

Abstract
Investigator initiated randomized clinical trials (IITs) are the backbone of academic clinical research. IITs complement the large clinical studies sponsored by industry and address questions, which are usually not the main focus of a commercially directed research but have the purpose to confirm, improve or refute clinically important questions with regard to diagnostic and therapeutic approaches in patient care. The aim of this review is to illustrate the necessary steps to start and complete an IIT in the field of inflammatory bowel diseases (IBD) in the US. The initial milestones for an investigator include structuring a protocol, planning and building of the trial infrastructure, accurately estimating the costs of the trial and gauging the time span for recruitment. Once the trial has begun it is important to keep patient recruitment on target, monitor of the data quality, and document treatment emergent adverse events. This article provides a framework for the different phases of an IIT and outlines potential hurdles, which could hinder a successful execution.
Keywords: Inflammatory bowel disease, investigator initiated clinical trial, Crohn’s disease, ulcerative colitis, therapy
Introduction
Clinical trials conducted by pharmaceutical companies are designed to improve or expand available medical treatments and are aligned with the sponsor’s commercial interest. “Investigator initiated trials” (IITs) typically have the same objectives while being independent of commercial interests. In the context of IITs, the Food and Drug Administration (FDA) uses a specific nomenclature and distinguishes between an “investigator” and a “sponsor”. An “investigator” is defined as the individual who conducts the clinical investigation, whereas a “sponsor” is the individual person or funding entity or pharmaceutical organization that takes responsibility for and initiates the clinical investigation.(1) A sponsor does not perform the clinical trial unless the sponsor is simultaneously the investigator, who both initiates and conducts an investigation, taking on the regulatory obligations of both. The term sponsor-investigator refers only to an individual. A pharmaceutical company, hospital, or an academic institution cannot be a sponsor-investigator because it is not an individual person.
Conducting an IIT as a sponsor-investigator commands considerable resources and infrastructure planning (see figure 1). This review points out important steps and requirements to start up an IIT. Based on the authors’ experience in running a multicenter trial in patients with ulcerative colitis, this review points out the critical steps required to set up an IIT. The trial with the acronym MERIT-UC (Randomized, double blind, prospective trial investigating the efficacy of Methotrexate in induction and maintenance of steroid-free remission in ulcerative colitis; ClinicalTrials.gov Identifier: NCT01393405) was funded by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), initially in 2010 with a Multi-Center Clinical Study Implementation Planning Grant (U34) followed in 2011 by a cooperative agreement (U01) to conduct the study.(2) The results of this maintenance trial are expected in late 2017. The study recruited at 42 U.S. sites. All of these sites belong to the Clinical Research Alliance (CRA) of the Crohn’s and Colitis Foundation of America (CCFA), a group of academic medical centers, multi-specialty group practices, and private practices that conduct clinical trials in inflammatory bowel disease (www.CCFACRA.org).
Figure 1.
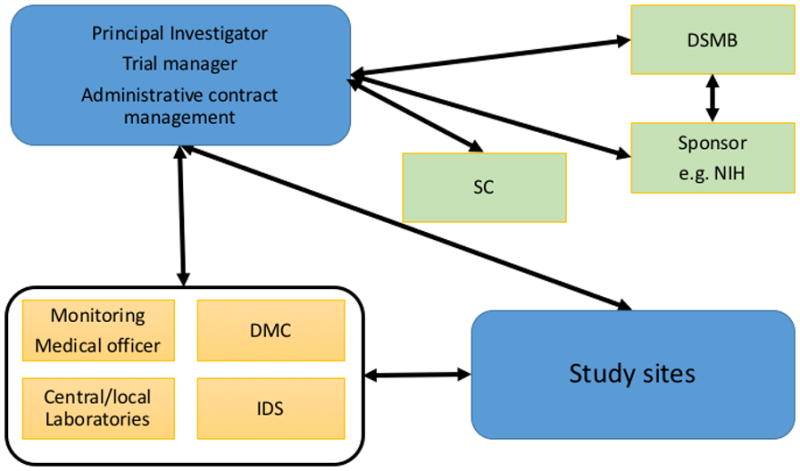
Multi-center clinical trial infrastructure.
IDS, Investigational drug service; DMC, data management center; DSM, data and safety monitoring board; SC, steering committee;
From trial idea to starting a trial – necessary documents and regulatory requirements
At the core of a successful clinical trial is the generation of a compelling research question, the choice of the target population, intervention, comparison group and the measure and timing of the appropriate outcome criterion.(3) These factors have to be translated into a detailed clinical trial protocol. Several rubrics have been proposed for the development of a good research question and the construction of a trial protocol (table 1). The trial protocol comprises several essential sections ranging from the study population, the intervention, the outcome criteria, both favorable and harmful, and the statistical plan (table 2).
Table 1.
