

Abstract
Endothelial cell (EC) activation and inflammation is a key step in the initiation and progression of many cardiovascular diseases. Targeted delivery of therapeutic reagents to inflamed EC using nanoparticles is challenging as nanoparticles do not arrest on EC efficiently under high shear stress. In this study, we developed a novel polymeric platelet-mimicking nanoparticle for strong particle adhesion onto ECs and enhanced particle internalization by ECs. This nanoparticle was encapsulated with dexamethasone as the anti-inflammatory drug, and conjugated with polyethylene glycol, glycoprotein 1b, and trans-activating transcriptional peptide. The multi-ligand nanoparticle showed significantly greater adhesion on P-selectin, von Willebrand Factor, than the unmodified particles, and activated EC in vitro under both static and flow conditions. Treatment of injured rat carotid arteries with these multi-ligand nanoparticles suppressed neointimal stenosis more than unconjugated nanoparticles did. These results indicate that this novel multi-ligand nanoparticle is efficient to target inflamed EC and inhibit inflammation and subsequent stenosis.
Keywords: Endothelial cell, Nanoparticle, Glycoprotein 1b, Trans-activating transcriptional peptide
Introduction
Cardiovascular diseases and diabetes mellitus continue to be the major causes of death. The activation of vascular endothelial cells (EC) during inflammation results in the expression of adhesion molecules, leukocyte adhesion and activation, and release of inflammatory cytokines, all of which are key steps in the initiation and progression of cardiovascular diseases [1–3]. Drugs such as dexamethasone (Dx) and nitric oxide donor have been demonstrated to inhibit EC inflammation [4, 5]. Dx is a potent anti-inflammatory steroid and its efficacy in reducing post-stent restenosis has been demonstrated when it is administered either systemically or through drug-eluting stents (DES) [6, 7]. However, its prolonged systemic use is associated with a number of side effects such as muscle atrophy [8], osteoporosis [9], and diabetes [10]. There are also situations where drug-eluting stents are not appropriate treatment options; including at locations exposed to mechanical flexion, at large vascular bifurcations, or in patients unable to take potent blood thinning medications. Therefore, a targeted delivery of anti-inflammatory drugs such as Dx to inflamed EC is ideal to achieve therapeutic goals with minimized potential for systemic complications.
Recently, nanoparticles (NPs) have increasingly been tested as vehicles for drug delivery as they provide more therapeutic payloads to pathologic tissues with specific “targeting ligands” attached to the particles, leading to less systemic side effects and toxicity [11, 12]. Although NPs can be easily taken up by ECs at static condition or low shear stress flow, they do not sufficiently arrest on ECs under high shear flow [13]. In addition, internalizing these NPs, an essential step for the drug delivery, is also greatly reduced under arterial level shear stress [14]. Hence, enhancing NPs adhesion and uptake by inflamed ECs under arterial flow conditions (>10 dyn/cm2) is critical for these NPs drug carriers.
Platelets adhere to inflamed ECs and sub-endothelial layer at sites of vascular injury, triggering platelet activation and thrombus formation. This platelet-EC adhesion under flow conditions is mediated by the binding of platelet glycoprotein 1b (GP1b) to P-selectin and von Willebrand Factor (vWF) expressed on the surface of inflamed EC membrane and sub-endothelium [15]. Cell penetrating peptides having the sequences of membrane-interacting proteins such as transmembrane domains, signal peptide, or fusion proteins, have been found to facilitate the transport of large biomolecules into the cells [16, 17]. Inspired by these observations, we developed a novel type of platelet-mimicking multi-ligand NPs that have enhanced cellular internalization property and tested them for delivering anti-inflammatory drugs to inflamed ECs and sub-endothelium (Fig. 1). Poly(L-lactic-co-glycolic acid) (PLGA), an FDA-approved biodegradable polymer was used to fabricate these NPs. Dx was chosen as the prototype drug for its potent anti-inflammatory effect. Polyethylene glycol (PEG) was used to inhibit the opsonization of NPs and extend their life-span in the circulation. PEG also works as a spacer for conjugation of GP1b and trans-activating transcriptional peptide (TAT), one of the cell penetrating peptides facilitating cellular internalization. We hypothesized that such multi-ligand NPs could have enhanced adhesion to and internalization by inflamed ECs under high shear force, and effectively inhibit endothelial cell inflammation.
Fig. 1.
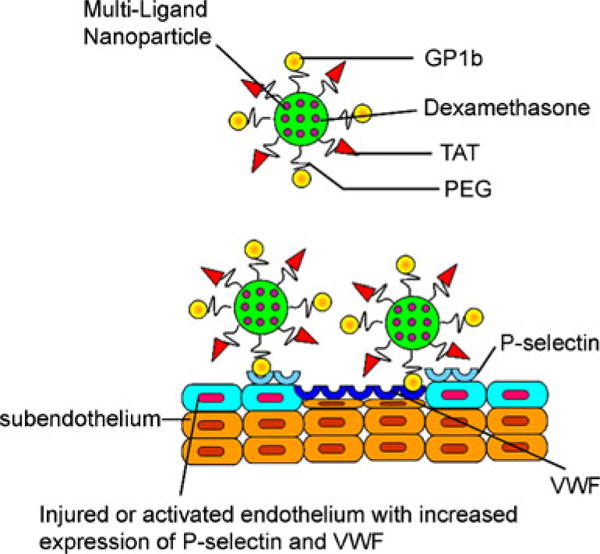
Schematic of multi-ligand NPs interacting with injured blood vessel for targeted drug delivery.
