

Abstract
Fibroblast growth factors (FGFs) regulate several cellular developmental processes by interacting with cell surface heparan proteoglycans and transmembrane cell surface receptors (FGFR). The interaction of FGF with heparan sulfate (HS) is known to induce protein oligomerization, increase the affinity of FGF towards its receptor FGFR, promoting the formation of the HS–FGF–FGFR signaling complex. Although the role of HS in the signaling pathways is well recognized, the details of FGF oligomerization and formation of the ternary signaling complex are still not clear, with several conflicting models proposed in literature. Here, we examine the effect of size and sulfation pattern of HS upon FGF1 oligomerization, binding stoichiometry and conformational stability, through a combination of ion mobility (IM) and theoretical modeling approaches. Ion mobility-mass spectrometry (IMMS) of FGF1 in the presence of several HS fragments ranging from tetrasaccharide (dp4) to dodecasaccharide (dp12) in length was performed. A comparison of the binding stoichiometry of variably sulfated dp4 HS to FGF1 confirmed the significance of the previously known high-affinity binding motif in FGF1 dimerization, and demonstrated that certain tetrasaccharide-length fragments are also capable of inducing dimerization of FGF1. The degree of oligomerization was found to increase in the presence of dp12 HS, and a general lack of specificity for longer HS was observed. Additionally, collision cross-sections (CCSs) of several FGF1–HS complexes were calculated, and were found to be in close agreement with experimental results. Based on the (CCSs) a number of plausible binding modes of 2:1 and 3:1 FGF1–HS are proposed.
Keywords: Glycosaminoglycan, Ion mobility, TWIMS, Native mass spectrometry, Fibroblast growth factor
Graphical abstract

Introduction
Fibroblast growth factors (FGFs) are mitogenic polypeptide growth factors expressed in all multicellular organisms [1]. Eighteen human FGFs have been identified so far, which share 13%–71% sequence similarity and a homology core domain that consists of three copies of a basic four-stranded antiparallel beta sheet. These FGFs differ in their size, their interaction with receptors, and their biological functions [2, 3]. FGFs are potent mitogens and mediate a multitude of biological processes, including cell proliferation, cell differentiation, cell migration, morphogenesis, and angiogenesis [4]. Aberrant FGF signaling promotes tumor development and tumor resistance to anti-angiogenic and other chemotherapies by directly driving cancer cell proliferation and survival [5]. FGF1 (along with FGF2) were the first FGFs to be identified and extensively studied [6].
The growth stimulatory activity of FGFs is achieved through direct interaction with two types of receptors on the cell surface [7]. The high affinity receptor FGFR are transmembrane proteins, four isoforms of which have been identified in mammals. Three extracellular immunoglobulin (Ig)-like domains and an intracellular cytoplasmic tyrosine kinase domain are linked though a single transmembrane helix to form the structure of FGFRs [8]. The ligand binding activity and specificity of FGFRs manifest through the Ig-domains II and III and the linker connecting the two domains [9]. Alternative splicing in Ig domain III dramatically alters the affinity of the FGFRs for FGFs and introduces the first level of specificity in FGF signaling for all FGF ligands except for FGF1, which serves as a universal ligand and activates all of the FGFRs [10].
The low affinity receptor heparan sulfates (HS) are linear sulfated glycosaminoglycan (GAG) polysaccharides comprising of alternating N-acetyl glucosamine and uronic acid disaccharide units linked through a 1–4 glycosidic linkage. HS are abundant components found on cell surfaces, for example, syndecans and glypicans, or in the extracellular matrix, such as perlecan, agrin, or collagen XVIII [11]. The structural polydispersity and heterogeneity of HS is a result of incomplete enzymatic modifications of HS in Golgi apparatus, including: (1) replacing N-acetyl with N-sulfation of glucosamine by N-deacetylase/N-sulfotranferases (NDSTs), (2) epimerization of glucuronic acid to iduronic acid by C5-epimerase, (3) addition of sulfo group to the 2-O position of iduronic acid by 2-O sulfotransferase, (4) addition of sulfo group to the 6-O and, rarely, the 3-O positions of glucosamine by 6-O and 3-O sulfotranferases [12]. These non-template enzymatic modifications produce domains that are either highly N-sulfated, highly N-acetylated, or mixed domains of HS, which serve as potential binding site for a large array of HS binding proteins [13].
It has been generally appreciated that HS plays an essential role in transmitting extracellular signals to intracellular signaling pathways through direct interactions with both FGF and FGFR [14]. The binding of FGF to HS induces FGF oligomerization, increases the affinity of FGF to the extracellular part of FGFR, and further enables activation of FGFR and the formation of the highly stable HS–FGF–FGFR trimeric signaling complex [15]. The kinase domains from activated FGFRs are activated through transphosphorylation, followed by recruiting adapter protein to the activated receptors and triggering the downstream intracellular signaling cascades [16].
Although the role of HS in FGF signaling has been well elucidated, the structural details of how the FGFs oligomerize in the presence of HS, and how the trimeric signaling complex of FGF, FGFR, and HS are integrated is still a matter of debate. Two physiologically relevant FGF dimerization configurations with HS have been proposed [17]: The trans-oriented form features two FGF molecules arranged on opposite sides of one HS domain, and is characterized by a complete absence of FGF–FGF interactions. The cis-oriented form features two FGF molecules present on the same side of the HS domain, requiring a HS with sufficient length. Many studies suggest that FGF1 favors the formation of trans form dimeric complex, and so far the crystallographic structure of only the trans form has been observed for FGF1 [18]. In contrast, for FGF2, a cis and a trans dimeric form may coexist [19]. In the presence of FGFR, two crystallographic ternary complex structures have been resolved, the 2:2:1 FGF1–FGFR2–HS dodecasaccharide model (or Pellegrini model [20]) and the 2:2:2 FGF2–FGFR1–HS dodecasaccharide model (or Schlessinger model [21]), differing in their preparation, binding stoichiometry, intermolecular interactions, as well as the protein species used.
It has been long disputed whether FGF1 recognizes a consensus sequence with a specific sulfation pattern and minimum length, similar to the specific pentasaccharide sequences recognized by Antithrombin III (ATIII) [22]. HS octasaccharide and decasaccharide have been suggested to be the minimum length required for signaling [23], and a trisaccharide HS motif of IdoA2S-α-(1-4)-GlcNS6S-α-(1-4)-IdoA2S on HS oligosaccharides has been recognized to show strong binding affinity for FGF1 [24], but the minimum length of HS required to activate FGF signaling is still uncertain. There is considerable inconsistency in the literature about the binding specificity of FGF1–HS interaction, where some results suggest that an overall degree of sulfation rather than a distinct sulfation pattern mediates this interactions [25, 26], whereas others propose binding selectivity that is highly susceptible to subtle changes to the fine structure of HS regarding the particular location of sulfation [27–30]. Studies of the binding sequence for FGFs show that they each have different preferences for the sulfation pattern of HS [29]. Additionally, some other studies indicate that specific FGF-FGFR pairs, rather than FGF itself, dictate binding specificity and affinity [31]. For example, HS deficient in 2-O- and 6-O- sulfation still maintains its activating effect on FGF1-FGFR2 signaling but not FGF1-FGFR1 or FGF7-FGFR2b signaling [32].
A wide array of biophysical techniques, including X-ray crystallography [18], NMR [33], size exclusion chromatography [34], size exclusion chromatography followed by hydrophobic trapping [35], isothermal calorimetry [36], surface plasmon resonance [37], and affinity co-electrophoresis [38], have been applied to elucidate the nature of these molecular interactions. Recently, ion mobility-mass spectrometry (IMMS), a gas-phased biophysical technique that separates biomolecules based on their mobilities has emerged and found numerous applications such as measuring conformational change in proteins upon ligand-binding [39], determining binding affinity and specificity [40], monitoring real-time conformational dynamics [41], detecting intermediate structures [42], and elucidating the architecture of protein assemblies [43]. Furthermore, with the commercialization of Synapt instruments from Waters Corporation, traveling wave ion mobility spectrometry (TWIMS) has become a widely used approach for the study of biomolecular structure and dynamics [44]. IMMS studies are often supported by molecular modeling studies, which provide computational predictions of molecular structure, binding orientations, binding affinities, as well as theoretical estimation of the molecular shape collision cross-sections (CCSs), and serve a critical role for the correct interpretation of experimental findings.
We recently applied TWIMS to examine the well-characterized, highly specific interaction between ATIII and a synthetic heparin (Hp) pentasaccharide [40]. We successfully measured and theoretically validated the conformational change within ATIII induced by the binding of Hp while maintaining the solution structure of ATIII and its complexes as well as the binding selectivity and specificity of Hp–ATIII interaction. In this study, we extended this method to a more complicated system of FGF1 signaling, in order to investigate the conformational and stoichiometric details of the binding of FGF1 and HS. TWIMS experiments and molecular modeling techniques were combined in order to address the minimum length of HS required to induce FGF1 dimerization, the binding stoichiometry for the interaction of FGF1 and HS of varying sizes and sulfation patterns, the selectivity of the interaction between FGF1 and HS, and the conformational stability of FGF1 and its HS bound complexes. This work provides more details to the model of the manner in which HS interacts with FGF1, as well as additional validation for the approach of using gas-phase measurements of GAG–protein interactions to derive biologically relevant structural details.