FINER criteria for a good research question and PICOT criteria to construct the framework of the study and to aid protocol development. Adapted from (22, 23)
| FINER Criteria | PICOT criteria | ||
|---|---|---|---|
| Feasible |
|
Population |
|
| Interesting |
|
Intervention |
|
| Novel |
|
Comparison group |
|
| Ethical |
|
Outcome |
|
| Relevant |
|
Time |
|
Table 2.
Required contents of a Clinical Trial Protocol. Additional sections describing the clinical necessity for this trial and a description of how the results of the trial will impact the future care in IBD is often required by the potential funding agencies.
|
A separate statistical analysis plan might be required
The adverse event section has to outline how these events are captured and reported. The focus is on the capture of treatment emergent adverse events (TEAE), which are defined as first to appear during treatment and were absent before treatment or which worsened relative to the pre-treatment state. The reporting of TEAE can be simplified if a known drug with a defined adverse event profile is tested. In this case a separation between expected TEAE, which are based on the drug profile and unexpected TEAE can be made. This separation simplifies the reporting of TEAEs to institutional review boards (IRBs) and to data and safety monitoring boards (DSMBs). For the categorization of adverse events multiple classification systems have been described, but the most commonly used system is the Medical Dictionary for Regulatory Activities (MedDRA), which is also endorsed by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).(4)
Another essential part of the protocol is the statistical plan, which describes how the study results are to be analyzed and reported. The plan should include hypotheses being tested, a sample size calculation, the level of significance and the procedures planned for missing data. The sample size calculation is an essential component of a randomized trial.(5, 6) The projected sample size significantly influences both the feasibility and the budget of the proposed trial. In IITs, sample sizes are often estimated using the maximum effect size with the aim of reducing the required sample size, which might improve the likelihood of funding by enhancing feasibility and minimizing budget requirements. However, this tends to increase the risk of a Type II error (the probability of making a false negative conclusion) due to underpowering of the study for an effect size less than the maximum. In contrast, sample size calculations commonly used in industry trials are designed to detect smaller differences in order to specifically reduce the Type II error probability, or equivalently, to increase the power of the study.
In case of a randomized, blinded IIT, all details of the randomization plan must be pre-specified in the protocol. This includes the following detailed descriptions: who will construct the randomization table (i.e., study statistician) and what randomization schema will be used; who will be blinded; how will the randomization assignment be securely communicated to the pharmacy; how and when unblinding occurs if required for patient safety. For trials where it is anticipated that a large number of sites will each have a small number of patients, as it often occurs in IBD trials, permuted block randomization schemas help prevent drifting too far from designed balance of treatment assignment. For instance, using permuted blocks of size two or four ensures that every patient from sites having only 2 or 4 patients will be distributed according to the design. Many small trials have inadvertently lost statistical power due to imbalanced treatment assignments when using simple randomization within site without use of blocking.
The establishment of a trial protocol is only a small part of the total work necessary to start a trial. The process can very broadly be categorized into 4 phases as shown in figure 2. After the protocol is finalized, several other documents need to be created. These include the informed consent form (ICF) and the case report forms (CRF). Both should be based on a finalized protocol to prevent unnecessary repeated adjustments of both documents. Another essential document is a manual of procedures (MOP) containing standard operating procedures that describe how to conduct every process at each study visit, including recruitment, consent, procedures for intervention and data gathering, data entry, and data analysis. This manual guides the local investigators and coordinators in the performance of each patient encounter in an orderly and consistent fashion.
Figure 2.

Milestones of trial management.
*Essential documents: Financial disclosure forms, investigators curriculum vitae, FDA form 1572, protocol signature pages, Good clinical practice (GCP) training certificates. Abbreviations: ICF, informed consent form, CRF, case report form; MOP, Manual of procedures; IB, Investigator brochure; PP, publication policy; IND/IDE, Investigational New Drug Application or Investigational Device Exemption.
The finalized trial protocol, the ICF and the CRF need approval by the IRBs of all participating sites or in an optimal situation by a single central IRB.(7) A review of a multi-site study by the IRB of each participating site involves significant administrative burden in terms of IRB staff and members’ time to perform duplicative reviews, which can take many months and significantly delay the launch of a trial. Use of single IRBs in multi-site studies has been shown to decrease approval times for clinical protocols and may be more cost effective than local IRB review.(8) However, not all sites will acknowledge or permit the involvement of a central IRB, presenting challenges in study organization.
In regard to forecasting the necessary timeframe for contract negotiations with the sites, longer contractual negotiations periods especially with academic sites should be expected. We suggest planning a time frame of at least 6–8 months from start of contract negotiation until final contract approval with an individual site. This time frame can be accelerated if master contracts between each site and a central organization (e.g. a pharmaceutical sponsor or a CRO) have previously been established.