Materials and Methods
Materials
PLGA (inherent viscosity 0.15–0.25 dl/g, copolymer ratio 50:50) with a carboxyl end group was purchased from Lakeshore Biomaterials (Birmingham, AL). Bi-functional PEG (NH2-PEG-COOH) was a product of Laysan Bio inc. (Arab, AL). Human aorta endothelial cells (HAEC) were purchased from PromoCell Corp., Germany. Medium 199, fetal bovine serum, and low serum growth supplement, FM® 4–64 FX dye were purchased from Invitrogen, Inc. (Carlsbad, CA). Glycocalicin (MW 145 kDa), which consists of the extracellular part of GP1b alpha, was prepared from platelets as previously described [18]. Biotin-TAT (sequence: GRKKRRQRRR) was purchased from Anaspec inc. (Fremont, CA). All other chemicals if not specified were purchased from Sigma-Aldrich, Inc. (St. Louis, MO).
Multi-Ligand NPs Preparation
We first synthesized Dx-encapsulated PLGA NPs using a double emulsion method [19], followed by the conjugation of PEG, glycocalicin (GP1b), and TAT on the particle surface [20] (supplementary materials). These particles are named PLGA-PEG-GP1b, PLGA-PEG-TAT, or PLGA-PEG-GP1b/TAT NPs, in accordance with the ligands conjugated on these NPs.
Characterization of Multi-Ligand NPs
The NPs size, polydispersity, and surface charge (zeta potential) before and after ligand conjugation were measured using dynamic light scattering (ZetaPLUS, Brookhaven Instruments, NY). The morphology of NPs was annotated under transmission electron microscopy (FEI Tecnai G2 Spirit, OR). The conjugation of PEG on PLGA NPs was verified using Fourier-transform infrared spectroscopy (FTIR) (Nicolet 6700 FTIR spectrometer, Thermo Fisher Scientific). The loading efficiency and dynamic release profile of Dx from synthesized multi-ligand NPs were also determined using methods described in the Supplementary materials.
Adhesion of Multi-Ligand NPs on P-Selectin or vWF Under Flow
P-selectin or vWF-coated surfaces were used as the substrates in flow systems to test the adhesion of PLGA-PEG, PLGA-PEG-GP1b, or PLGA-PEG-GP1b/TAT NPs, as previously described [21]. The amounts of attached NPs were quantified after 30 min of perfusion at a flow that generated 10 dyne/cm2 shear stress (see detail in supplementary materials).
Uptake of Multi-Ligand NPs by Inflamed EC Under Static or Flow Conditions
HAEC grown on glass slides were first activated by histamine as previously described [22]. They were incubated with coumarin-incorporated PLGA-PEG, PLGA-PEG-GP1b, or PLGA-PEG-GP1b/TAT NPs (200 μg/ml) at static or under flow condition by mounting glass slides on a customized flow system [23] that was perfused with NPs suspension at an arterial level of shear stress (10 dyne/cm2). After 30 min, the cells were gently washed and lysed with 1 % Triton X-100. The fluorescence intensity of cell lysates (from coumarin-incorporated NPs) was measured to determine the amount of NPs uptaken by the cells, by comparing it to a standard curve established between the NP concentration and fluorescence intensity. The amount of cells was quantified by measuring the total cell DNA using the Quant-iT™ PicoGreen® dsDNA Assay (Invitrogen Inc). The amount of NPs uptaken by the cells was then normalized to the amount total cell DNA, and the uptake of ligand-conjugated nanoparticles was compared with that of control (unconjugated) nanoparticles (presented as a percentage of the PLGA-PEG control group).
In addition, particles uptaken by ECs were visualized under a confocal microscope after staining the cells with cell membrane dye FM® 4–64 FX (5 μg/ml) for 5 min immediately after incubation with the NPs.
Localization of Multi-Ligand NPs on Injured Rat Artery Ex Vivo
Animal studies were performed in accordance with the animal welfare policy and IACUC-approved protocol by the University of Texas at Arlington. In brief, the right common carotid artery of Sprague–Dawley rats (300–500 g) was injured by three repeated inflations of angioplasty balloon catheter (1.5 mm×8 mm, PTCA Dilatation Catheter, Boston Scientific Corp.) to 8 psi and maintained for 30 s. For visualization and quantification of NPs uptaken and accumulated by injured arteries, near-infrared dye NIR-797 was loaded inside the multi-ligand NPs. Immediately after vessel injury, 50 μl of PLGA-PEG, PLGA-PEG-GP1b, PLGA-PEG-TAT, or PLGA-PEG-GP1b/TAT NPs suspension (200 μg/ml suspended in normal saline) was injected into the injured artery segments with both ends temporarily clamped. After a 30-min incubation, the blood flow was restored. The animals were killed after another 30 min. The treated carotid artery was quickly dissected, rinsed to remove blood, then measured of the near-infrared intensity using Kodak In Vivo FX Pro imaging system at a wavelength of Ex 760 nm/Em 830 nm to quantify the amount of NPs retained in the artery wall.