Experimental
Reagents
All chemicals and solvents (ammonium acetate, methanol, water, and formic acid) were of HPLC grade and purchased from Sigma-Alldrich, (St. Louis, MO). Human recombinant FGF 1 expressed in Escherichia coli was a gift from Amgen (Thousand Oaks, CA, USA). Protein calibrants (myoglobin from equine heart, cytochrome c from equine heart, avidin from egg white, concanavalin A from Canavalia ensiformis, and bovine serum albumin) were purchased from Sigma-Aldrich. HS dodecasaccharides were chemo-enzymatically synthesized as previously described [45]. HS tetrasaccharides were synthesized as previously described based by fluorous supported modular synthesis [46].
Sample Preparation
For MS analyses under nondenaturing conditions, FGF1 was diluted in 20 mM ammonium acetate buffer, pH 6.8, to a final concentration of 10 μM. FGF1–HS complex was obtained by incubating FGF1 with HS oligosaccharides at room temperature for 60 min. Protein calibrants were diluted in either denaturing solution or nondenaturing solution to a final concentration of 10 μM.
IMMS Measurement and Data Analysis
NanoESI-IMMS experiments were performed using a quadrupole-TWIMS-TOF hybrid mass spectrometer (Synapt G2 HDMS; Waters Corp., Manchester, UK) in positive ionization mode. Protein samples were injected into the nanoESI source through a fused-silica emitter (PicoTip; New Objective, Woburn, MA, USA) with a flow rate varying from 0.2 to 0.5 uL/min. Experimental parameters were carefully tuned to prevent the protein and protein complex from unfolding or losing integrity due to extensive activation, while keeping substantial ion transmission, including controlling the collisional energy in the ion guides and keeping the source temperature and desolvation energy low. The applied experimental parameters were capillary voltage, 1.4 kV; sampling cone voltage, 20 V; extraction cone voltage, 5 V; source temperature, 30 °C; flow rate of nitrogen in the IM ion guide, 50 mL/min; flow rate of helium in the helium cell, 180 mL/ min; transfer collision energy, 0 V. Different sets of wave height and corresponding wave velocity were examined to optimize the mobility separation. The drift times of the calibrants and FGF1 samples were measured and identical experimental conditions were stringently applied.
For monitoring the collisional induced unfolding (CIU) of FGF1 and FGF1–HS complexes, protein ions of a selected charge state were isolated in a quadrupole mass filter and activated in the trap ion guide where subsequent collisional induced activation of ions takes place, followed by separation in the TWIMS ion mobility stage, and detection by TOF-MS.
Data analysis was performed using MassLynx 4.1 and Driftscope (Waters Corp., Manchester, UK). The CIU data was analyzed using CIUSuite [47]. CIU fingerprints of protein ions, which records the relative ion intensity (normalized and smoothed using Savitsky-Golay filter) as a function of collision energy and drift times, were shown in a 2-D contour plot using the CIUSuite plot function. The CIUsuite detect function was applied to identify and extract the most intense CIU features of the fingerprint based on a first derivative analysis, providing centroid drift times, transitional voltage, and stability range of each detected feature.
Converting Drift Times into CCSs
The CCS measurements were calibrated based on an empirical relationship between the drift times of protein calibrants and their known CCS obtained previously by DTIMS [48]. Briefly, a selected set of native and denatured protein calibrants, with a mass range from 12 to 102 KDa and a CCS range from 2303 to 5550 Å2 were employed. The drift times of these calibrates were corrected for mass-dependent flight time spent in the transfer ion guide and TOF mass analyzer and mass-independent flight time spent in the transfer ion guide. The CCSs of calibrants were corrected for their charge state and reduced mass with respect to the buffer gas. The natural logarithm of corrected CCSs were plotted against the natural logarithm of corrected drift times and a mathematical formula (LnΩ′ = A × Lndt′ + B) was derived. The calibration coefficient A was extracted to calculate the effective drift times dt″: . A calibration curve was generated by plotting the literature CCSs as a function of dt″. The experimental CCS of the analyte ion can be derived from this calibration curve based on the measured drift time [49, 50].
Preparation of Theoretical Models
FGF1–dp4 Complex
The Protein Data Bank (PDB) contains several models of wild-type human FGF1 in complex with heparin oligosaccharide, and each of them displays slightly differing contacts with the protein. The interaction energy between each protein–HS tetrasaccharide complex was calculated using a short minimization in implicit solvent followed by single frame binding free energy calculation to select one model for further theoretical investigation (see details in Supplementary Data and Supplementary Table 1). The complex that shows the strongest interaction (2AXM chain B) [18] was used for all further calculations.
FGF1–HS Complexes for d6, dp8, dp10, and dp12
2AXM chain B bound to Hp hexasaccharide was used to build the model for 1:1 and 2:1 FGF1–HS hexasaccharide complex. In order to avoid disrupting the contacts that the hexasaccharide made with the two FGF1 molecules, the heparin sequence from the crystal structure was kept, which is different from the experimentally used dp6 fragment.
The chosen dp4 complex was used and the heparin sequence was extended on either side using tleap to build the FGF1–dp8 complexes. The FGF1–dp6 complex was used and additional residues were added on either side of the existing dp6, according to the experimentally used sequences (Table 1), using tleap to build the FGF1–dp10 and the FGF1–dp12 complexes.
Table 1. Experimental and Theoretical CCS (Å2) for FGF1–HS Complexes in 2:1 Stoichiometry.
| Collision cross section (Å2) | |||||
|---|---|---|---|---|---|
|
|
|||||
| ID | HS sequencea | Experimental | PA | Scaled PA | TM |
| FGF1-dp4 | IdoA2S-GlcNS6S-IdoA2S-GlcNS6S | 2486.1 ± 6.3 | 2117.2 ± 5.2 | 2413.6 ± 6.0 | 2682.2 ±7.1 |
| FGF1-dp6 | GlcNS6S-GlcA-GlcNS6S-IdoA2S-GlcNS6S-GlcAb | 2520.2 ± 10.2 | 2350.0 ± 2.5 | 2679.0 ± 2.8 | 2990.0 ± 6.6 |
| FGF1-dp8 | GlcNS6S-GlcA-[GlcNS6S-IdoA2S]2-GlcNS6S-GlcA | 2536.0 ± 8.0 | 2191.9 ± 2.5 | 2498.8 ± 2.9 | 2784.8 ± 6.2 |
| FGF1-dp10 | GlcNS6S-GlcA-[GlcNS6S-IdoA2S]3-GlcNS6S-GlcA | 2543.9 ± 8.0 | 2253.6 ± 3.8 | 2569.2 ± 4.4 | 2871.9 ±7.1 |
| FGF1-dp12 | GlcNS6S-GlcA-[GlcNS6S-IdoA2S]4-GlcNS6S-GlcA | 2581.5 ± 7.9 | 2271.7 ± 5.3 | 2589.8 ± 6.0 | 2885.4 ± 8.8 |
All but dp4 HS oligosaccharides contained p-nitrophenol linker at the reducing end (dp4 contained OMe instead). Theoretical modeling employed OMe as the terminus for all HS
For modeling, the dp6 present in the FGF1-HS crystal structure (PDB id: 2AXM) was employed
2:1 FGF1–dp12 cis Model and 3:1 FGF–HS–dp12 Model
Two models were built for the 2:1 FGF1-fully sulfated dp12 in the cis conformation. Cis-model-1 was modeled using two FGF1–dp6 molecules connected through the GAG chain using tleap. First chain D of the crystal structure of apo-FGF1 dimer (PDB id: 2AFG [51]) was superimposed on chain A of FGF1– dp6 crystal structure (PDB id: 2AXM) and the coordinates for the dp6 were transferred to make 2AFG–dp6 complex to build the second 2:1 cis complex (Cis-model-2). This dp6 structure was extended to match the experimentally used fully sulfated dp12 by adding three residues on either side, using tleap.
3:1 FGF1 A fully sulfated dp12 complex
was built by connecting the available crystal structure for 2:1 FGF1–dp6 dimeric complex with a 1:1 FGF1–dp6 monomeric complex through the HS chain.
Molecular Dynamics Simulations
Topology and coordinate files for each system were generated using the tleap program, employing the ff99SB [52] and GLYCAM06 (version j) [53, 54] parameters for the protein and GAGs, respectively. Each system underwent energy minimization (1000 steps) in implicit solvent (IGB = 2). The net charge on each system, after energy minimization, was neutralized by addition of an appropriate number of counter ions (Na+ or Cl−). This was followed by solvation with TIP3P [55] water molecules in a cubic box extending at least 12 Å from any atom of the solute.