In trials testing a new drug or device an Investigational New Drug Application or Investigational Device Exemption (IND/IDE) is required. Together with a finalized protocol a formal application needs to be submitted to the FDA. The submission to the FDA may additionally require an investigator brochure (IB). If the IIT will study medications that are already approved by the FDA, the approved package insert can serve as the core of the IB. If the IIT will test a new compound in development by an industry partner, the investigator will need to gather relevant data from the manufacturer. Lastly, if the IIT will test a new compound developed by the investigator-sponsor, the responsibility for documenting appropriate preclinical and early clinical data will fall on the study team. The FDA provides guidance for this process.(9). In the event the investigator is not sure whether or not an IND is necessary a short formal application for an IND exemption should be submitted. The FDA responds in a timeframe of 30 days if the exemption is granted or a full IND submission needs to be submitted.(10)
To prevent future acrimonious discussions about authorship at the publication stage of the trial, a document detailing the publication policy before recruitment begins is strongly recommended.(11) Guidance for a policy for multi-center studies in IBD can be found on www.ccfacra.org.
Clinical trial data capture
Clinical study data can be collected on paper CRFs, which are then transcribed into an electronic data capture system or by direct electronic collection and storage using an electronic record system (ERS). Implementation of an ERS has many significant advantages. First, with an ERS, data are entered in real time at each clinical site using web forms developed to replicate paper case report forms, resulting in increased speed of data collection and exchange. Secondly, the ERS enables extensive data validation at the point of capture, reducing format errors, permissible value errors, and missing data, and ensuring a significantly more accurate and complete data set. Thirdly, web entry of data allows for web access to data, namely in reports that give up-to-date feedback about data quality and progress in the trial. Fourthly, a well-engineered ERS will facilitate adherence to protocol in all activities throughout the trial. In addition, ERS permits economical storage not only of clinical study data but other essential trial documents such as local IRB approvals and training documents thus easing the accessibility for the site coordinators and investigators. The ERS can be a convenient source of information to the sites when the study documents (protocol, CRFs, IB, MOP, newsletters, etc.) are stored and available for download. Finally, ERS facilitates real-time monitoring of the clinical study data.
It is important to work with your collaborating informatics team to complete engineering of study processes. This step, which includes the development of flow charts, a data dictionary, and validation specifications, is often overlooked because it is erroneously thought to delay the start of the trial. However, good engineering of the system ensures that your system will accurately reflect the protocol requirements, timely and effective capture high quality data, and roll out in the most cost effective manner. Making significant system changes after the trial is underway is both very costly and risky for adherence to the study protocol
The MERIT-UC study used an ERS system, which stored and archived all study relevant documents as well as the blinded patient data in one place. This system provided continuous support for the trial manager, the monitoring team, the site coordinators and the site investigators, thus allowing trial related issues to be resolved quickly, before these became major problems. However, one has to keep in mind that ERS still requires in person monitoring visits, but the monitoring process changes, since the monitor knows about the site's performance before visiting. Each audit is performed more to document what is already known rather than to learn for the first time how the site is performing. The result is a greatly enhanced relationship between site investigators and the monitors. The construction of a central ERS can be costly. The CCFA provides a central ERS in the setting of CRA studies as a service; however if studies are sponsored by NIH or other entities the PI of the study must create an ERS by e.g. using REDCap (http://projectredcap.org) for simple protocols, or by engaging with a research computing collaborator or a commercial vendor for more complex studies.(12–14)
Steering committee and Data and Safety Monitoring Board (DSMB)
Early in the planning phase the investigator should consider creating a steering committee. This committee should consist of specialists in the therapeutic area or key participants in the trial and if possible also a biostatistician. The steering committee will be charged with assisting in designing the study, maintaining the quality of study management, ongoing monitoring of individual toxicities and adverse events and writing the study publications. In addition to a steering committee, the NIH and FDA often recommend or require a DSMB for the appropriate oversight and monitoring of the conduct of clinical trials to ensure the safety of participants and the validity and integrity of the data. The establishment of a DSMB and a DSMB charter detailing the tasks of the DSMB is requested for multi-site clinical trials involving interventions that entail potential risk to the participants. However, a DSMB may be also required for single-center phase 1 studies of a new drug or procedure that involves relatively high risk to a small number of participants. The DSMB functions and supervision of such activities are distinct from the requirement for study review and approval by an Institutional Review Board (IRB).(15)
Clinical trial infrastructure and trial budget
The PI has to build the trial infrastructure and coordinate with several committees including the steering committee, the DSMB, the sponsor, IRBs, necessary trial services (data managing center (DMC), trial monitoring, investigational drug services (IDS), central and/or local laboratories) (see figure 1). The steering committee, the IRB (central and local IRB’s), and the DSMB each have roles in the oversight of the trial. For data capture and analyses, the PI must coordinate the DMC, the data monitoring unit and the medical officer. In larger multi-center trials a medical officer for the independent evaluation and adjudication of TEAE and serious adverse events is often required.