Anti-Restenosis Effects of Multi-Ligand NPs on the Injured Rat Artery In Vivo
A rat carotid artery was injured as described above. Fifty microliters of either normal saline (NS), Dx-loaded PLGA-PEG NPs suspension (200 μg/ml suspended in NS), Dx-loaded PLGA-PEG-GP1b/TAT NPs suspension (200 μg/ml suspended in NS), or Dx solution (3 % w/v in NS) were injected into the injured carotid artery segments via infusion catheter as described previously [24]. Five rats were used in each experimental group. Since previous studies had found no significant differences between ligand-conjugated PLGA NPs with the sham (saline) control group in neointima stenosis [25, 26], we omitted NP groups without drug loading in our animal studies. These animals were killed, and the carotid arteries were collected and processed for histological analysis 14 days after arterial injury. Ten cross-sections along each treated artery segment were cut (6 μm thick) and stained with HE and Hart’s elastin stain. Video microscopy with computerized digital planimetry was used to measure areas of the lumen, intima (limited by the internal elastic lamina), and arterial wall (limited by the external elastic lamina). The intimal hyperplasia was evaluated by the intimal area (in square millimeter) and ratio of intima area normalized to total wall area [27].
Statistical Analysis
Results were analyzed using StatView 5.0 software (SAS Institute). Data were presented as mean ± stand error of the mean. p<0.05 presented the existence of significant statistical differences between compared groups (n=3–6 experiments).
Results
Characterization of Multi-Ligand NPs
The synthesized PLGA NPs were spherical with diameters of ∼200 nm or less as illustrated by TEM (Fig. 2b). The mean hydrodynamic diameter of these NPs measured by DLS increased slightly from 188 to 197 nm after incorporating of PEG, and further to 241 nm after GP1b and TAT conjugation (Fig. 2a). The homogenicity of these NPs decreased after surface modifications, as reflected by the increased polydispersity index from 0.14 to 0.31. The surface charge was reduced from −26 mV of unmodified NPs to −6.5 mV after GP1b and TAT conjugation. The conjugation of PEG to PLGA NPs was verified using FTIR analysis. As shown in Fig. 2c, unmodified PLGA NPs have a sharp peak at 1750 cm−1, a characteristic for carbonyl groups of an ester bond. The peaks at around 3000, 1670, and 1560 cm−1 are all attributed to amine groups of PEG. After PEG conjugation, the carbonyl amide peaks shrank, with the appearance of a new prominent peak at 2215–2240 cm−1. This is a characteristic for the formation of a ═C–N bond. Together, these changes of reflection spectrum indicate the successful binding between PLGA and PEG.
Fig. 2.
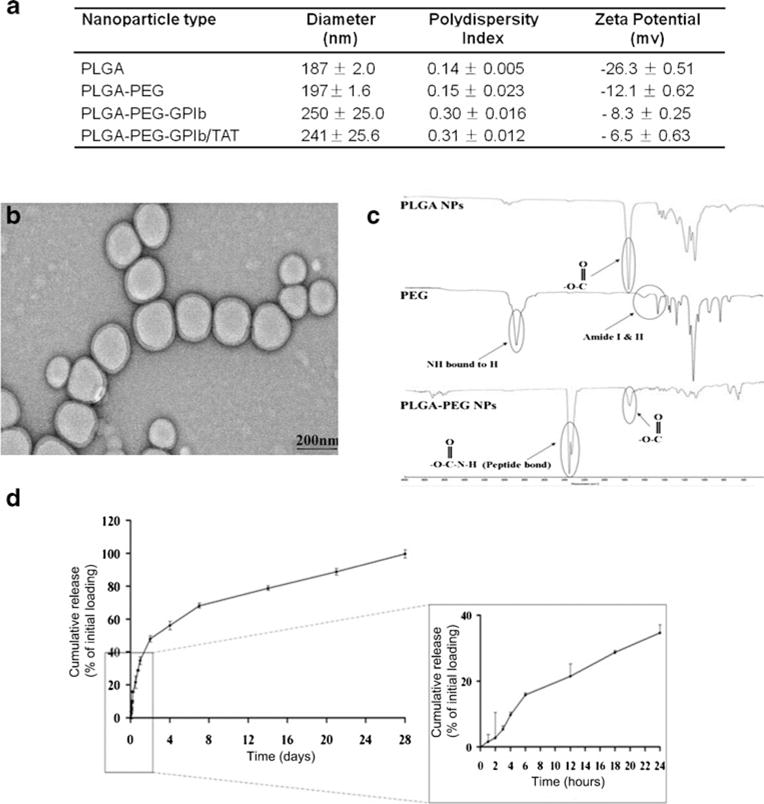
Characterization of multi-ligand NPs. a Dynamic light scattering measurement of NPs size, polydispersity, and zeta potential. b Representative transmission electron microscopy image of the NPs. c FTIR spectra of the PLGA-PEG NPs. d In vitro release of Dx from multi-ligand NPs at 37 °C (N=4)
The loading efficiency of Dx in multi-ligand NPs was calculated to be 63.5 %. The in vitro release of Dx from these NPs exhibited a biphasic profile (Fig. 2d). Up to 35 % of loaded Dx was quickly released within the first 24 h and the rest at a sustained rate that lasted for 4 weeks.
Adhesion of Multi-Ligand NPs to P-Selectin or vWF Under Flow
The adhesion of PLGA-PEG-GP1b NPs to a P-selectin or vWF-coated surface under 10 dyn/cm2 of shear stress was significantly greater (>85 %) compared to PLGA-PEG particles. A double conjugation of GP1b and TAT (PLGA-PEF-GP1b/TAT) did not further increase particle adhesion (Fig. 3).
Fig. 3.
