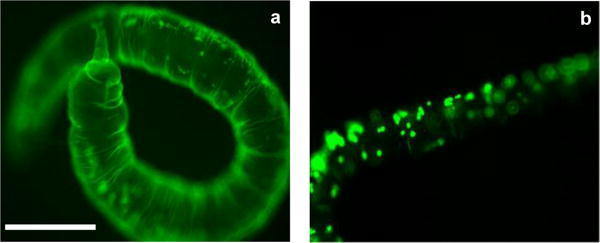
Fluorescent microscopy image of Pristina leidyi (a) incubated with FL7V1 for 15 min at 19 °C and (b) control, no peptide present; bar represents 200 μm.
The animals were subsequently observed for 36 h and remain mobile without any adverse effects detected. The fluorescence from the skin persisted during observation time. These results suggest that the nanocarriers were localized within the epidermal cells. If FL7V1 nanocarriers have been localized within the cuticle, but not uptaken by the epidermal cells, the nanocarrier would have been either present within the skin at both tested temperatures (19 °C and 37 °C) or removed during the post experimental washing at both temperatures.
The cell selection in vivo was performed by positioning P. leidyi within the temperature gradient where part of the animal was exposed to cold and part to warm temperatures. The animal was immobilized by “sandwich” coating in 4% agar gel (about 3 mm thick) to prevent movements. After 15 min of incubation with FL7V1 nanocarrier, the animals were removed from the gel, washed and imaged under fluorescent microscope (Figure 7). The comparison were in plane, thus the intensity of the fluorescence is representative throughout the image. The thermally selected epidermal cells (cold) show intense fluorescence from nanocarrier. The auto fluorescence from coelomocytes was well visible in the FITC images of animals exposed to FL7V1 nanocarrier (Figure 7 a,b), as well as for those with no nanocarrier (control) present during incubation (Figure 7 c,d). The area of the animal that was exposed to temperatures above 28 °C (NOT selected) shows very little fluorescence from the epidermal cells, thus we concluded that there was very little FL7V1 nanocarrier uptake at higher temperatures.
Figure 7.
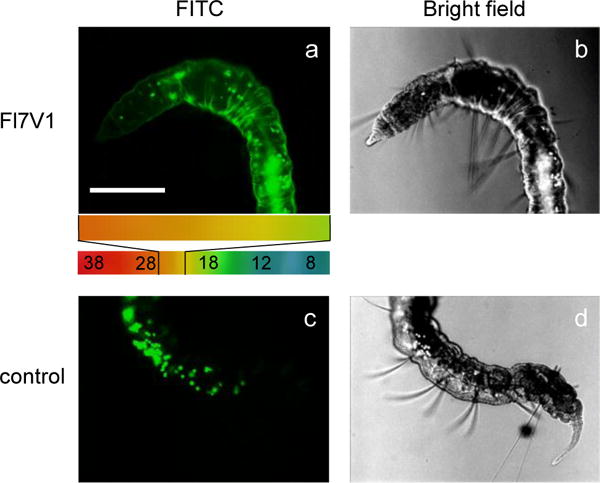
Fluorescence and bright field images of Pristina leidyi subjected to temperature gradient (indicated in color) and (a,b) incubated with FL6V1 for 15 min, (c,d) control, no peptide present; bar represents 200μm.
Both in vitro and in vivo results point to the same conclusion that by controlling the temperature within the specimen (either cell culture or animal) the cells can be thermally selected for uptake. When exposed to temperatures above 28 °C cells are unable to uptake significant amounts of thermo-responsive peptide carriers FL6V1 and FL7V1 (OFF switch), while the cells exposed to lower temperatures show significant nanocarrier uptake (ON switch).
In this report we presented a general idea of thermally controlled cellular uptake. The described peptide carrier has Tm equal to 15°C, but there is still visible uptake to the cells at 24°C (Figure 4), while no uptake at 28°C. Taking physiological conditions into account, the peptide carrier should show lack of uptake at 31°C (peripheral body temperature), and uptake at slightly lower temperature. Assuming that ON/OFF uptake depends on ΔT= 4°C in our case, the ideal “ON” temperature could be as high as 27°C. This temperature is low for whole body therapeutic hypothermia, but appears manageable in case of local, small area cooling, especially that this low temperature will be required for a short period of time (<30 min). Thus the reported peptide might have potential applications in clinical setting. Moreover, relatively small changes in the peptide sequence could allow efficient peptide carrier uptake at temperatures more convenient for clinical applications.
CONCLUSIONS
In this work, we have developed a generic, targeting-free method of effective cell selection using temperature-responsive peptide nanocarriers. The nanocarriers belong to the family of hybrid peptides, composed of collagen and cell-penetrating domains and are capable of discriminating delivery to identical cells using local thermal control both in vitro and in vivo. Unlike other reported thermo-responsive systems based on the premise of polymer swelling at locally higher temperature and releasing the cargo, our system is activated at low temperatures. The selectivity can be potentially further enhanced by improving spatial resolution of temperature gradient used for cell selection. Additionally, the cellular uptake is very fast and can be accomplished under 15 minutes. This fast uptake is encouraging in view of future clinical applications, where fast renal clearance of short peptides often prevents their therapeutic applications by lowering their bioavailability. Moreover, the peptides are non-cytotoxic and can be easily modified to include various cargos by simply exchanging one (or several) amino acids to lysine or cysteine and employ it as convenient modification site. We are currently working on bioconjugation of Paclitaxel to peptide carrier and measuring the prodrug potency on multiple cancer cell lines.