Packaging, dispensing and tracking study medications and placebo is typically handled by an investigational drug service (IDS). In many IITs, the pharmaceutical company that manufactures the study drug supplies this directly to the IDS. When possible, the manufacturer may also supply matching placebo. However, when this is not possible, it may be necessary to have the IDS develop a placebo formulation.
There are many possible methods to create placebos, which are beyond the scope of this review; nevertheless, one needs to be carefully considered. If a placebo is to be developed or the study drug repackage, the IDS will need to support the PI in the IND process. In comparative effectiveness trials, these challenges may influence the choice between a blinded versus open label study. Since study drugs often have to be shipped across state borders, the drug shipments are subject to the legislation of the receiving state.(16) The IDS often requires licensure for each state to which it will need to ship drug. Therefore it is crucial for the PI and IDS to know about the legislative requirements surrounding the shipment and delivery processes of interstate shipment of investigational product to maintain compliance with all regulations. At the end of the study, the IDS will also be responsible for destroying any residual study medication.
Beyond the science and infrastructure planning a detailed budget estimate for the trail is crucial to determine if the study is financially feasible. The budget is also an essential part of any application process to a funding agency. The following expenditures for efforts of study personnel should be included on a per patient/per visit basis: a) obtaining medical history including current and past medications, b) evaluating disease activity (e.g. evaluation of clinical Mayo score), c) performing physical exam, and d) coordinating study procedures.
Additionally, every study should consider patient reimbursement e.g. for travel costs and lost work time. Procedures which are performed in the context of the trial (e.g. blood draw, radiological or endoscopic evaluations) also need to be also covered. A recent survey of 15 academic centers active in the CCFA CRA revealed a wide range of procedural costs as outlined in table 3. It is important to proactively contact sites upfront to evaluate the costs for specific procedures with which to estimate an optimal reimbursement scheme. Moreover, a straightforward cost analysis will help prevent situations in which sites fail to recruit due to financial burdens. Sites often waive the fees for IRB approval in the setting of an IIT.
Table 3.
Procedural costs and fees for an IBD IIT in the US. Survey conducted in 2014/2015 at 15 academic IBD sites of the CCFA CRA.
| Costs ($) | ||
|---|---|---|
| Procedure/Fees | Mean | Max |
| Upper-endoscopy + biopsy | 2682 | 5500 |
| Colonoscopy + biopsy | 3235 | 6000 |
| Sigmoidoscopy + biopsy | 1983 | 3851 |
| MR-Enterography | 2312 | 3851 |
| CT-Enterography | 1745 | 6032 |
| Chest x-ray | 187 | 491 |
| Coordinator/hr | 94 | 250 |
| PI/hr | 177 | 300 |
| Clinic space/hr | 119 | 200 |
| Pharmacy start up | 2037 | 2893 |
| Site start up | 4698 | 8000 |
Abbreviations: PI, principal investigator; MR, magnet resonance tomography; CT, computed tomography.
Other factors that need to be considered in a trial budget are the charges of an IDS and expenses for monitoring of the trial. An IDS is necessary for acquisition and distribution of study and placebo drug to the sites. If the IRB permits an IDS to mail the study drug directly to the patient, no additional fees need to be calculated. However, if it is necessary for the local study site to dispense the drug or placebo to the patient, IDSs at each individual site also have to be reimbursed to provide these services. The monitoring budget includes cost estimates for the time and effort to train monitors for the specific trial, the required hours and fees to perform a monitoring site visit including the generation of a monitoring report (e.g. 8 hours/site/visit) and estimates of travel time and travel costs for the monitor to visit the sites. Collaboration with an informatics and data management group at the trial planning stage will allow for more accurate estimate of the costs for trial system development and/or customization and data management support. Given all the above cost factors and the procedural expenses outlined above and in table 3, the MERIT-UC budget exceeded $5 million in direct costs, which is the current reality for a moderate-sized multi-center IIT in IBD. The amount of necessary funding also significantly limits the number of agencies able to support these types of trials.
Time and momentum are significant considerations in completing a multi-center trial. It is important to consider the local site perspective in recruitment rates. A slow-recruiting trial costs less to the study budget, but the local site still has to pay the study coordinator. If funds are not coming in regularly, the local PI and study coordinator will focus on other studies with easier recruitment or more lucrative payments. If recruitment slows, it is important to incentivize momentum, and it may be wise to allocate a portion of the budget for future incentives, or increasing the payment per subject in later years of the trial to compensate for inflation.
How to project recruitment into the trial?