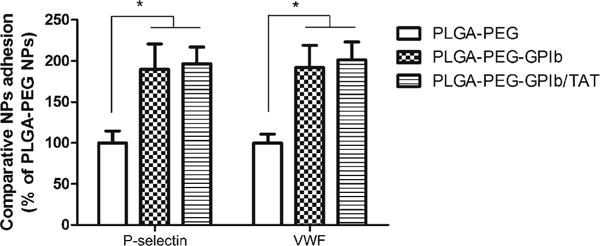
Adhesion of control NPs (PLGA-PEG), GP1b-conjugated NPs (PLGA-PEG-GP1b), and both GP1b and TAT-conjugated NPs (PLGA-PEG-GP1b/TAT) on P-selectin or vWF-coated substrates under parallel flow conditions (10 dyne/cm2). N=4. *p<0.05
Cellular Uptake of Multi-Ligand NPs
PLGA-PEG-GP1b NPs taken by activated HAECs were significantly increased (>80 %) compared to PLGA-PEG NPs (control group) under static conditions (I of Fig. 4). In addition, the conjugation of TAT (PLGA-PEG-GP1b/TAT) further significantly enhanced NPs uptake. Under a shear stress of 10 dyn/cm2, the uptake of the PLGA-PEG NPs and PLGA-PEG-GP1b NPs were reduced to ∼40 % compared to their relative uptake under static conditions. However, the uptake of PLGA-PEG-GP1b/TAT NPs remained more than 70 %. As shown in II of Fig. 4, more NPs (green dots) were internalized by activated ECs when they were decorated with GP1b and TAT (II-c of Fig. 4) than those which were conjugated with GP1b alone (II-b of Fig. 4). On the other hand, only a few PLGA-PEG NPs were taken by the cells (II-a of Fig. 4).
Fig. 4.
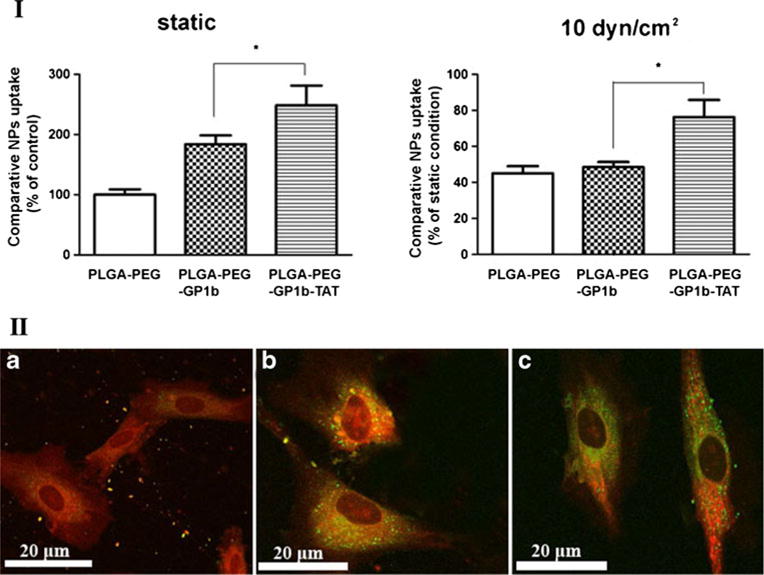
Uptake of multi-ligand NPs by activated endothelial cells. (I) Uptake of control NPs (PLGA-PEG), GP1b-conjugated NPs (PLGA-PEG-GP1b), and both GP1b and TAT-conjugated NPs (PLGA-PEG-GP1b/TAT) by activated HAECs at static or flow condition (10 dyne/cm2). N=4, *p<0.05. (II) Confocal microscopy images of HAECs uptaken of PLGA-PEG (a), PLGA-PEG-GP1b (b), or PLGA-PEG-GP1b/TAT (c) NPs. Scale bar 20 μm
Retention of Multi-Ligand PLGA NPs in Injured EC in Vivo
By visualizing and quantifying fluorescence intensity, we found that the amount of PLGA-PEG-GP1b NPs retained in injured carotid arteries was more than four times of that with PLGA-PEG NPs (Fig. 5). PLGA-PEG-GP1b/TAT NPs further increased particle retention to more than ten times as compared to PLGA-PEG NPs. PLGA-PEG-TAT NPs had a similar level of particle retention as PLGA-PEG NPs.
Fig. 5.
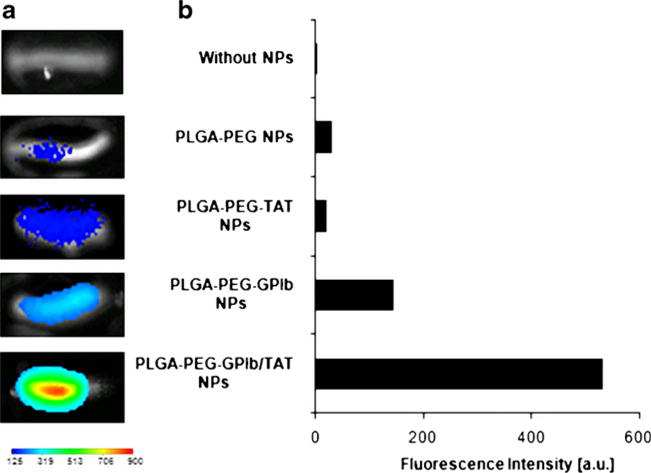
Multi-ligand NPs retention in the injured rat carotid artery wall. a Near infrared (NIR) fluorescence images of NPs retention in injured rat carotid artery wall. b Quantification of fluorescence intensities of particles retention in the arteries
Inhibition of Intima Stenosis In Vivo
Intima stenosis (intima area/total wall area=0.55) resulting from endothelial injury was significantly reduced by the treatment with Dx either alone or loaded in multi-ligand NPs (intima area/total wall area=0.21 or 0.18, respectively) as shown in Fig. 6. In contrast, Dx-incorporated PLGA-PEG NPs only slightly reduced the intima stenosis (intima area/total wall area=0.44, p=0.08 vs NS (normal saline)).