The development of the targeting-free methods of cell selection for drug delivery should be explored further in view of potential applications in mixed cell differentiation carcinoma encountered in breast and ovarian cancers. These types of cancer often exhibit variability in gene expression in cells that are located within the tumor. The difference in gene expression can lead to differences in physiological properties of the cells, which in turn can result in different drug sensitivity profiles. We envision that the described method has a potential of selecting all types of cancer cells within the tumor, even if the cells are not identical.
Supplementary Material
Acknowledgments
We are grateful to Dr. Pernet for his help with P. leidyi culture and Dr. Stutts for helpful comments. This work was funded by the National Institutes of Health GM099594 and National Science Foundation MRI DBI0722757 (confocal microscopy). Selina F. Urfano was supported by the BUILD (Building Infrastructure Leading to Diversity) Program (National Institutes of Health GM118979-02; GM118980-02; GM118978-02).
Footnotes
Supporting Information
Schemes of device generating temperature gradient including temperature distribution. Confocal microscopy and flow cytometry of 3T3 cells incubated with peptide nanocarrier at variable temperatures and incubation time. The Supporting Information is available free of charge on the ACS Publications website.
Notes
The authors declare no competing financial interests.
References
- 1.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Nature Nanotech. 2008;3:242–244. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 2.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nature Nanotech. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 3.Srinivasarao M, Galliford CV, Low PS. Nature Rev Drug Disc. 2015;14:203–219. doi: 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- 4.Teicher BA. Drug Resistance Updates. 2000;3:67–73. doi: 10.1054/drup.2000.0123. [DOI] [PubMed] [Google Scholar]
- 5.Chari RVJ. Adv Drug Deliv Rev. 1998;31:89–104. doi: 10.1016/s0169-409x(97)00095-1. [DOI] [PubMed] [Google Scholar]
- 6.Van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, Van Gaal EV. Adv Drug Deliv Rev. 2013;65:1284–1298. doi: 10.1016/j.addr.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 7.Chari RVJ, Miller ML, Widdison WC. Angew Chem Int Ed. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 8.Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. J Clin Oncol. 1993;11:1936–1942. doi: 10.1200/JCO.1993.11.10.1936. [DOI] [PubMed] [Google Scholar]
- 9.Brune V, Tiacci E, Pfeil I, Döring C, Eckerle S, van Noesel CJ, Klapper W, Falini B, von Heydebreck A, Metzler D, Bräuninger A, Hansmann ML, Küppers R. J Exp Med. 2008;205:2251–2268. doi: 10.1084/jem.20080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckerle S, Brune V, Döring C, Tiacci E, Bohle V, Sundström C, Kodet R, Paulli M, Falini B, Klapper W, Chaubert AB, Willenbrock K, Metzler D, Bräuninger A, Küppers R, Hansmann ML. Leukemia. 2009;23:2129–2138. doi: 10.1038/leu.2009.161. [DOI] [PubMed] [Google Scholar]
- 11.Rose AA, Pepin F, Russo C, Abou Khalil JE, Hallett M, Siegel PM. Mol Cancer Res. 2007;5:1001–1014. doi: 10.1158/1541-7786.MCR-07-0119. [DOI] [PubMed] [Google Scholar]
- 12.Zhou LT, Liu FY, Li Y, Peng YM, Liu YH, Li J. Neoplasm. 2012;59:1–5. doi: 10.4149/neo_2012_001. [DOI] [PubMed] [Google Scholar]
- 13.Abou-Bakr AA, Elbasmi A. Gulf J Oncology. 2013;1:28–34. [PubMed] [Google Scholar]
- 14.Mesteri I, Schoppmann SF, Preusser M, Birner P. Eur J Cancer. 2014;50:1354–1360. doi: 10.1016/j.ejca.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Nat Clin Pract Oncol. 2006;3:269–80. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 16.Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY. Cancer Informat. 2011;10:149–157. doi: 10.4137/CIN.S7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurman RJ, Shih IM. Am J Surg Path. 2010;34:433–443. doi: 10.1097/PAS.0b013e3181cf3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganta S, Devalapally H, Shahiwala A, Amiji M. J Control Release. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 21.Gerweck LE, Seetharaman K. Cancer Res. 1996;56:1194–1198. [PubMed] [Google Scholar]