IITs often fail due to insufficient enrollment of patients. Recruitment into IBD trials is increasingly difficult as the number of available therapies has expanded. One must consider all factors influencing trial recruitment including type of disease (e.g. early CD or UC), the allowed or excluded current or previous medications, the mode of application of the study drug, the frequency of endoscopies and study visits or the design of the trial with for example, the possibility for open label extension in case of no response or absence of a placebo arm in comparative effectiveness trials.(17–19) The number of active competing trials, especially industry sponsored trials should be evaluated, because reimbursements for sites are generally more generous in industry sponsored studies. Foreseeing approvals of new drugs for UC or CD might be helpful to estimate a realistic recruitment time, since approved new therapies influence the willingness of patients to participate in a trial and the clinical perspective of site investigators to recruit into the study.
A guideline to estimate the number of patients a single site might recruit in 1 year can be based on previously published IITs in Crohn’s disease (CD) or ulcerative colitis (UC) (see table 4). The recruitment in these IIT, which targeted different disease stages (early, uncomplicated and late and complicated disease) and employed different study designs ranged from 0.7–2.5 patients/site/year. Globally conducted industry sponsored trials fair not much better in enrolling patients into trials. The CD PREVENT study (Prospective, Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial Comparing REMICADE® and Placebo in the Prevention of Recurrence in Crohn’s Disease Patients Undergoing Surgical Resection Who Are at an Increased Risk of Recurrence), recruited 297 patients at 104 sites in 18 months (2010–2012 1.8 patients/site/year).(20)
Table 4.
Patient recruitment in IIT’s in IBD (* study stopped prematurely due to futility)
| Author or study acronym | Recruitment period (months) | Disease | Design | Drug | Patients enrolled | Number of study sites | Location of sites | Patients/site /year |
|---|---|---|---|---|---|---|---|---|
| Lewis(21) | 2002–2006 (39) | UC | Randomized, double blind | Rosiglitazone vs placebo | 105 | 15 | U.S. | 2.2 |
| Osterman(24) | 2008–2012 (41) | UC | Open-label, randomized | Increase 5-ASA vs stable 5-ASA in patients with increased fecal calprotectin | 119 | 14 | U.S. | 2.5 |
| Carbonnel(25) | 2007–2013 (72) | UC | Randomized, double blind | Methotrexate vs placebo | 111 | 26 | France, Austria, Belgium, Italy Finland, Greece. | 0.7 |
| MERIT-UC | 2012–2016 (52) | UC | Randomized, double blind | Methotrexate vs placebo | 179 | 42 | U.S. | 1.0 |
| Dassopoulos(26) | 2005–2007 (29) | CD | Randomized, double blind | weight-based vs. individualized azathioprine dosing | 50 | 12 | U.S., Canada | 1.7* |
| Cosnes(27) | 2005–2010 (66) | CD | Open-label, randomized | Azathioprine vs conventional management | 132 | 24 | France | 1.0 |
| Panes(28) | 2006–2009 (42) | CD | Randomized, double blind | Azathioprine vs placebo | 131 | 31 | Spain | 1.2* |
Site management, recruitment, and enrollment
A crucial issue in IIT is the regular communication with sites to solve study related problems and to encourage enrollment into the study. For the startup of sites the local investigators and coordinators have to be trained on the protocol. This can be in the setting of a study meeting either in person, which is costly and often tedious to organize or via webinars. Regular newsletters sent by email or mail to the sites to address study related problems and encourage enrollment are a tool to keep the sites involved. The investigator or trial manager should also contact each site frequently and to solve recruitment questions. In a survey of 18 sites in the MERIT-UC trial, individual contact with the sites was unanimously the preferred method of communications, but a combination of web based meetings, regular newsletter and personal contact are most likely the most effective ways to manage trial problems and stimulate recruitment. Individual contact with sites is even more imperative due to a high turnover of study personnel.
Monitoring of study sites
Monitoring of a clinical trial assures a high quality of trial conduct. The tasks of "on site" monitoring are the evaluation of individual case histories of study participants, the assessment of site adherence to the protocol, the review of the ongoing implementation of appropriate data entry and quality control procedures, and essentially to assess adherence to good clinical practices. On-site monitoring might be required at the initial site selection to confirm that the site meets all study requirements. Monitoring represents a significant cost expenditure due to travel expenses and length of study visit. Thus some monitoring tasks such as review of regulatory files can be performed electronically if an ERS with such functions is in place (see table 5). However, only a physical site visit can establish the adequacy of study staff and facilities to conduct the study, to verify source documentation of eligibility of study subjects, to review the adherence to protocol and treatment plan, to confirm the drug accountability, and to validate source documentation of all data in the ERS. While the frequency of site visits is based on the study protocol, nonetheless at least 2 visits/site should be planned: one shortly after the enrolment of the first or second patient to confirm the adherence to study procedures and another at study closure to review all enrolled subjects since the initial visit, resolution of queries from the DMC, and preparation for study storage. Budgeting for additional as-needed visits for sites with high enrollment to identify potential problems early in the study is wise.