Fig. 6.
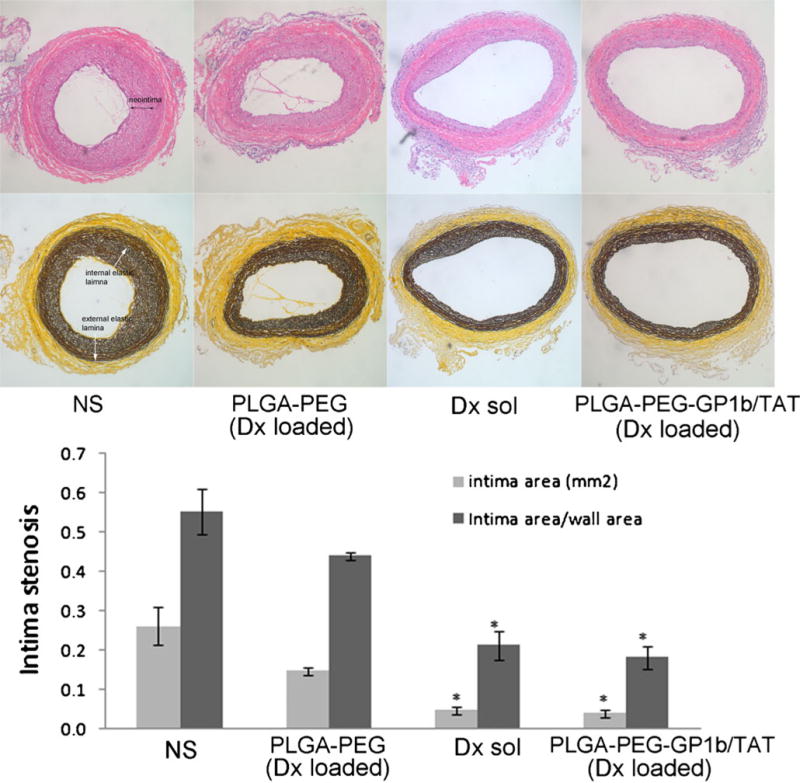
Cohort balloon-injured rat carotid artery intima stenosis after treatment with normal saline (NS), PLGA NPs conjugated with PEG (PLGA-PEG) and Dx-loaded, Dx solution, or PLGA NPs conjugated with PEG, GP1b, and TAT (PLGA-PEG-GP1b/TAT) and Dx-loaded. Top row HE staining; Middle row elastin staining; Bottom row calculated intima stenosis. N=5, *p<0.05 vs. NS
Discussion
Developing an NP carrier to deliver anti-inflammatory drugs to inflamed arterial wall has long been a challenge as high shear stress in the blood flow impedes the particle adhesion and uptake by the endothelium. Inspired by the platelet-EC adhesion during blood coagulation under physiological flow conditions, we developed a novel type of multi-ligand (PEG, GP1b, and TAT)-coated NPs for a targeted delivery of therapeutic reagents (Dx as the model drug in this study) to inflamed or injured endothelial cells on the arterial wall. These multi-ligand NPs have a significantly greater rate of adhesion to a surface that was coated with P-selectin or vWF as well as to activated ECs in vitro under static and flow conditions as compared to unmodified particles. As a result of these enhanced adhesions and uptake, these Dx packed multi-ligand NPs significantly suppressed the intima stenosis of an injured artery, at a level that was only achieved with a significantly higher concentration of Dx than what was packed in these NPs. These results demonstrate that these novel multi-ligand NPs improved the efficiency of delivering Dx to inflamed ECs.
Many factors including materials, particle size, and surface-bond moieties can affect the retention of NPs in the circulation. We used PLGA to formulate these NPs because they have been approved by the FDA for drug delivery. PLGA NPs loaded with paclitaxel or Dx have been used to inhibit the proliferation of smooth muscle cells during endovascular interventions [26, 28, 29]. PLGA NPs formulated in our study burst released 35 % of loaded Dx within the first day, followed by a sustained release over a month. This drug-releasing rate is similar to other contemporary studies [30]. The drug release rate of PLGA NPs can also be altered by varying the ratio of lactide and glycolide of the PLGA materials [31]. We synthesized NPs with a 200-nm diameter because NPs with a diameter between 100 and 200 nm have a comparatively longer half-life in the circulation due to their sizes being large enough to avoid uptake in the liver, but small enough to avoid filtration in the spleen [32].
The surface modification that we opted for has been widely used for optimizing the rate of NPs adhesion and retention. PEG is one of the most widely used materials to decorate NPs that provide a “stealth effect” to improve particle stability, solubility, and reduce immunogenicity in the blood [33]. Three common methods for PEG decoration are: covalent conjugation of PEG on PLGA particle surfaces after synthesis, conjugation of PEG to PLGA before particle synthesis, and ring opening-copolymerization of PEG with PLA/PGA [34]. The direct conjugation of PEG to PLGA NPs after particle synthesis, as used in this study, does not render the highest PEG surface density. However, it does not disturb the drug–polymer hydrophobic or charge interactions. Excessive surfactants and toxic soluble byproducts generated during conjugation can also be easily removed using this method.