All MD simulations were performed with the GPU implementation of pmemd, pmemd.cuda_SPDP [56] in Amber14 [57]. Energy minimization of the solvent was performed in an NVT ensemble (1000 steps of steepest descent, 24,000 steps of conjugate gradient), followed by a full system energy minimization (1000 steps of steepest descent, 24,000 steps of conjugate gradient). The systems were heated from 5 to 300 K over 60 ps in an NVT ensemble, with a weak positional restraint (10 kcal/mol-Å2) on the atoms in the solute. A Berendsen-type thermostat [58] with a time coupling constant of 1 ps was utilized for temperature regulation. Equilibration and production were performed at constant pressure (NPT ensemble; 1 atm) with a pressure relaxation time of 1 ps. After the heating step, the restraints were removed from the solute atoms, and the entire system was allowed to equilibrate at 300 K for 1 ns. All covalent bonds involving hydrogen atoms were constrained using the SHAKE [59] algorithm, allowing a simulation time step of 2 fs. Scaling factors for 1–4 interactions were set to the recommended values of 1.0 and 1.2 for the GAG [53] and protein [52], respectively, and a non-bonded interaction cutoff of 8.0 Å was employed. Long-range electrostatics were computed with the particle mesh Ewald (PME) method. Data were collected for 200 ns for each system. Post-processing of the MD simulations was performed using CPPTRAJ [60] module of Amber. The graphical representations were generated using VMD [61].
Binding Energy Calculations
Models of the gas-phase complexes were generated from the MD simulations by removing the water molecules and subjecting the biomolecules to in vacuo minimization (IMIN = 5) with the SANDER module of AMBER14. This was performed for 5000 frames selected at even steps from the last 100 ns of the solvated simulation, and the resultant gas-phase trajectory was used to calculate the binding free energy in the gas phase using the MMPBSA.py script [62].
CCS Calculations
CCSs were calculated using 50 frames from the last 100 ns of the simulation using MOBCAL [63]. Each of these frames was minimized in vacuo, followed by the calculation of CCS using the projection approximation and trajectory methods.
Results and Discussion
IMMS of Unbound FGF1 and HS Tetrasaccharide-Bound FGF1
Under nondenaturing conditions, in the absence of HS, FGF1 exists mainly in the form of a monomer with a charge state distribution ranging from +9 to +7, accompanied by a lower abundance by the form of dimer with a charge state distribution ranging from +13 to +11 (Figure 1). This self-association of FGF1 has been reported before [51, 64], but the biological relevance of the self-association that we observed is not clear because of its low abundance. The narrow distribution of lower charge states indicates that FGF1 ions adapt a folded and compact conformation with fewer basic sites exposed for protonation [65]. Each observed charge state displays three peaks, corresponding to the sequence mass of FGF1 and masses corresponding to variants with one less amino acid from either the N-terminus or C-terminus. Owing to the compositional complexity, only the peaks of lowest mass were selected for further ion mobility analysis.
Figure 1.
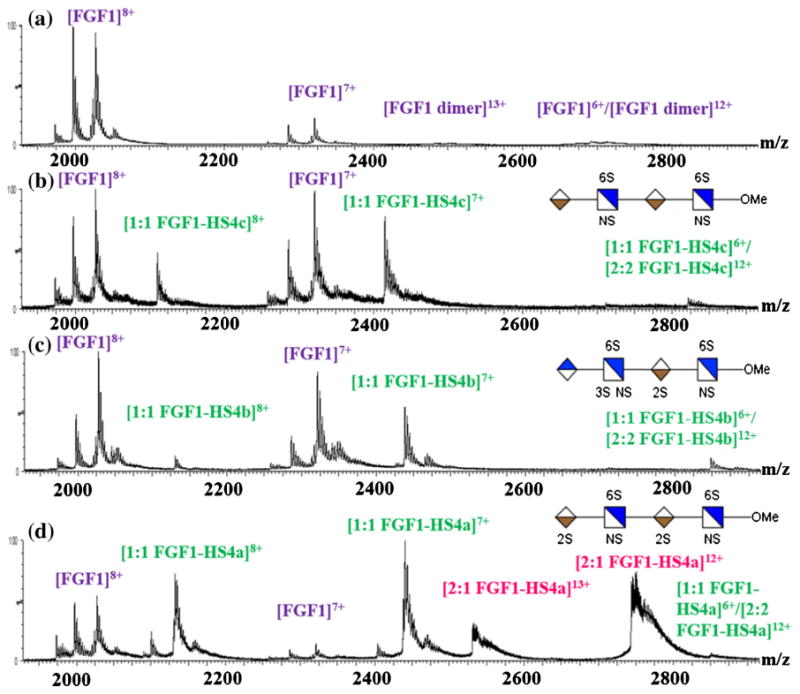
ESI mass spectra, obtained under nondenaturing conditions, of (a) FGF1 itself; (b) FGF1 incubated with HS dp4c at 2:1 ratio; (c) FGF1 incubated with HS dp4b at 2:1 ratio; (d) FGF1 incubated with HS dp4a at 2:1 ratio. Associating of two FGF1s by one HS (2:1 FGF1–HS complex) were observed only for the HS dp4a containing the high affinity binding motif
In an effort to examine the binding stoichiometry for the interaction of FGF1 and HS, a series of tetrasaccharides (dp4) with different sulfation patterns were incubated with FGF1 in a molar ratio of 1:2. The HS dp4a contains the high affinity binding motif for FGF1 IdoA2S-α-(1-4)-GlcNS6S-α-(1-4)-IdoA2S [24]. HS dp4b has the same overall level of sulfation as the dp4a, but the 2-O sulfation on the first residue from the nonreducing end was shifted to 3-O sulfation on the second residue, accompanied by the change of uronic acid stereochemistry (GlcA instead of IdoA) at the non-educing end. The HS dp4c lacks two 2-O sulfo groups compared with dp4-1. In the presence of HS, the formation of the 1:1 FGF1–HS complex was observed (Figure 1) without altering the charge state distributions of apo-FGF1. It is noteworthy that two peaks corresponding to a complex of two FGF1s with one HS (2:1 FGF1–HS complex) were detected only for the HS dp4a containing the high affinity binding motif (Figure 1d).
A majority of previous studies suggest that dimerization of FGF1 or complexation of FGF1 with HS and FGFR requires a long-chain HS oligosaccharides (at least a hexasaccharide or octasaccharide) [23, 66]. Other studies showed that a fully sulfated dp4 is sufficient for a high-affinity interaction with FGF [67, 68], for stabilizing FGF1 against thermal unfolding or digestion [69], and for initiating mitogenic activity [70]. A dimeric complex of FGF1 and HS as well as trimeric complex of HS, FGF1, and FGFR1 produced in the presence of tetrasaccharides containing the critical binding motif have been detected using MALDI mass spectrometry [71] and gel mobility shift assay [68]. Our observations highlight the importance of this critical binding motif, which imparts upon HS oligosaccharides as short as a tetrasaccharide the potential to induce FGF1 dimerization.
The arrival time distributions of the +7 charge state of FGF1 alone, the +7 charge state of 1:1 FGF1–HS dp4a complex, and the +12 charge state of 2:1 FGF1–HS dp4a complex are shown in Figure 2a, recorded at a wave height of 17 V. One narrow arrival time distribution was observed for each species, indicating the presence of one compact and folded conformation for the protein ion and the noncovalently associated protein– HS assemblies at both binding stoichiometries. These observations suggest that the solution structure of FGF1 survived in the gas phase environment of TWIMS, maintaining its capability to bind to HS.
Figure 2.
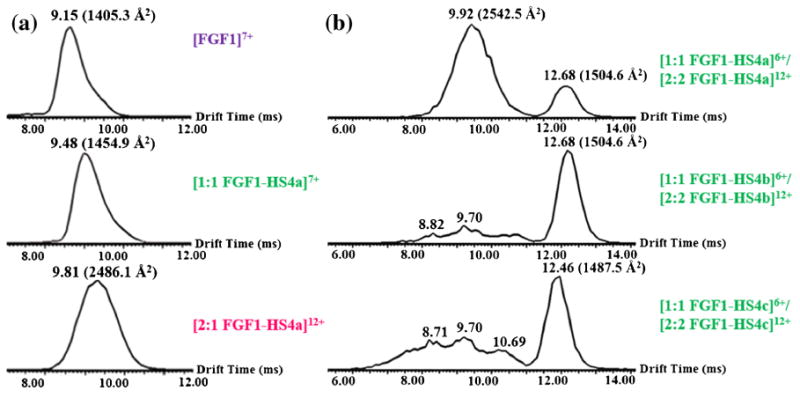
(a) Arrival time distributions (ms) of +7 charge state of FGF1 (upper), +7 charge state of 1:1 FGF1–HS dp4a complex (middle) and +12 charge state of 2:1 FGF1–HS dp4a complex (lower). The narrow arrival time distribution observed for each species indicates that the native structure of FGF1 and the noncovalent complex of FGF1 with HS are stable in the TWIMS experiment. (b) Arrival time distributions (ms) of +6 charge state of 1:1 FGF1–HS complex and +12 charge state of 2:2 FGF1–HS complex, with HS4a (upper), HS4b (middle), and HS4c (lower). The comparison between the narrow, well-defined arrival distribution of 2:1 FGF1–HS dp4a complex and the broad arrival time distribution of 2:1 FGF1–HS dp4b complex or 2:1 FGF1–HS dp4c complex indicates that the high affinity binding motif is responsible for the level of specificity in the dimerization of FGF1 upon the binding of HS
The peak corresponding to the even charge state of the 1:1 FGF1–HS complex overlaps in the mass spectrum with that corresponding to the 2:2 FGF1-HS complex with a doubled charge. However, the dimeric complex has a decreased charge-normalized cross section [72] so that it can be separated from the monomeric complex on the basis of a difference in drift times in a TWIMS experiment (Figure 2b). The feature with the longer drift time corresponds to the 1:1 FGF1–HS dp4 complexes, each of which exhibits a narrow drift time distribution as observed for the same species in a different charge state (Figure 2a). The features with shorter drift times correspond to 2:2 FGF1–HS complexes. One narrow arrival time distribution was detected for the FGF1–HS dp4a mixture, implying the presence of a single compact, ordered gas-phase conformation, in strong contrast to the broader peak with many features detected for the FGF1–HS dp4b mixture or the FGF1–HS dp4c mixture, implying the existence of a range of less specific binding modes.