Table 5.
Necessary documents in an Investigator’s File (Essential Documents):
|
Challenges and lessons learned from the MERIT-UC trial
During the 8 years of planning, applying of funding and finally conducting the MERIT-UC study, the authors of this paper, who were all involved in the trial learned several important lessons, which are outlined in the following paragraphs:
If you conduct an IIT similar rules apply as in industry studies, meaning a lot of regulatory and infrastructural work has to be done. For this you need a large pool of knowledgeable co-investigators and co-workers
For MERIT-UC a considerable number of people were necessary to build the trial infrastructure. The core infrastructure comprised 25 persons. In addition to the PI and a fulltime study manager, 5 members in the steering committee and 2 biostatisticians helped to finalize the protocol and statistical plan. Six data specialists established the ERS, 2 monitors performed the monitoring tasks and travelled to the sites, 1 medical monitor oversaw the TEAE and SAE reporting and 2 administrators managed the contracts. Finally, a DSMB with 5 members had the oversight of the trial. Also, a single IDS at the University of Pennsylvania belonged to the core infrastructure MERIT-UC, which had the regulatory requirements in place to ship drug and placebo across state lines.
You need to be flexible to adapt the protocol (and often several other regulatory documents) if new medical knowledge emerges
One month before the planned start of recruitment, the FDA issued a warning for ondansentron and alerted of the risk of fatal arrhythmia. To comply with this warning we had to change the original plan to send each patient ondansetron to prevent MTX induced nausea. With the help of the IDS we replaced ondansetron with promethazine, but had to amend the protocol. This amendment, which had to be reapproved by IRBs of all participating sites, allowed for the dispensing of ondansetron if promethazine failed to alleviate the nausea but required the performance a normal electrocardiogram (ECG) by the sites. The costs for the ECG had to be added to the budget and the contracts had to be revised.
You need to be innovative and flexible in your budget and contract planning
Despite a provisional agreement with all local PIs for reimbursement for each enrolled patient, the actual contract negotiations could only be started once the U01 grant was presented. These negotiations took much longer than anticipated resulting in a launch delay of by 24 months after the initial funds were awarded. In fact, at the start of recruitment only 6 sites were activated and it took an additional 12 months until finally all sites were recruiting patients. Also because recruitment time spanned over 4 years, reimbursement costs changed at many sites consequently the budget had to be adapted to prevent sites from halting recruitment since they were losing money on the study.
Recruitment in the realm of IBD is extremely difficult
MERIT-UC was initially conceptualized in 2007/2008 and recruitment was estimated based on the rosiglitazone in active ulcerative colitis study, which recruited between 2002–2006 (table 4).(21)The plan for the trial was to activate 35 sites with the aim to include 6–8 patient/site in 3 years (around 2 patients/site/year). Only few clinical trials for patients with UC were available in the US at the time of conceptualization of MERIT-UC and the therapeutic alternatives for patients with steroid-dependent or mesalamine-resistant ulcerative colitis were very limited (azathioprine or infliximab). However, by the time the trial finally started in 2012, multiple industry sponsored phase 2 and 3 studies had been initiated (figure 3). Additionally, 3 biologics to treat UC were approved during the first 3 years of the study (adalimumab in 2012, golimumab in 2013 and vedolizumab in 2014), which affected recruitment. Despite the considerable number of sites, and uncomplicated in- and ex-clusion criteria as well as an open label induction period, recruitment for MERIT-UC was a challenge resulting in an average recruitment pace of only 1 patients/site/year. Eight sites had to be closed during the trial due to problems in recruiting for the trial or departure of the local principal investigator. Thus additional 7 new sites had to be found and activated, which were not easy tasks due to competing industry trials. Moreover, in contrast to professional research organizations (such as Parexel, Quintiles, Icon, Robarts and others), which have years of experience in finding reliable trial sites, IITs may have more problems in identifying new investigators outside of defined clinical research clusters such as the CCFA CRA.
Figure 3.
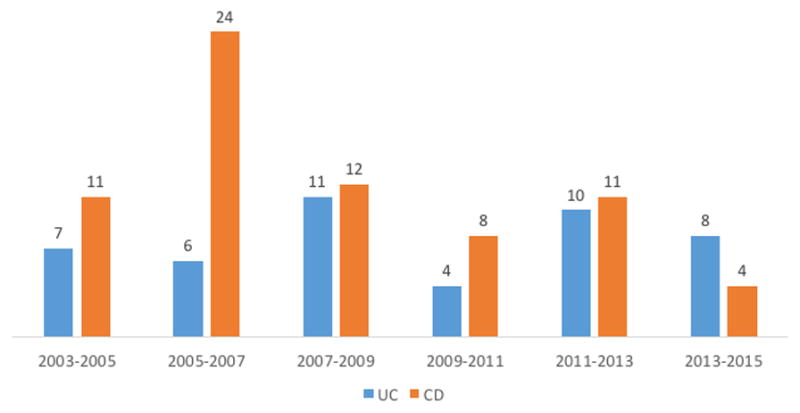
Industry sponsored interventional phase 2 and phase 3 studies in CD and UC in the US between 01/01/2005-01/01/2015 as listed on ClinicalTrials.gov. Only studies in adult patients with the following known statuses were counted: recruitment, completed, (with or without results), terminated with results. Trials evaluating the efficacy of mesalamine products in UC were excluded from the analyses.