The ligands GP1b and TAT were conjugated on the NPs surface to facilitate particle adhesion and internalization by inflamed ECs. GP1b is the ligand binding subunit of the glycoprotein Ib-IX-V complex that is almost exclusively expressed on platelets. It binds P-selectin and vWF expressed on inflamed endothelial cells and sub-endothelium [35]. In comparison to adhesion molecules such as integrins that mediate cell adhesion primarily at low shear stress, GP1b can effectively interact with P-selectin and vWF at a high level of shear stress found in arterioles and small arteries [15, 36]. The selectin-counter-receptor P-selectin glycoprotein ligand-1 [37], biotinylated-Sialyl Lewis (X) [38], and monoclonal antibodies against E- and P-selectin [39] have also been reported to enhance NPs adhesion onto ECs under flow conditions.
Cell penetrating peptides (CPP) have been used to assist the cellular internalization of NPs [40, 41]. The HIV TAT was the first CPP discovered in 1988 and now has many naturally derived peptide analogs [42, 43]. We found that TAT did not favor particle adhesion (Fig. 3), but greatly increased NPs internalization by activating ECs under flow conditions (Fig. 4). The internalization of NPs involves several endocytotic pathways, including caveolar-mediated endocytosis, clathrin-medicated endocytosis, and macropinocytosis. TAT primarily assists in macropinocytosis [44]. Here, we found that the cellular uptake of these multi-ligand NPs was reduced to 50, 80, and 60 % when chlorpromazine (clathrin inhibitor), filipin (caveolae inhibitor), and amiloride (macropinocytosis inhibitor) were used, respectively (data not shown), suggesting that multiple pathways were involved simultaneously in the internalization of the multi-ligand NPs.
Using a rat surgery model, we show that these multi-ligands significantly enhanced the adhesion and uptake of Dx packed NPs by injured carotid arteries under arterial blood flow (Fig. 5). The treatment of an injured carotid artery with these Dx-incorporated particles inhibited stenosis to a level similar to that achieved by significantly higher concentrations of Dx without being packed in these NPs (Fig. 6). These NPs could sustain therapeutic dosages of a given drug to the targeted cells, with potential reduction in systemic side effects.
One limitation of this research is that we were not able to observe the particle retention in the arteries when the wound was closed in the animal study, possibly because only a small amount of particles were used and the NIR signals of these NPs were not intense enough to penetrate the vessel wall. More advanced techniques, such as magnetic resonance imaging combined with magnetic material-loaded NPs might be more sensitive for real-time detection [45].
Conclusion
In the present study, we developed a novel multi-ligand (GP1b/TAT) decorated PLGA NPs for targeted delivery of Dx to inflamed ECs. Both in vitro and in vivo data demonstrated that these particles improved adhesion and cellular internalization by inflamed ECs compared to unmodified NPs, even under high fluidic shear stress. Treatment of injured carotid artery with these NPs suppressed neointimal stenosis. These NPs provide a new tool for treating vascular inflammation.
Supplementary Material
Clinical Relevance.
This study was conducted to assess the effect of a novel targeted drug-delivery nanoparticle developed in our laboratories for treatments of complications after transluminal angioplasty and vascular stenting.
Acknowledgments
The authors would like to acknowledge the assistance provided by members of the Core Imaging Facility at UTSW and the Characterization Center for Materials and Biology at UTA. We also thank Alicia J. Sisemore for her help with the manuscript editing. We acknowledge the financial support from the American Heart Association Scientist Development Award 0735270N (K.N.), NIH grants HL091232 (K.N.) and EB007271 (L.T.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12265-013-9460-5) contains supplementary material, which is available to authorized users.
Contributor Information
Hao Xu, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Soujanya Kona, Department of Bioengineering, University of Texas at Arlington, 500 UTA Blvd, ERB-241, Arlington, TX 76010, USA.
Lee-Chun Su, Department of Bioengineering, University of Texas at Arlington, 500 UTA Blvd, ERB-241, Arlington, TX 76010, USA.
Yi-Ting Tsai, Department of Bioengineering, University of Texas at Arlington, 500 UTA Blvd, ERB-241, Arlington, TX 76010, USA.
Jing-Fei Dong, Puget Sound Blood Center and University of Washington, Seattle, WA, USA.
Emmanouil S. Brilakis, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas, TX, USA
Liping Tang, Department of Bioengineering, University of Texas at Arlington, 500 UTA Blvd, ERB-241, Arlington, TX 76010, USA.
Subhash Banerjee, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas, TX, USA.