- 22.Wike-Hooley JL, Haveman J, Reinhold HS. Radiother Oncol. 1984;2:343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed M, Lukyanov AN, Torchilin V, Tournier H, Schneider AN, Goldberg SN. J Vasc Interv Radiol. 2005;16:1365–1371. doi: 10.1097/01.RVI.0000175324.63304.25. [DOI] [PubMed] [Google Scholar]
- 24.Ponce AM, Vujaskovic Z, Yuan F, Needham D, Dewhirst MW. Int J Hyperthon. 2006;22:205–213. doi: 10.1080/02656730600582956. [DOI] [PubMed] [Google Scholar]
- 25.Meyer DE, Shin BC, Kong GA, Dewhirst MW, Chilkoti A. J Control Release. 2001;74:213–224. doi: 10.1016/s0168-3659(01)00319-4. [DOI] [PubMed] [Google Scholar]
- 26.Rohren EM, Turkington TG, Coleman RE. Radiology. 2004;231:305–332. doi: 10.1148/radiol.2312021185. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Li H, He H, Wu H, Jin Y. Anal Chem. 2015;87:6868–6874. doi: 10.1021/acs.analchem.5b01260. [DOI] [PubMed] [Google Scholar]
- 28.Kostakoglu L, Agress H, Goldsmith SJ. Radiographics. 2003;23:315–340. doi: 10.1148/rg.232025705. [DOI] [PubMed] [Google Scholar]
- 29.Shinde A, Feher KM, Hu C, Slowinska K. J Pep Science. 2015;21:77–84. doi: 10.1002/psc.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamazaki CM, Nakase I, Endo H, Kishimoto S, Mashiyama Y, Masuda R, Futaki S, Koide T. Angew Chem Int Ed. 2013;52:5497–5500. doi: 10.1002/anie.201301266. [DOI] [PubMed] [Google Scholar]
- 31.Slotboom J, Kiefer C, Brekenfeld C, Ozdoba C, Remonda L, Nedeltchev K, Arnold M, Mattle H, Schroth G. Neuroradiology. 2004;46:923–934. doi: 10.1007/s00234-004-1286-z. [DOI] [PubMed] [Google Scholar]
- 32.Nolan JP, Morley PT, Van den Hoek TL, Hickey RW. Circulation. 2003;108:118–121. doi: 10.1161/01.CIR.0000079019.02601.90. [DOI] [PubMed] [Google Scholar]
- 33.Polderman KH, Callaghan J Critical Care. 2006;10:234. doi: 10.1186/cc5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venjakob AJ, Vogt S, Stockl K, Tischer T, Jost PJ, Thein E, Imhoff AB, Anetzberger H. J Orthop Res. 2013;31:1820–1827. doi: 10.1002/jor.22417. [DOI] [PubMed] [Google Scholar]
- 35.Gladen A, Iaizzo PA, Bischof JC, Erdman AG, Divani AA. J Med Devices. 2014;8:011002–011002-9. doi: 10.1115/1.4025448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ning J, Mo L, Zhao H, Lu K, Wang L, Lai X, Yang B, Zhao H, Sanders RD, Ma D. Critical Care Med. 2014;42:68–78. doi: 10.1097/CCM.0b013e3182a84daa. [DOI] [PubMed] [Google Scholar]
- 37.Black JH, Davison JK, Cambria RP. Semin Thorac Cardiovasc Surg. 2003;15:345–352. doi: 10.1053/s1043-0679(03)00086-8. [DOI] [PubMed] [Google Scholar]
- 38.Bely AE, Wray GA. Development. 2001;128:2781–2791. doi: 10.1242/dev.128.14.2781. [DOI] [PubMed] [Google Scholar]
- 39.Zattara EE, Bely AE. Evolution and Development. 2011;13:80–95. doi: 10.1111/j.1525-142X.2010.00458.x. [DOI] [PubMed] [Google Scholar]
- 40.Ky D, Liu CK, Marumoto C, Castaneda L, Slowinska K. J Contr Release. 2006;112:214–222. doi: 10.1016/j.jconrel.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Hwang E, Brodsky B. J Biol Chem. 2012;287:4368–4375. doi: 10.1074/jbc.M111.269084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer RZ, Bella J, Mayville P, Brodsky B, Berman HM. Nature Struc Biol. 1999;6:454–457. doi: 10.1038/8259. [DOI] [PubMed] [Google Scholar]
- 43.Persikov AV, Ramshaw JAM, Brodsky B. J Biol Chem. 2005;280:19343–19349. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- 44.Milletti F. Drug Discovery Today. 2012;17:850–860. doi: 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Bechara C, Sagan S. FEBS Letters. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 46.Futaki S, Nakase I, Tadokoro A, Takeuchi T, Jones A. Biochem Soc Trans. 2007;35:784–787. doi: 10.1042/BST0350784. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt N, Mishra A, Lai GH, Wong GC. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 48.Futaki S. Int J Pharm. 2002;245:1–7. doi: 10.1016/s0378-5173(02)00337-x. [DOI] [PubMed] [Google Scholar]
- 49.Nakase I, Takeuchi T, Tanaka G, Futaki S. Adv Drug Deliv Rev. 2008;60:598–607. doi: 10.1016/j.addr.2007.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.