You need to watch your sites
We observed a high turnover of study coordinators at many sites necessitating repeated training on the study protocol. Over the 4 years of the recruitment period, we experienced a turnover of >140 coordinators and 6 principal site investigators. To keep track of all the necessary regulatory changes in the setting of these staff changes, the electronic monitoring of the study documents was extremely helpful. Initially 3 monitoring visits were planned at each site, but ultimately 24 additional visits were necessary to underperforming sites or sites with a higher than expected volume.
Conducting a clinical trial is highly rewarding
There are several aspects, which make the successful completion of an IIT rewarding. Clinical research is conducted to improve or expand available medical treatments. Thus the outcome of an IIT can have a considerable impact on the care of patients. The established infrastructure of a large trial will strengthen the site of the PI and facilitate the funding of future studies not only of the PI but also of coworkers. IIT provide an entry point into the clinical trial landscape and as the PI of an IIT you regularly come in contact with numerous investigators within your research community. Thus you may find new collaborations for novel diagnostic or therapeutic approach and ultimately all these interactions will improve the lives of your patients. In our case a study investigating the therapeutic value of different diets, planned to start sometime 2017–2018, was also conceptualized based on experiences collected in MERIT-UC.
Conclusion
Due to numerous legal, contractual and infrastructural requirements, the conduct of IITs in the US in the context of current regulatory, research and clinical practices, is extremely complex. Investigators should allocate sufficient time to achieve milestones in each phase of a startup of a multi-center clinical trial. In addition to the protocol and multiple other mandated documents, a well-balanced and realistic budget builds the foundation for eventual success. Furthermore, recruitment into IBD trials is extremely challenging and a sufficient number of sites should be engaged. Once the trial is in the recruitment phase the most critical part is to maintain study momentum, stimulate the eagerness of the investigators to actively participate in the trial and assure ongoing data quality. To better navigate these complex processes and master all the known and unknown hurdles in the trial process the PI requires a reliable trial infrastructure guided by a core team of committed support staff with in depth knowledge of the regulatory requirements and practical aspects of implementing a clinical trial.
Acknowledgments
HHE, SJ, BGS, CM, KA, RSS, MDL, KLI, BES, JDL are supported by 5U01DK092239. HHE, PDH, CM, KA, RSS are also supported by the CCFA.
References
- 1.U.S. Government Publishing Office. [Accessed May 2016];Electronic code of federal regulations: Title 21, Chapter I, Subchapter D, Part 312. Available at: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.3.
- 2.Herfarth HH. Methotrexate for Inflammatory Bowel Diseases - New Developments. Dig Dis. 2016;34:140–146. doi: 10.1159/000443129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sands BE, Abreu MT, Ferry GD, et al. Design issues and outcomes in IBD clinical trials. Inflamm Bowel Dis. 2005;11(Suppl 1):S22–28. doi: 10.1097/01.mib.0000184849.38816.39. [DOI] [PubMed] [Google Scholar]
- 4.Bailey C, Peddie D, Wickham ME, et al. Adverse Drug Event Reporting Systems-a Systematic Review. Br J Clin Pharmacol. 2016 doi: 10.1111/bcp.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freiman JA, Chalmers TC, Smith H, Jr, et al. The importance of beta, the type II error and sample size in the design and interpretation of the randomized control trial. Survey of 71 "negative" trials. N Engl J Med. 1978;299:690–694. doi: 10.1056/NEJM197809282991304. [DOI] [PubMed] [Google Scholar]
- 6.Charles P, Giraudeau B, Dechartres A, et al. Reporting of sample size calculation in randomised controlled trials: review. Bmj. 2009;338:b1732. doi: 10.1136/bmj.b1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gold JL, Dewa CS. Institutional review boards and multisite studies in health services research: is there a better way? Health services research. 2005;40:291–308. doi: 10.1111/j.1475-6773.2005.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner TH, Murray C, Goldberg J, et al. Costs and benefits of the national cancer institute central institutional review board. J Clin Oncol. 2010;28:662–666. doi: 10.1200/JCO.2009.23.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. [Accessed May 2016];Investigational New Drug (IND) Application. Available at: http://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/investigationalnewdrugindapplication/default.htm.