Kytai T. Nguyen, Department of Bioengineering, University of Texas at Arlington, 500 UTA Blvd, ERB-241, Arlington, TX 76010, USA
References
- 1.Schillinger M, Minar E. Restenosis after percutaneous angioplasty: the role of vascular inflammation. Vascular Health and Risk Management. 2005;1:73–78. doi: 10.2147/vhrm.1.1.73.58932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clinical Science (London, England) 2007;112:375–384. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- 3.Munro JM, Cotran RS. The pathogenesis of atherosclerosis: atherogenesis and inflammation. Laboratory Investigation. 1988;58:249–261. [PubMed] [Google Scholar]
- 4.Zampolli A, Basta G, Lazzerini G, Feelisch M, De Caterina R. Inhibition of endothelial cell activation by nitric oxide donors. Journal of Pharmacology and Experimental Therapeutics. 2000;295:818–823. [PubMed] [Google Scholar]
- 5.Everts M, Kok RJ, Asgeirsdottir SA, Melgert BN, Moolenaar TJM, Koning GA, et al. Selective intracellular delivery of dexamethasone into activated endothelial cells using an e-selectin-directed immunoconjugate. Journal of Immunology. 2002;168:883–889. doi: 10.4049/jimmunol.168.2.883. [DOI] [PubMed] [Google Scholar]
- 6.Versaci F, Gaspardone A, Tomai F, Ribichini F, Russo P, Proietti I, et al. Immunosuppressive therapy for the prevention of restenosis after coronary artery stent implantation (impress study) Journal of the American College of Cardiology. 2002;40:1935–1942. doi: 10.1016/s0735-1097(02)02562-7. [DOI] [PubMed] [Google Scholar]
- 7.Park YM, Han SH, Lee K, Suh SY, Oh PC, Shin EK. Dexamethasone-eluting stents had sustained favorable ischemic driven target lesion revascularization rates over 5 years: a randomized controlled prospective study. International Journal of Cardiology. 2013 doi: 10.1016/j.ijcard.2012.08.033. in press. [DOI] [PubMed] [Google Scholar]
- 8.Qin J, Du R, Yang YQ, Zhang HQ, Li Q, Liu L, et al. Dexamethasone-induced skeletal muscle atrophy was associated with upregulation of myostatin promoter activity. Research Veterinary Science. 2013;94:84–89. doi: 10.1016/j.rvsc.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 9.Kim MH, Lee GS, Jung EM, Choi KC, Jeung EB. The negative effect of dexamethasone on calcium-processing gene expressions is associated with a glucocorticoid-induced calcium-absorbing disorder. Life Sciences. 2009;85:146–152. doi: 10.1016/j.lfs.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Ranta F, Avram D, Berchtold S, Dufer M, Drews G, Lang F, et al. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. 2006;55:1380–1390. doi: 10.2337/db05-1220. [DOI] [PubMed] [Google Scholar]
- 11.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nature Reviews. Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clinical Pharmacology and Therapeutics. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 13.Dickerson JB, Blackwell JE, Ou JJ, Shinde Patil VR, Goetz DJ. Limited adhesion of biodegradable microspheres to e-and p-selectin under flow. Biotechnology and Bioengineering. 2001;73:500–509. doi: 10.1002/bit.1085. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen KT, Shukla KP, Moctezuma M, Braden AR, Zhou J, Hu Z, et al. Studies of the cellular uptake of hydrogel nanospheres and microspheres by phagocytes, vascular endothelial cells, and smooth muscle cells. Journal of Biomedical Materials Research. Part A. 2009;88:1022–1030. doi: 10.1002/jbm.a.31734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowenberg EC, Meijers JC, Levi M. Platelet-vessel wall interaction in health and disease. The Netherlands Journal of Medicine. 2010;68:242–251. [PubMed] [Google Scholar]
- 16.Deshayes S, Morris MC, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cellular and Molecular Life Sciences. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biology of the Cell. 2008;100:201–217. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]
- 18.Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, et al. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for p-selectin. The Journal of Experimental Medicine. 1999;190:803–813. doi: 10.1084/jem.190.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DH, Martin DC. Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials. 2006;27:3031–3037. doi: 10.1016/j.biomaterials.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Deshmukh R, Timmons R, Nguyen KT. Enhanced endothelialization on surface modified poly(L-lactic acid) substrates. Tissue Engineering. Part A. 2011;17:865–876. doi: 10.1089/ten.tea.2010.0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kona S, Dong JF, Liu YL, Tan JF, Nguyen KT. Biodegradable nanoparticles mimicking platelet binding as a targeted and controlled drug delivery system. International Journal of Pharmaceutical. 2012;423:516–524. doi: 10.1016/j.ijpharm.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin A, Sabnis A, Kona S, Nattama S, Patel H, Dong JF, et al. Shear-regulated uptake of nanoparticles by endothelial cells and development of endothelial-targeting nanoparticles. Journal of Biomedical Materials Research. Part A. 2010;93:833–842. doi: 10.1002/jbm.a.32592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu H, Nguyen KT, Brilakis ES, Yang J, Fuh E, Banerjee S. Enhanced endothelialization of a new stent polymer through surface enhancement and incorporation of growth factor-delivering microparticles. Journal of Cardiovascular Translation. 2012;5:519–527. doi: 10.1007/s12265-012-9381-8. [DOI] [PubMed] [Google Scholar]