The fact that subtle alterations of the HS sequence with regard to the sulfation pattern or epimerization lead to noticeable changes in both the binding stoichiometry and the binding mode of the FGF1–HS interaction suggest that the interactions between FGF1 and HS has certain degree of selectivity. These observations strengthen the importance of the critical binding motif in modulating the assembly of FGF1 and HS through a specific dimerization mode.
IMMS of HS Dodecasaccharide Bound FGF1 Complexes
A series of dodecasaccharides (dp12) with varying sulfation patterns were incubated with FGF1 at a molar ratio of 1:4 (GAG to protein) (Figure 3). An excess of FGF1was used to saturate the FGF1 binding epitopes on the dp12s. In the presence of the fully sulfated HS dp12, the binding of one HS dp12 to three FGF1 (3:1 FGF1–HS complex) were detected with a charge state distribution ranging from +15 to +13, accompanied by the binding of one HS dp12 to two FGF1 (2:1 FGF1–HS complex) with a charge state distribution ranging from +13 to +11. Peaks corresponding to apo-FGF1 were also detected, but in very low abundance compared with that for the complexes. Previous studies showed that the interaction between FGF1 and HS is driven by positive cooperativity [34, 73], so that FGF1 oligomerizes in preference to staying in monomeric state when bound with long-chain HS, and the formation of higher order of FGF1 oligomers was observed as the length of HS increases [74]. Our observations are consistent with these previous studies and highlight the biological function of HS as a storage reservoir for FGFs on the cell surface [23].
Figure 3.
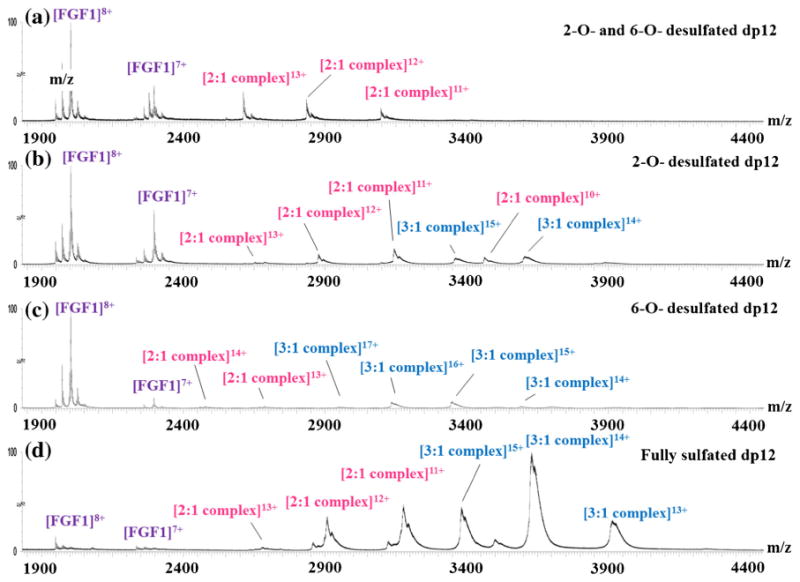
Native ESI mass spectra of (a) FGF1 with de-2-O- and de-6-O-sulfated dp12; (b) FGF1 with de-2-O-sulfated dp12; (c) FGF1 with de-6-O-sulfated dp12; (d) FGF1 with the dp12 that contains the critical binding motif. Structures of the dp12s are shown in Supplementary Data
The presence of de-2-O sulfated or de-6-O sulfated dp12 also resulted in two types of binding stereochemistry to FGF1, but their abundance was much lower than that of the fully sulfated dp12 for FGF1. In the presence of de-2-O and 6-O sulfated dp12, only a small fraction of FGF1 formed 2:1 FGF1–HS complex, whereas mostof FGF1 stayed inapo form. The marked difference in binding stoichiometry implies that the absence of 2-O and 6-O sulfo groups dramatically reduces the binding affinity.
Although the modified dp12s exhibit varying binding stoichiometry to FGF1, the arrival time distribution of the 3:1 FGF1–HS dp12 complexes all exhibited a single, narrow peak. The same observation has been reported for a different GAG– protein binding system, chemokine CCL2 and HS [75]. The fact that the ion mobility profiles of FGF1-modified HS dp12 complexes are similar implies that changes in HS structure lead to altered interactions with FGFs, but these alterations may not dramatically affect the compact, gas-phase conformation adapted by these complexes.
Our observations suggest that long-chain HS may not place as stringent requirement on the fine structure of HS sequence as short-chain HS, indicating a relative lack of specificity of binding for long-chain HS. Instead of presenting a specific binding motif for FGF1, the sequence of HS may be composed of many domains with different sulfation patterns and binding affinities toward FGF1, forming an affinity gradient on the cell surface that regulates FGFs signaling by directing them to the site of interaction [76].
CCSs of FGF1-HS Complexes
A comparison of the experimental and theoretical CCSs for the HS bound FGF1 complexes were carried out to determine their binding mode as well as the higher order oligomerization behavior of FGF1. The experimental CCSs for the complexes (Table 1) were calibrated based on an empirical relationship between the drift times of protein calibrants and their known CCSs obtained previously by conventional drift time IMS. For theoretical estimation, we used both the projection approximation (PA) [77] and the trajectory (TM) [63] method. PA estimates the orientationally-averaged CCS by projecting the molecule onto a randomly chosen plane in space, drawing a circle with the corresponding collision radius around the projection of each atom, and estimating the area of the “shadow” from all faces [78]. This method is known to underestimate CCS since it fails to consider the influence of long-range interactions and the scattering between the ion and neutral gas. TM represent the ions as a collection of atoms defined by their Lennard-Jones (12-6-4) potential, and relates the scattering angle of the buffer gas molecules before and after collision with the molecule's geometry. An orientationally-averaged CCS can be obtained by integrating over all possible collision geometries of the ion; however, this method is computationally much more intensive than the PA method [63]. The experimental CCSs are generally found to be larger than those computed using the PA method, and smaller than the TM results [79]. A previous study [80] have shown that there is a direct scaling relationship between experimental CCS and values calculated using PA methods, so calibrating the PA CCSs though this scaling relationship should provide a good standard for correct interpretation of experimental data.
Each model of the 2:1 FGF1–HS complex (Figure 4a) was based on the crystal structure 2AXM, which contains a heparin hexasaccharide bound to two molecules of FGF1 in trans orientation. The FGF1–dp4 complex was built by removing two residues from the hexasaccharide. For the complexes formed with oligosaccharides with chain length longer than dp6, we extended the crystal hexasaccharide sequence to build models for dp8 and higher in a relatively straightforward manner. However, considering the fact that changing the interactions at the binding interface between the two FGF1 molecules to accommodate GlcA in place of IdoA would affect the interaction dramatically, we chose to use the crystal structure for the FGF1–dp6 modeling.
Figure 4.