- 10. [Accessed 5/16/2016, 2016];IND Application Procedures: Exemptions from IND Requirements. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/InvestigationalNewDrugINDApplication/ucm362743.htm.
- 11.Strange K. Authorship: why not just toss a coin? American Journal of Physiology - Cell Physiology. 2008;295:C567–C575. doi: 10.1152/ajpcell.00208.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandborn WJ, Present DH, Isaacs KL, et al. Tacrolimus for the treatment of fistulas in patients with Crohn's disease: a randomized, placebo-controlled trial. Gastroenterology. 2003;125:380–388. doi: 10.1016/s0016-5085(03)00877-1. [DOI] [PubMed] [Google Scholar]
- 14.Herfarth HH, Katz JA, Hanauer SB, et al. Ciprofloxacin for the Prevention of Postoperative Recurrence in Patients with Crohn's Disease: A Randomized, Double-blind, Placebo-controlled Pilot Study. Inflamm Bowel Dis. 2013;19:1073–1079. doi: 10.1097/01.MIB.0000428910.36091.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. [Accessed 6/13/2016, 2016];Data and Safety Monitoring Board (DSMB) Guidelines. Available at: http://www.nidcr.nih.gov/Research/ToolsforResearchers/Toolkit/DSMBGuidelines.htm.
- 16.McIntyre C. Regulations guiding the interstate shipment of investigational product. Journal of pharmacy practice. 2014;27:101–105. doi: 10.1177/0897190013504958. [DOI] [PubMed] [Google Scholar]
- 17.D'Haens G, Feagan B, Colombel JF, et al. Challenges to the design, execution, and analysis of randomized controlled trials for inflammatory bowel disease. Gastroenterology. 2012;143:1461–1469. doi: 10.1053/j.gastro.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 18.Ravikoff JE, Cole EB, Korzenik JR. Barriers to enrollment in inflammatory bowel disease randomized controlled trials: an investigation of patient perspectives. Inflamm Bowel Dis. 2012;18:2092–2098. doi: 10.1002/ibd.22872. [DOI] [PubMed] [Google Scholar]
- 19.Ha C, Ullman TA, Siegel CA, et al. Patients enrolled in randomized controlled trials do not represent the inflammatory bowel disease patient population. Clin Gastroenterol Hepatol. 2012;10:1002–1007. doi: 10.1016/j.cgh.2012.02.004. quiz e1078. [DOI] [PubMed] [Google Scholar]
- 20.Regueiro M, Feagan BG, Zou B, et al. Infliximab Reduces Endoscopic, but Not Clinical, Recurrence of Crohn's Disease After Ileocolonic Resection. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.02.072. [DOI] [PubMed] [Google Scholar]
- 21.Lewis JD, Lichtenstein GR, Deren JJ, et al. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. 2008;134:688–695. doi: 10.1053/j.gastro.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brian Haynes R. Forming research questions. Journal of Clinical Epidemiology. 2006;59:881–886. doi: 10.1016/j.jclinepi.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farrugia P, Petrisor BA, Farrokhyar F, et al. Research questions, hypotheses and objectives. Canadian Journal of Surgery. 2010;53:278–281. [PMC free article] [PubMed] [Google Scholar]
- 24.Osterman MT, Aberra FN, Cross R, et al. Mesalamine dose escalation reduces fecal calprotectin in patients with quiescent ulcerative colitis. Clin Gastroenterol Hepatol. 2014;12:1887–1893. e1883. doi: 10.1016/j.cgh.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbonnel F, Colombel JF, Filippi J, et al. Methotrexate Is Not Superior to Placebo for Inducing Steroid-Free Remission, but Induces Steroid-Free Clinical Remission in a Larger Proportion of Patients With Ulcerative Colitis. Gastroenterology. 2016;150:380–388. e384. doi: 10.1053/j.gastro.2015.10.050. [DOI] [PubMed] [Google Scholar]
- 26.Dassopoulos T, Dubinsky MC, Bentsen JL, et al. Randomised clinical trial: individualised vs. weight-based dosing of azathioprine in Crohn's disease. Aliment Pharmacol Ther. 2014;39:163–175. doi: 10.1111/apt.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosnes J, Bourrier A, Laharie D, et al. Early administration of azathioprine vs conventional management of Crohn's Disease: a randomized controlled trial. Gastroenterology. 2013;145:758–765. doi: 10.1053/j.gastro.2013.04.048. [DOI] [PubMed] [Google Scholar]
- 28.Panes J, Lopez-Sanroman A, Bermejo F, et al. Early azathioprine therapy is no more effective than placebo for newly diagnosed Crohn's disease. Gastroenterology. 2013;145:766–774. doi: 10.1053/j.gastro.2013.06.009. [DOI] [PubMed] [Google Scholar]