- 24.Larifla L, Deprez I, Pham I, Rideau D, Louzier V, Adam M, et al. Inhibition of vascular smooth muscle cell proliferation and migration in vitro and neointimal hyperplasia in vivo by adenoviral-mediated atrial natriuretic peptide delivery. The Journal of Gene Medicine. 2012;14:459–467. doi: 10.1002/jgm.2639. [DOI] [PubMed] [Google Scholar]
- 25.Kimura S, Egashira K, Nakano K, Iwata E, Miyagawa M, Tsujimoto H, et al. Local delivery of imatinib mesylate (sti571)-incorporated nanoparticle ex vivo suppresses vein graft neointima formation. Circulation. 2008;118:S65–S70. doi: 10.1161/CIRCULATIONAHA.107.740613. [DOI] [PubMed] [Google Scholar]
- 26.Mei L, Sun H, Jin X, Zhu D, Sun R, Zhang M, et al. Modified paclitaxel-loaded nanoparticles for inhibition of hyperplasia in a rabbit arterial balloon injury model. Pharmaceutical Research. 2007;24:955–962. doi: 10.1007/s11095-006-9214-z. [DOI] [PubMed] [Google Scholar]
- 27.Bonan R, Paiement P, Scortichini D, Cloutier MJ, Leung TK. Coronary restenosis: evaluation of a restenosis injury index in a swine model. American Heart Journal. 1993;126:1334–1340. doi: 10.1016/0002-8703(93)90531-d. [DOI] [PubMed] [Google Scholar]
- 28.Feng SS, Zeng W, Teng Lim Y, Zhao L, Yin Win K, Oakley R, et al. Vitamin E TPGS-emulsified poly(lactic-co-glycolic acid) nanoparticles for cardiovascular restenosis treatment. Nanomedicine (London, England) 2007;2:333–344. doi: 10.2217/17435889.2.3.333. [DOI] [PubMed] [Google Scholar]
- 29.Westedt U, Kalinowski M, Wittmar M, Merdan T, Unger F, Fuchs J, et al. Poly(vinyl alcohol)-graft-poly(lactide-co-glycolide) nanoparticles for local delivery of paclitaxel for restenosis treatment. Journal of Controlled Release. 2007;119:41–51. doi: 10.1016/j.jconrel.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Kamath KR, Barry JJ, Miller KM. The taxus drug-eluting stent: a new paradigm in controlled drug delivery. Advanced Drug Delivery Reviews. 2006;58:412–436. doi: 10.1016/j.addr.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 31.Jain AK, Das M, Swarnakar NK, Jain S. Engineered PLGA nanoparticles: an emerging delivery tool in cancer therapeutics. Critical Reviews in Therapeutic Drug Carrier Systems. 2011;28:1–45. doi: 10.1615/critrevtherdrugcarriersyst.v28.i1.10. [DOI] [PubMed] [Google Scholar]
- 32.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nature Reviews. Drug Discovery. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 33.Peracchia MT, Vauthier C, Puisieux F, Couvreur P. Development of sterically stabilized poly(isobutyl 2-cyanoacrylate) nanoparticles by chemical coupling of poly(ethylene glycol) Journal of Biomedical Materials Research. 1997;34:17–26. doi: 10.1002/(sici)1097-4636(19970305)34:3<317::aid-jbm6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 34.Betancourt T, Byrne JD, Sunaryo N, Crowder SW, Kadapakkam M, Patel S, et al. Pegylation strategies for active targeting of PLA/PLGA nanoparticles. Journal of Biomedical Materials Research. Part A. 2009;91:263–276. doi: 10.1002/jbm.a.32247. [DOI] [PubMed] [Google Scholar]
- 35.Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thrombosis Research. 2004;114:447–453. doi: 10.1016/j.thromres.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 36.Ni H, Freedman J. Platelets in hemostasis and thrombosis: role of integrins and their ligands. Transfusion and Apheresis Science. 2003;28:257–264. doi: 10.1016/S1473-0502(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 37.Zou XY, Patil VRS, Dagia NM, Smith LA, Wargo MJ, Interliggi KA, et al. Psgl-1 derived from human neutrophils is a high-efficiency ligand for endothelium-expressed e-selectin under flow. American Journal Physiological-Cell Ph. 2005;289:C415–C424. doi: 10.1152/ajpcell.00289.2004. [DOI] [PubMed] [Google Scholar]
- 38.Eniola AO, Hammer DA. Characterization of biodegradable drug delivery vehicles with the adhesive properties of leukocytes II: effect of degradation on targeting activity. Biomaterials. 2005;26:661–670. doi: 10.1016/j.biomaterials.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Blackwell JE, Dagia NM, Dickerson JB, Berg EL, Goetz DJ. Ligand coated nanosphere adhesion to e-and p-selectin under static and flow conditions. Annals of Biomedical Engineering. 2001;29:523–533. doi: 10.1114/1.1376697. [DOI] [PubMed] [Google Scholar]
- 40.Smith CA, de la Fuente J, Pelaz B, Furlani EP, Mullin M, Berry CC. The effect of static magnetic fields and tat peptides on cellular and nuclear uptake of magnetic nanoparticles. Biomaterials. 2010;31:4392–4400. doi: 10.1016/j.biomaterials.2010.01.096. [DOI] [PubMed] [Google Scholar]
- 41.Medintz IL, Pons T, Delehanty JB, Susumu K, Brunel FM, Dawson PE, et al. Intracellular delivery of quantum dot-protein cargos mediated by cell penetrating peptides. Bioconjugate Chemistry. 2008;19:1785–1795. doi: 10.1021/bc800089r. [DOI] [PubMed] [Google Scholar]
- 42.Zhang K, Fang H, Chen Z, Taylor JS, Wooley KL. Shape effects of nanoparticles conjugated with cell-penetrating peptides (HIV Tat PTD) on CHO cell uptake. Bioconjugate Chemistry. 2008;19:1880–1887. doi: 10.1021/bc800160b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foerg C, Merkle HP. On the biomedical promise of cell penetrating peptides: limits versus prospects. Journal of Pharmaceutical Sciences. 2008;97:144–162. doi: 10.1002/jps.21117. [DOI] [PubMed] [Google Scholar]
- 44.Torchilin VP. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers. 2008;90:604–610. doi: 10.1002/bip.20989. [DOI] [PubMed] [Google Scholar]
- 45.Tsourkas A, Shinde-Patil VR, Kelly KA, Patel P, Wolley A, Allport JR, et al. In vivo imaging of activated endothelium using an anti-VCAM-1 magnetooptical probe. Bioconjugate Chemistry. 2005;16:576–581. doi: 10.1021/bc050002e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.