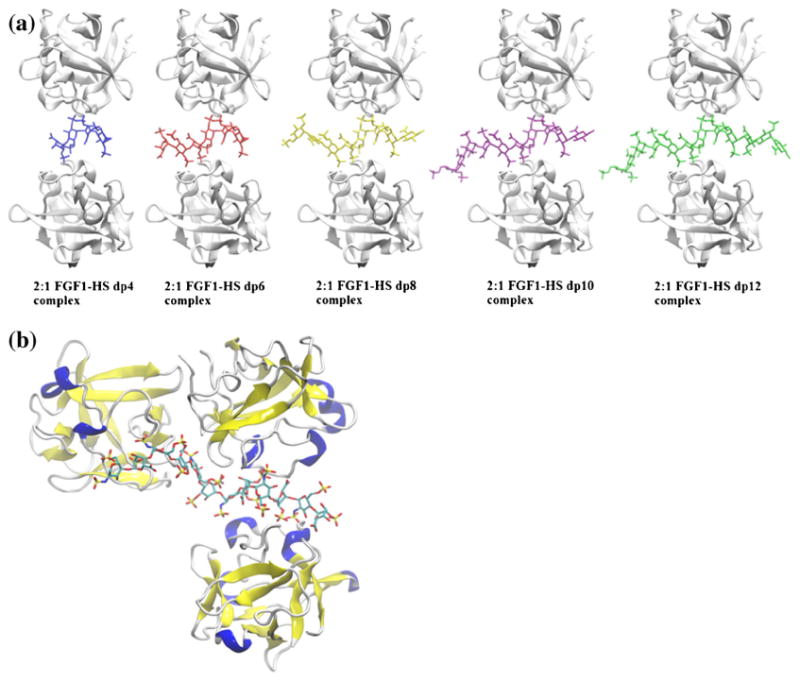
(a) Structural representation of 2:1 FGF1–HS complexes, displaying the binding of dp4-dp12 (left to right). The protein and HS are shown in ribbon and stick representation, respectively. (b) Structural representation of the hypothesized 3:1 FGF1–HS dodecasaccharide complex, obtained by connecting the available crystal structure for 2:1 FGF1–dp6 dimeric complex with a 1:1 FGF1–dp6 monomeric complex through the HS chain
Molecular dynamics (MD) simulations of these complexes were carried out in explicit solvent, followed by in vacuo minimization of a subset of the frames from the MD trajectories and CCS calculations. The theoretical CCSs for the FGF–dp6 complex do not follow the same trends as the other complexes, and the PA and TM numbers offer a slightly larger range due to the difference between the experimental and theoretically modeled dp6. A comparison of the experimental and theoretical CCSs for the five complexes shows that the experimental numbers lie between the PA and TM numbers, as expected. The experimental CCSs are on an average 12% lower than the TM numbers, which is as expected because of the partial collapse of the protein structure as a result of desolvation [81, 82]. The experimental CCSs are on an average 13% higher than the PA numbers, which is in agreement with previous studies [40, 83], which report experimental CCS to be about 15% higher than PA estimates. We have also reported the scaled PA numbers, which showed a close agreement with the experimental numbers. Moreover, we have compared the representative structure of the dimeric protein complex during the MD simulation and after the in vacuo minimization (as shown in Supplementary Data, Figure 6). The protein backbones RMSD between the two structures is 2.07Å, suggesting that there is no dramatic change of the overall shape of the protein, and the most relevant features of the protein structure are still retained. These results imply that the desolvation process in the TWIMS experiment did not significantly alter the solvated structure of FGF1 and its complexes, and their biological function to bind with HS was still retained.
Figure 6.
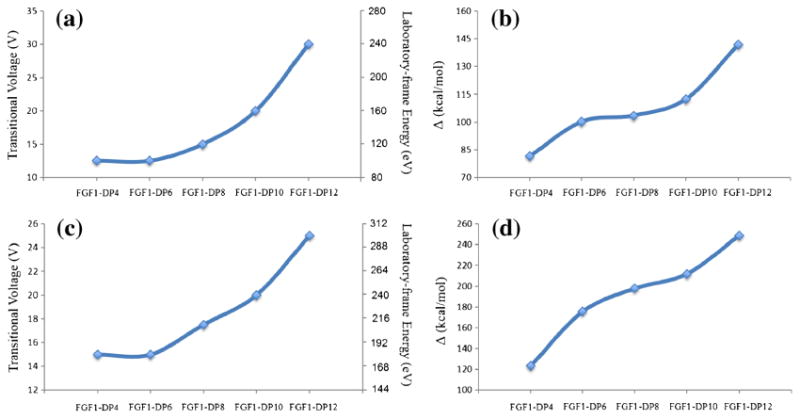
Trends for the experimental transitional voltages required for inducing unfolding for the stability of the FGF1–HS complexes in the 1:1 (a) and 2:1 (c) binding stoichiometry compared with the trends for theoretical binding free energies for the 1:1 (b) and 2:1 (d) complexes. The two trends are comparable, indicating that the binding of longer heparin oligosaccharides imparts greater stability to the protein against unfolding
In terms of the gas-phase stability of these complexes, it is noteworthy that the experimentally derived CCSs for the five complexes formed with HS of varying lengths lie very close to each other, with the difference between FGF1–dp4 and FGF1– dp12 being 100 Å2. This difference in the CCSs appears to arise only due to the size of the HS fragment, implying that the compact and folded conformations of the FGF1 complexes were not disturbed and remained stable during the TWIMS experiments.
A comparison of the CCS of the protein from the crystal structure (PDB id: 2AFG) and the protein after removal of the HS tetrasaccharide from the 1:1 FGF1–dp4 complex was made to determine any conformational changes induced in the protein upon HS binding. The conformational difference in the CCS of the two protein-forms was calculated to be less than 1% using both the TM and PA methods (data in Supplementary Table 2). We can therefore conclude that there is no major conformational change brought about in the protein as a result of HS binding, and the overall increase in the CCS can be attributed to the presence of HS itself, which is in agreement with previous studies [76, 84].
Two different dimerization modes for FGF2 have been previously proposed [17]. In the presence of long-chain HS fragments, FGF2 molecules oligomerize to form side-by-side cis dimers, whereas with short-chain HS disaccharides they form head-to-head trans dimers [17]. Similar observations have not been made for FGF1, and the crystal structures available in the PDB for 2:1 FGF1–HS complex only indicate a trans binding mode. We generated two models for the plausible cis forms of 2:1 FGF1–HS dp12 complexes (Supplementary Figure 1). Cis-model-1 used two FGF1–dp6 molecules joined through the GAG chain, with a cis-like starting conformation, to examine whether formation of cis form for FGF1 may be feasible, and if it could be differentiated from the trans model on the basis of differences in the CCS. However, during the course of MD simulations, it adopted a more trans-like conformation, likely due to the lack of stable protein–protein interactions between the two FGF1 molecules. In contrast, the Cis-model-2, based on the crystal structure of apo-FGF1 dimer (PDB id: 2AFG) with stable protein–protein interactions, was able to maintain the cis conformation during MD simulations. The models differed significantly in their starting and end conformations; however, both formed compact structures that lead to very similar theoretical CCS (data in Supplementary Table 3), making them indistinguishable from each other, and also from the trans form (Table 1). On the basis of this data, therefore, it is difficult to determine whether the cis form exists in solution, but if it does, it may be difficult to distinguish the cis and the trans binding conformations solely based on the CCS.
The TWIMS experiments also detected a 3:1 FGF1–HS complex in the presence of select sulfated HS–dp12. A plausible model, with three molecules of FGF1 in contact with HS– dp12, was computationally examined, using a model that melds the available crystal structure for 2:1 FGF1–dp6 dimeric complex (Figure 4b) with a 1:1 FGF1–dp6 monomeric complex through the HS chain. For this study, it was ensured that each of the three FGF1 molecules interacts directly with the HS since the formation of this higher order oligomer seems to be induced only in the presence of HS. The MD simulation of the complex was followed by calculation of CCS, and the experimentally calculated CCS was found to lie between the theoretically obtained CCS, using the PA and TM methods. Specifically, the experimental number was 15.9% higher than the PA and 11% lower than the TM methods. Although this is only one of the plausible 3:1 FGF1–HS models, the agreement with experimental CCS values provides reasonable confidence in believing that a 3:1 complex would adopt a similar oligomeric conformation.
Gas-Phase Stability of FGF1 and Its Complexes
The impact of GAG binding upon the stability of the three-dimensional structure of FGF1 was examined using collision induced unfolding (CIU) experiments, an approach developed by Ruotolo and co-workers [47]. We performed a series of collision-induced activation experiments with incremental changes in trapping collisional energy (CE) applied to selected protein and protein complex ions. Their conformational transition from a fully folded to a fully unfolded state was tracked, and was used to assess the ligand-induced influence on the gas-phase stability of these ions. A range of HS oligosaccharides of increasing lengths, containing the critical binding motif of IdoA2S-α-(1-4)-GlcNS6S-α-(1-4)-IdoA2S, were used to form dimeric and monomeric complexes with FGF1. The +8 charge state of monomeric protein complex ions was selected for activation, since this charge state provides higher ion intensity, sufficiently stability against low-energy dissociation, and distinguishable unfolding features within a relatively narrow collision energy window. The resulting CIU fingerprints of FGF1 and FGF1–HS complexes are shown in Figure 5, which record the drift time and intensity of each conformation feature that emerged during the unfolding process as the trap CE was ramped from 0 to 60 V in a 2.5 V increment.
Figure 5.
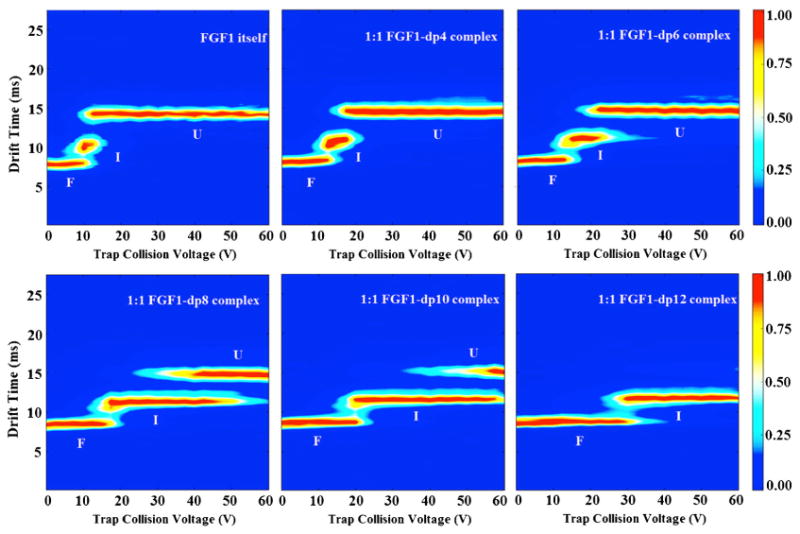
The 2D-contour CIU fingerprint of the monomeric apo-FGF1 and the 1:1 FGF1-HS, with HS of increasing length from dp4-dp12. The conformations were labeled as F, I, and U, indicating folded, intermediate, and unfolded states, respectively. The ion intensities are indicated by a color axis. As the length of the HS increases, they form complexes with FGF1 and increase the conformational stability of the complexes accordingly
The CIU fingerprint of unbound, apo-FGF1 exhibits three features. The first feature, which has the lowest drift time at 7.9 ms, corresponded to the most compact conformation of the protein ion. As the CE was raised to 10 V, this compact conformation diminished while the second feature appeared at 10.2 ms, corresponding to the intermediate state with a more extended conformation. Beyond the CE of 12.5 V, the third feature started to emerge and stabilized at 14.3 ms, indicating that the three-dimensional protein structure had unfolded completely.
The CIU fingerprints of 1:1 FGF1–HSdp4/HSdp6 complexes showed similar unfolding pathways as the apo-FGF1, for which the most extended conformation was dominant across the investigated voltage range. However, for both complexes, not only their most compact feature but also their partially unfolded feature showed a more elongated shape than those for the apo-FGF1. An increase of 2.5 V in the voltage at which the most compact feature starts to unfold was observed. A similar trend was observed for the voltage at which the intermediate feature further extends, shifting the voltages by 5 and 10 V, for the dp4-bound FGF1 and dp6-bound FGF1, respectively.
The CIU fingerprints of 1:1 FGF1–HSdp8 and –HSdp10 complexes show an even more substantial elevation of the voltage at which the most compact feature starts to unfold, shifting the voltage by 5 and 10 V, respectively. More importantly, the intermediate conformation gained more stability and became dominant for over 66.7% of the investigated voltage range. It is notable that the intermediate state and the extended state stabilize simultaneously for a longer voltage range, indicating that they have a different unfolding pathway from the species discussed above.
In significant contrast to the 1:1 short-chain HS complex bound FGF1 complex, the CIU fingerprint of the 1:1 FGF1–HSdp12 complex exhibited only two principal conformational features, corresponding to the fully folded conformation at 8.7 ms and the intermediate conformation at 11.7 ms, respectively. The survival of the relatively compact conformations at high voltages and the absence of the most extended conformation clearly suggest an even higher resistance towards collision induced unfolding than the apo-FGF1 or short HS bound FGF1 complexes.
The effect of collisional activation was also measured for 2:1 FGF1–HS complexes with HS oligosaccharides of different lengths, and a similar trend as the 1:1 FGF1–HS complexes was observed (Supplementary Figure 2). Comparing the fingerprints clearly shows a significant enhancement of the gas-phase stability of FGF1 upon the binding of HS oligosaccharides, regardless of the stoichiometry of the complexes formed. These observations are consistent with the solution-phase behavior of the FGF1 signaling system, which show that HS improves the conformational stability of FGF–FGFR complex against thermal, enzymatic, or pH-dependent inactivation and degradation [85, 86].
CIU fingerprints of the +12 charge state of the 2:1 FGF1-selected sulfated HS dp12s were also compared (Supplementary Figure 3). At low collision energy, their ion mobility profiles all exhibited single, narrow-distribution peaks, indicating that they exist in their compact conformation in the gas phase. As the trap CE was ramped from 0 to 40 V, the 2:1 FGF1–fully sulfated dp12 complex showed a slightly higher stability. Nevertheless, all four complexes exhibited similar behavior in their unfolding mode, with one compact and one unfolded state with similar centroid drift times and stability ranges. These observations indicate that length of the HS oligomer may be more important than its sulfation pattern in determining the gas-phase stability of these protein complexes.
Previous studies proposed that the stability of FGF complexes depends more on the overall degree of sulfation rather than a specific arrangement of the sulfation groups [25]. Our observations suggest a more complicated mechanism than that: four selectively sulfated dp12s display similar conformational stability and gas-phase unfolding behavior despite their different overall extent of sulfation; The de 2-O and 6-O sulfated dp12 with only six sulfo groups have a much higher gas-phase stability than the fully sulfated dp4 to dp8 (which have six or more sulfo groups). Apparently, both the length of the HS oligosaccharide and the distribution of sulfo groups contribute to stabilizing the gas-phase conformation of HS-bound FGF1 complexes.
Since the most compact feature should resemble the native structure of a protein complex, the CE required for its transition to a more extended state should be an experimentally quantitative estimation of the binding affinity of the HS oligosaccharide towards FGF1 [87]. Thus, we measured and plotted the voltage at which the most compact feature starts to unfold for each HS-bound FGF1 complex, as shown in Figure 6. The degree of stabilization is observed to correlate with the length of the HS oligosaccharide, with long-chain HS oligosaccharides interacting with FGF1 and forming complexes that slowed down the initiation and the process of collision-induced unfolding more effectively than short-chain HS.
Theoretically, the stability of a complex can be measured as the strength of interaction between the binding partners, and it would be reasonable to believe that the greater the binding affinity, the higher will be the stability, and consequently a higher CE would be required to induce unfolding of the complex. Binding free energies of FGF1 in complex with HS of increasing lengths (dp4–dp12), in both 1:1 and 2:1 binding stoichiometry (Figure 4a) were calculated to provide theoretical quantification. In vacuo minimization of a subset of the frames from the MD trajectories was carried out, followed by energetic post-processing to obtain the gas-phase interaction energies to mimic the binding modes representative of these complexes during mass spectrometry experiments. We observed that the interaction energies for the monomeric and dimeric complexes both increase as the length of the HS chain increases (Figure 6), as expected. These results are in direct agreement with the observed experimental stability trends of the FGF1-HS complexes.
The increase in the stability of the complex with an increase in the length of the HS chain can be attributed to the additional number of saccharide residues which present more sulfo and carboxyl groups to interact with the protein [73]. It was also observed during the MD simulations that the longer HS chains fold onto the proteins, forming more interactions with proteins, further restraining the shape of these structures, and providing them with more resistance to collision-induced unfolding.
These observations demonstrate that while a HS oligosaccharide as short as a tetramer is capable of promoting the formation of a FGF1 dimer complex (2:1 protein:tetramer stoichiometry), its stability is generally low due to its low efficiency to counteract the charge repulsion between the two protein surfaces [18, 19]. This is consistent with the observation that lower concentrations of long-chain HS oligosaccharides display tighter binding and higher biological activity, whereas HS dp4 support FGF signaling only when present at a high concentration [67].
Conclusions
Although previous studies of FGF1–FGFR2–HS complexes have reported using mass spectrometry, relying upon size-exclusion chromatography to determine the dominant configuration of the ternary complex [88], less structural information was extracted about the influence of the variation of HS chain length and sulfation pattern on the FGF1–HS interactions. In the present work, we have applied IMMS to the FGF1–HS signaling complexes and used molecular modeling to interpret the results. This approach has led to a number of important new conclusions. Although some previous results suggest that HS octasaccharide is the minimal length required for FGF1 dimerization and activation, our results showed that a short tetrasaccharide containing the high affinity binding motif is capable of dimerizing FGF1, forming complexes with a compact, well-defined gas-phase conformation. These finding demonstrate the importance of high-affinity binding motif for promoting the correct assembly of FGF1, as well as dictating the binding selectivity of FGF1–HS binding. Moreover, we found that FGF1 can participate in high order oligomers when interacting with long-chain HSs (dodecasaccharides), and the degree of oligomerization increases as the chain length of HS increases. However, the ion mobility profiles of FGF1 in complex with dodecasaccharides with different patterns of sulfate modification did not show much difference in stability, indicating a relative lack of specificity of binding with long-chain HS.
Furthermore, the experiments of collisional activation of FGF1 and FGF1–HS complexes confirmed the stabilizing effect of HS binding on the folded structure of FGF1. The degree of stabilization correlates with the length of the HS oligosaccharide, with longer HS oligosaccharides forming a complex that has higher gas-phase stability against collision-induced unfolding. The stability of the FGF1–HS complexes correlate with the overall degree of sulfation rather than on the precise location of sulfo groups. Finally, collisional cross-sections of FGF1–HS complexes have been examined using theoretical models built using the available X-ray crystal structures of FGF1 in complex with HS. MD simulations with these models are in agreement with the experimental results, indicating that the folded gas-phase conformation of FGF1 and its binding activity survive the transition of the ions into the gas phase. We also proposed a plausible structure for the 3:1 FGF1–HS complex. The binding affinity of HS of varying lengths has also been studied using molecular modeling, which gave results consistent with experiments.
Collectively, these results extended the application of TWIMS for investigating GAG–protein interactions at the molecular level. Characterization of the interactions between FGF1 and HS will benefit the understanding of the full magnitude FGF signaling pathways and, eventually, the development of new medicines. The extension of the methodology will add further dimensions to the drug-development studies of a large array of other GAG-binding proteins.
Supplementary Material
Acknowledgments
The authors thank Jessica N. Rabuck, Joseph D. Eschweiler, and Dr. Brandon T. Ruotolo for sharing the CIUSuite, and for their considerable advice with the application of this software. The authors are grateful for funding from National Institutes of Health grant P41- GM103390 in support of this work.
Footnotes
Yuejie Zhao and Arunima Singh contributed equally to this work.
Electronic supplementary material The online version of this article (doi:10.1007/s13361-016-1496-8) contains supplementary material, which is available to authorized users.
References
- 1.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology, and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu X, Komiya H, Chirino A, Faham S, Fox G, Arakawa T, Hsu B, Rees D. Three-dimensional structures of acidic and basic fibroblast growth factors. Science. 1991;251:90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]
- 4.Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Bedussi F, Bottini A, Memo M, Fox SB, Sigala S, Generali D. Targeting fibroblast growth factor receptor in breast cancer: a promise or a pitfall? Expert Opin Ther Targets. 2014;18:665–678. doi: 10.1517/14728222.2014.898064. [DOI] [PubMed] [Google Scholar]
- 6.Böhlen P, Esch F, Baird A, Gospodarowicz D. Acidic fibroblast growth factor (FGF) from bovine brain: amino-terminal sequence and comparison with basic FGF. EMBO J. 1985;4:1951. doi: 10.1002/j.1460-2075.1985.tb03876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laestander C, Engström W. Role of fibroblast growth factors in elicitation of cell responses. Cell Prolif. 2014;47:3–11. doi: 10.1111/cpr.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eswarakumar V, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Hatch NE. FGF signaling in craniofacial biological control and pathological craniofacial development. Crit Rev Eukaryot Gene Expr. 2010;20:295–311. doi: 10.1615/critreveukargeneexpr.v20.i4.20. [DOI] [PubMed] [Google Scholar]
- 10.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:1–12. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Investig. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat Prod Rep. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 13.Garg HG, Linhardt RJ, Hales CA. Chemistry and biology of heparin and heparan sulfate. Elsevier; Oxford, UK: 2006. [Google Scholar]
- 14.Shimokawa K, Kimura-Yoshida C, Nagai N, Mukai K, Matsubara K, Watanabe H, Matsuda Y, Mochida K, Matsuo I. Cell surface heparan sulfate chains regulate local reception of FGF signaling in the mouse embryo. Dev Cell. 2011;21:257–272. doi: 10.1016/j.devcel.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 15.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Mohammadi M, Dikic I, Sorokin A, Burgess W, Jaye M, Schlessinger J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herr AB, Ornitz DM, Sasisekharan R, Venkataraman G, Waksman G. Heparin-induced self-association of fibroblast growth factor-2 evidence for two oligomerization processes. J Biol Chem. 1997;272:16382–16389. doi: 10.1074/jbc.272.26.16382. [DOI] [PubMed] [Google Scholar]
- 18.DiGabriele AD, Lax I, Chen DI, Svahn CM, Jaye M, Schlessinger J, Hendrickson WA. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature. 1998;393:812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- 19.Waksman G, Herr AB. New insights into heparin-induced FGF oligomerization. Nat Struct Biol. 1998;5:527–530. doi: 10.1038/778. [DOI] [PubMed] [Google Scholar]
- 20.Pellegrini L, Burke DF, von Delft F, Mulloy B, Blundell TL. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature. 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- 21.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. Crystal structure of a ternary FGF–FGFR–heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 22.Petitou M, Casu B, Lindahl U. 1976–1983, a critical period in the history of heparin: the discoveryof the antithrombin binding site. Biochimie. 2003;85:83–89. doi: 10.1016/s0300-9084(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 23.Plotnikov AN, Schlessinger J, Hubbard SR, Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 24.Kreuger J, Salmivirta M, Sturiale L, Giménez-Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- 25.Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kreuger J, Jemth P, Sanders-Lindberg E, Eliahu L, Dina R, Basilico C, Salmivirta M, Lindahl U. Fibroblast growth factors share binding sites in heparan sulphate. Biochem J. 2005;389:145–150. doi: 10.1042/BJ20042129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreuger J, Prydz K, Pettersson RF, Lindahl U, Salmivirta M. Characterization of fibroblast growth factor 1 binding heparan sulfate domain. Glycobiology. 1999;9:723–729. doi: 10.1093/glycob/9.7.723. [DOI] [PubMed] [Google Scholar]
- 28.Guimond SE, Turnbull JE. Fibroblast growth factor receptor signalling is dictated by specific heparan sulphate saccharides. Curr Biol. 1999;9:1343–1346. doi: 10.1016/s0960-9822(00)80060-3. [DOI] [PubMed] [Google Scholar]
- 29.Ashikari-Hada S, Habuchi H, Kariya Y, Itoh N, Reddi AH, Kimata K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J Biol Chem. 2004;279:12346–12354. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- 30.Matsuo I, Kimura-Yoshida C, Shimokawa K. Divergent roles of heparan sulfate in regulation of FGF signaling during mammalian embryogenesis. In: Kondoh H, Kuroiwa A, editors. New Principles in Developmental Processes. Springer; Japan: 2014. pp. 239–251. [Google Scholar]
- 31.Zhang F, Zhang Z, Lin X, Beenken A, Eliseenkova AV, Mohammadi M, Linhardt RJ. Compositional analysis of heparin/ heparan sulfate interacting with fibroblast growth factor–fibroblast growth factor receptor complexes. Biochemistry. 2009;48:8379–8386. doi: 10.1021/bi9006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrovsky O, Berman B, Gallagher J, Mulloy B, Fernig DG, Delehedde M, Ron D. Differential effects of heparin saccharides on the formation of specific fibroblast growth factor (FGF) and FGF receptor complexes. J Biol Chem. 2002;277:2444–2453. doi: 10.1074/jbc.M108540200. [DOI] [PubMed] [Google Scholar]
- 33.Moy FJ, Seddon AP, Campbell EB, Böhlen P, Powers R. 1H, 15N, 13C, and 13CO assignments and secondary structure determination of basic fibroblast growth factor using 3D heteronuclear NMR spectroscopy. J Biomol NMR. 1995;6:245–254. doi: 10.1007/BF00197806. [DOI] [PubMed] [Google Scholar]
- 34.Robinson CJ, Harmer NJ, Goodger SJ, Blundell TL, Gallagher JT. Cooperative dimerization of fibroblast growth factor 1 (FGF1) upon a single heparin saccharide may drive the formation of 2:2:1 FGF1– FGFR2c–heparin ternary complexes. J Biol Chem. 2005;280:42274–42282. doi: 10.1074/jbc.M505720200. [DOI] [PubMed] [Google Scholar]
- 35.Naimy H, Buczek-Thomas JA, Nugent MA, Leymarie N, Zaia J. Highly sulfated nonreducing end-derived heparan sulfate domains bind fibroblast growth factor-2 with high affinity and are enriched in biologically active fractions. J Biol Chem. 2011;286:19311–19319. doi: 10.1074/jbc.M110.204693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spivak-Kroizman T, Lemmon M, Dikic I, Ladbury J, Pinchasi D, Huang J, Jaye M, Crumley G, Schlessinger J, Lax I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell. 1994;79:1015–1024. doi: 10.1016/0092-8674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 37.Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum Mol Genet. 2004;13:69–78. doi: 10.1093/hmg/ddh011. [DOI] [PubMed] [Google Scholar]
- 38.Lee MK, Lander AD. Analysis of affinity and structural selectivity in the binding of proteins to glycosaminoglycans: development of a sensitive electrophoretic approach. Proc Natl Acad Sci U S A. 1991;88:2768–2772. doi: 10.1073/pnas.88.7.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atmanene CD, Petiot-Bécard SP, Zeyer D, Van Dorsselaer A, Vivat Hannah VR, Sanglier-Cianférani S. Exploring key parameters to detect subtle ligand-induced protein conformational changes using traveling wave ion mobility mass spectrometry. Anal Chem. 2012;84:4703–4710. doi: 10.1021/ac203223h. [DOI] [PubMed] [Google Scholar]
- 40.Zhao Y, Singh A, Li L, Linhardt RJ, Xu Y, Liu J, Woods RJ, Amster IJ. Investigating changes in the gas-phase conformation of anti-thrombin III upon binding of arixtra using traveling wave ion mobility spectrometry (TWIMS) Analyst. 2015;140:6980–6989. doi: 10.1039/c5an00908a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyung SJ, Robinson CV, Ruotolo BT. Gas-phase unfolding and disassembly reveals stability differences in ligand-bound multiprotein complexes. Chem Biol. 2009;16:382–390. doi: 10.1016/j.chembiol.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 42.Duijn EV, Barendregt A, Synowsky S, Versluis C, Heck AJ. Chaperonin complexes monitored by ion mobility mass spectrometry. J Am Chem Soc. 2009;131:1452–1459. doi: 10.1021/ja8055134. [DOI] [PubMed] [Google Scholar]
- 43.Politis A, Park AY, Hyung SJ, Barsky D, Ruotolo BT, Robinson CV. Integrating ion mobility mass spectrometry with molecular modelling to determine the architecture of multiprotein complexes. PLoS One. 2010;5:e12080. doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc Rev. 2010;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 45.Xu Y, Cai C, Chandarajoti K, Hsieh PH, Li L, Pham TQ, Sparkenbaugh EM, Sheng J, Key NS, Pawlinski R. Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat Chem Biol. 2014;10:248. doi: 10.1038/nchembio.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zong C, Venot A, Dhamale O, Boons GJ. Fluorous supported modular synthesis of heparan sulfate oligosaccharides. Org Lett. 2013;15:342–345. doi: 10.1021/ol303270v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eschweiler JD, Rabuck-Gibbons JN, Tian Y, Ruotolo BT. CIUSuite: a quantitative analysis package for collision induced unfolding measurements of gas-phase protein ions. Anal Chem. 2015;87:11516–11522. doi: 10.1021/acs.analchem.5b03292. [DOI] [PubMed] [Google Scholar]
- 48.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Collision cross-sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal Chem. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 49.Smith DP, Knapman TW, Campuzano I, Malham RW, Berryman JT, Radforda SE, Ashcrofta AE. Deciphering drift time measurements from travelling wave ion mobility spectrometry-mass spectrometry studies. Cell [Equation (1)] 2009;12:13. doi: 10.1255/ejms.947. [DOI] [PubMed] [Google Scholar]
- 50.Ruotolo BT, Benesch JL, Sandercock AM, Hyung SJ, Robinson CV. Ion mobility-mass spectrometry analysis of large protein complexes. Nat Protoc. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 51.Blaber M, DiSalvo J, Thomas KA. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry. 1996;35:2086–2094. doi: 10.1021/bi9521755. [DOI] [PubMed] [Google Scholar]
- 52.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct Funct Bioinformat. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydr J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh A, Tessier MB, Pederson K, Wang X, Venot AP, Boons GJ, Prestegard JH, Woods RJ. Extension and Validation of the GLYCAM Force Field Parameters for modeling Glycosaminoglycans. Can J Chem. 2016;94:1–9. doi: 10.1139/cjc-2015-0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 56.Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Case DA, VB, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER. 2014;14 [Google Scholar]
- 58.Berendsen HJ, Postma JPM, van Gunsteren WF, DiNola A, Haak J. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 59.Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 60.Roe DR, Cheatham TE., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 61.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 62.Miller BR, III, McGee TD, Jr, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA. py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 63.Mesleh M, Hunter J, Shvartsburg A, Schatz GC, Jarrold M. Structural information from ion mobility measurements: effects of the long-range potential. J Phys Chem. 1996;100:16082–16086. [Google Scholar]
- 64.Venkataraman G, Shriver Z, Davis JC, Sasisekharan R. Fibroblast growth factors 1 and 2 are distinct in oligomerization in the presence of heparin-like glycosaminoglycans. Proc Natl Acad Sci. 1999;96:1892–1897. doi: 10.1073/pnas.96.5.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chowdhury SK, Katta V, Chait BT. Probing conformational changes in proteins by mass spectrometry. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 66.Gambarini AG, Miyamoto CA, Lima GA, Nader HB, Dietrich CP. Mitogenic activity of acidic fibroblast growth factor is enhanced by highly sulfated oligosaccharides derived from heparin and heparan sulfate. Mol Cell Biochem. 1993;124:121–129. doi: 10.1007/BF00929204. [DOI] [PubMed] [Google Scholar]
- 67.Delehedde M, Malcolm L, Gallagher JT, Rudland PS, Fernig DG. Fibroblast growth factor-2 binds to small heparin-derived oligosaccharides and stimulates a sustained phosphorylation of p42/44 mitogen-activated protein kinase and proliferation of rat mammary fibroblasts. Biochem J. 2002;366:235–244. doi: 10.1042/BJ20011718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu ZL, Zhang L, Yabe T, Kuberan B, Beeler DL, Love A, Rosenberg RD. The involvement of heparan sulfate (HS) in FGF1/HS/ FGFR1 signaling complex. J Biol Chem. 2003;278:17121–17129. doi: 10.1074/jbc.M212590200. [DOI] [PubMed] [Google Scholar]
- 69.Mach H, Volkin DB, Burke CJ, Middaugh CR, Linhardt RJ, Fromm JR, Loganathan D, Mattsson L. Nature of the interaction of heparin with acidic fibroblast growth factor. Biochemistry. 1993;32:5480–5489. [Google Scholar]
- 70.Ornitz DM, Herr AB, Nilsson M, Westman J, Svahn CM, Waksman G. FGF binding and FGF receptor activation by synthetic heparanderived di- and trisaccharides. Science. 1995;268:432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- 71.Guerrini M, Agulles T, Bisio A, Hricovini M, Lay L, Naggi A, Poletti L, Sturiale L, Torri G, Casu B. Minimal heparin/heparan sulfate sequences for binding to fibroblast growth factor-1. Biochem Biophys Res Commun. 2002;292:222–230. doi: 10.1006/bbrc.2002.6634. [DOI] [PubMed] [Google Scholar]
- 72.Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, Bitan G, Teplow DB, Bowers MT. Amyloid β-protein: monomer structure and early aggregation states of Aβ42 and its Pro19 alloform. J Am Chem Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 73.Brown A, Robinson CJ, Gallagher JT, Blundell TL. Cooperative heparin-mediated oligomerization of fibroblast growth factor-1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys J. 2013;104:1720–1730. doi: 10.1016/j.bpj.2013.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faham S, Linhardt RJ, Rees DC. Diversity does make a difference: fibroblast growth factor-heparin interactions. Curr Opin Struct Biol. 1998;8:578–586. doi: 10.1016/s0959-440x(98)80147-4. [DOI] [PubMed] [Google Scholar]
- 75.Seo Y, Andaya A, Bleiholder C, Leary JA. Differentiation of CC versus CXC chemokine dimers with GAG octasaccharide binding partners: an ion mobility mass spectrometry approach. J Am Chem Soc. 2013;135:4325–4332. doi: 10.1021/ja310915m. [DOI] [PubMed] [Google Scholar]
- 76.Faham S, Hileman R, Fromm J, Linhardt R, Rees D. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 77.Mack E., Jr Average cross-sectional areas of molecules by gaseous diffusion methods. J Am Chem Soc. 1925;47:2468–2482. [Google Scholar]
- 78.Wyttenbach T, von Helden G, Batka JJ, Carlat D, Bowers MT. Effect of the long-range potential on ion mobility measurements. J Am Soc Mass Spectrom. 1997;8:275–282. [Google Scholar]
- 79.Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6:281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 80.Benesch JL, Ruotolo BT. Mass spectrometry: comes of age for structural and dynamical biology. Curr Opin Struct Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jurneczko E, Barran PE. How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross-sections in the gas phase. Analyst. 2011;136:20–28. doi: 10.1039/c0an00373e. [DOI] [PubMed] [Google Scholar]
- 82.Breuker K, McLafferty FW. Stepwise evolution of protein native structure with electrospray into the gas phase, 10-12 to 102 s. Proc Natl Acad Sci. 2008;105:18145–18152. doi: 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hall Z, Politis A, Bush MF, Smith LJ, Robinson CV. Charge-state dependent compaction and dissociation of protein complexes: insights from ion mobility and molecular dynamics. J Am Chem Soc. 2012;134:3429–3438. doi: 10.1021/ja2096859. [DOI] [PubMed] [Google Scholar]
- 84.Zhu X, Hsu BT, Rees DC. Structural studies of the binding of the anti-ulcer drug sucrose octasulfate to acidic fibroblast growth factor. Structure. 1993;1:27–34. doi: 10.1016/0969-2126(93)90006-3. [DOI] [PubMed] [Google Scholar]
- 85.Sommer A, Rifkin DB. Interaction of heparin with human basic fibroblast growth factor: protection of the angiogenic protein from proteolytic degradation by a glycosaminoglycan. J Cell Physiol. 1989;138:215–220. doi: 10.1002/jcp.1041380129. [DOI] [PubMed] [Google Scholar]
- 86.Zhoulli FY, Kanb M, Owensa RT, McKeehanl WL, Thompson JA, Linhardtd RJ, Hook M. Heparin-dependent fibroblast growth factor activities: effects of defined heparin oligosaccharides. Eur J Cell Biol. 1997;73:71–78D. [PubMed] [Google Scholar]
- 87.Niu S, Rabuck JN, Ruotolo BT. Ion mobility-mass spectrometry of intact protein–ligand complexes for pharmaceutical drug discovery and development. Curr Opin Chem Biol. 2013;17:809–817. doi: 10.1016/j.cbpa.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 88.Harmer NJ, Ilag LL, Mulloy B, Pellegrini L, Robinson CV, Blundell TL. Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)–FGF receptor–heparin complex. J Mol Biol. 2004;339:821–834. doi: 10.1016/j.jmb.2004.04.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.